Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics



Abstract
:1. Introduction
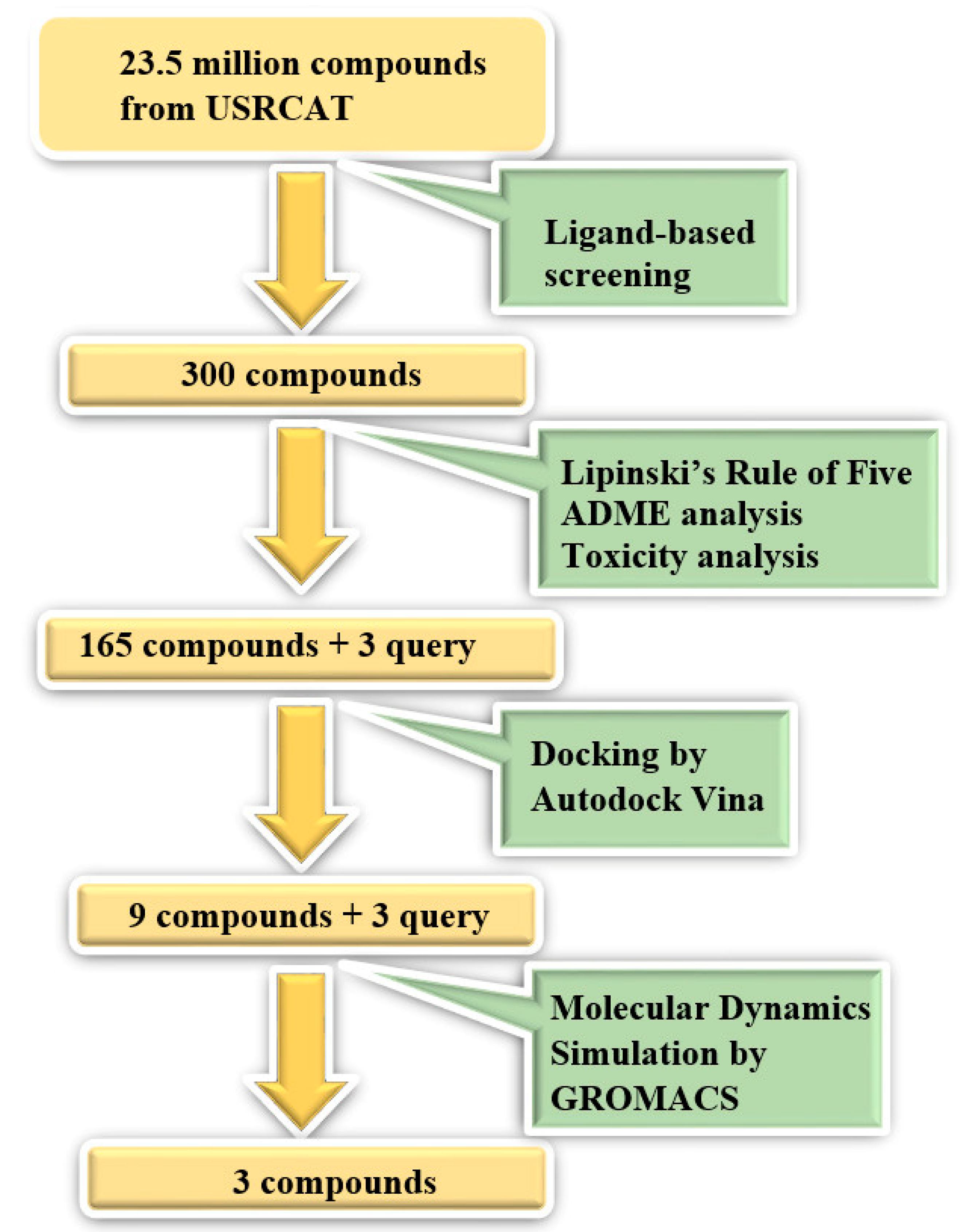
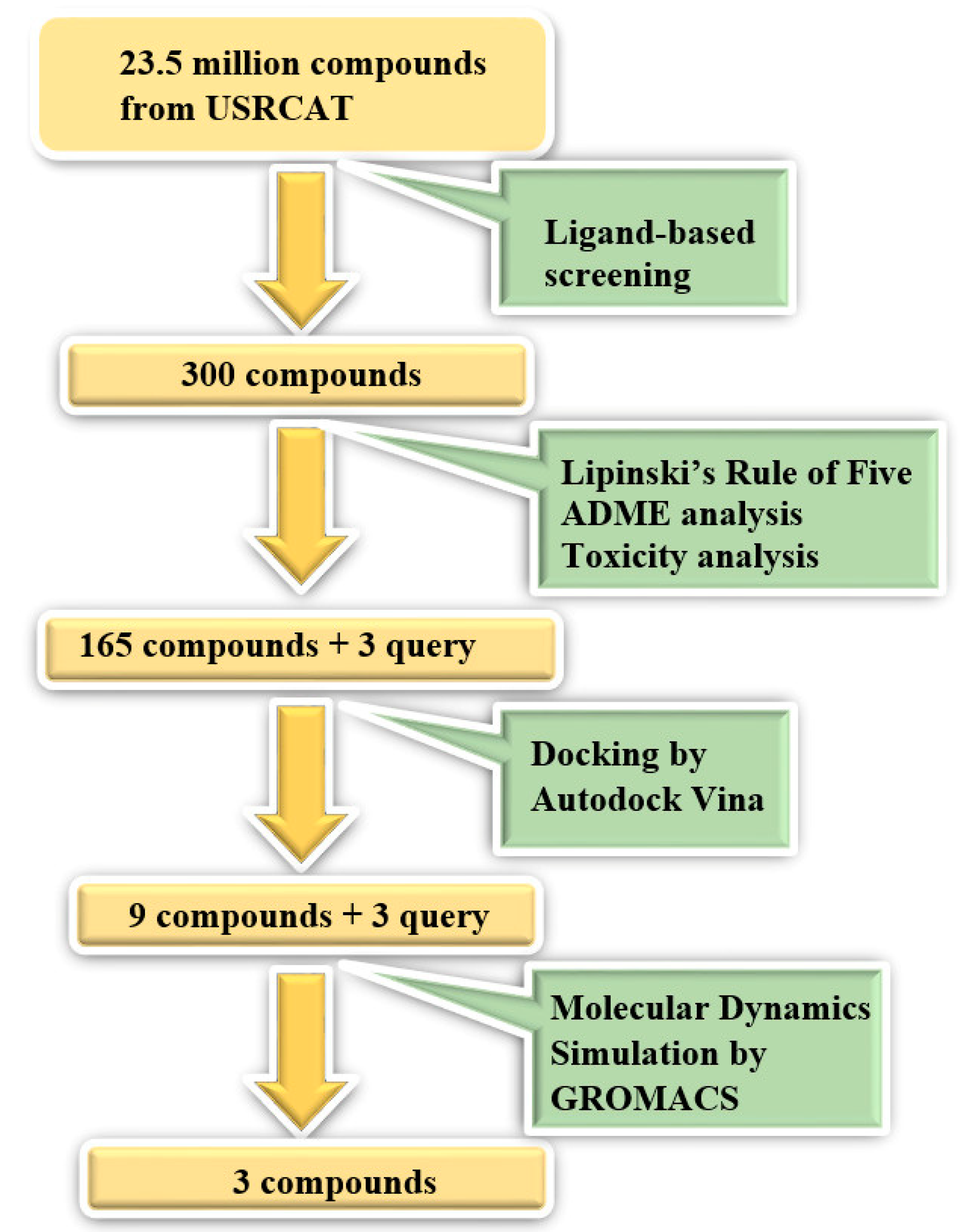
2. Materials and Methods
2.1. Ligand-Based Screening
2.1.1. Identification of Lead Molecules and Analogues
2.1.2. ADME Analysis
2.1.3. Toxicity Test
2.2. Structure-Based Screening
2.2.1. Retrieval and Structural Preparation of the Protein
2.2.2. Molecular Docking
2.3. Molecular Dynamics Simulation
3. Results and Discussion
3.1. Ligand-Based Screening
3.1.1. Identification of Analogues
3.1.2. Physiochemical Properties Analysis (ADME)
3.1.3. Toxicity Test
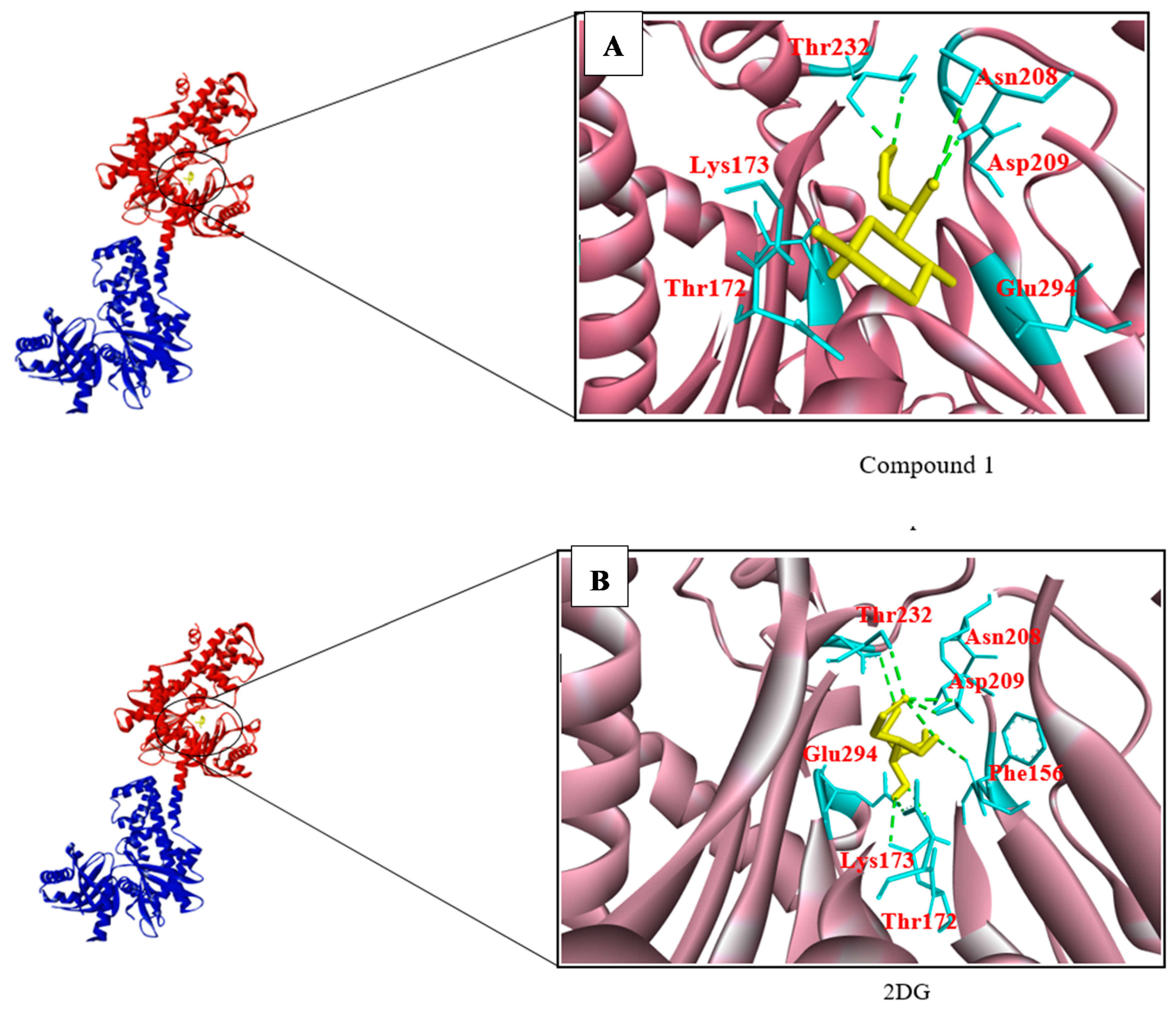
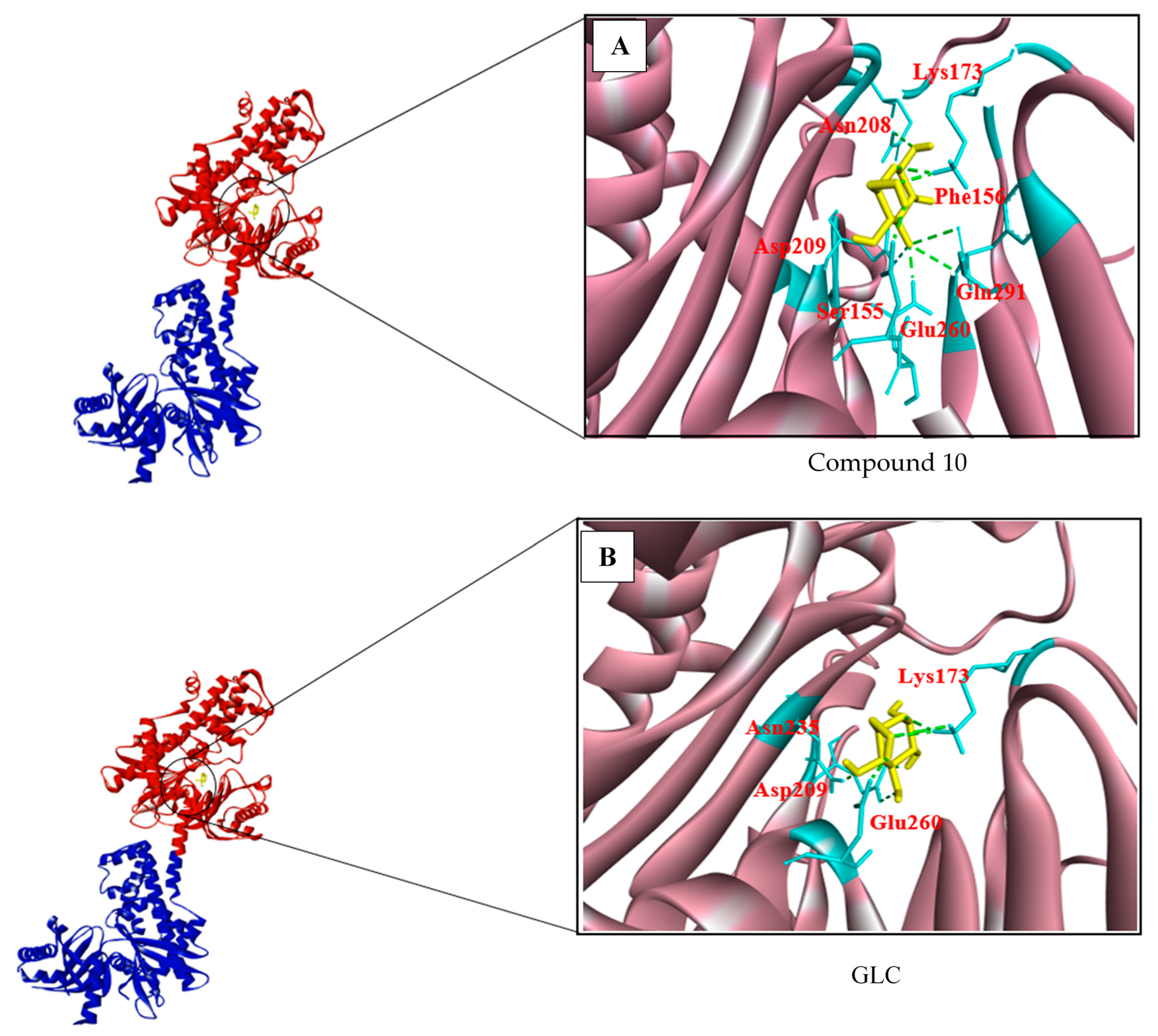
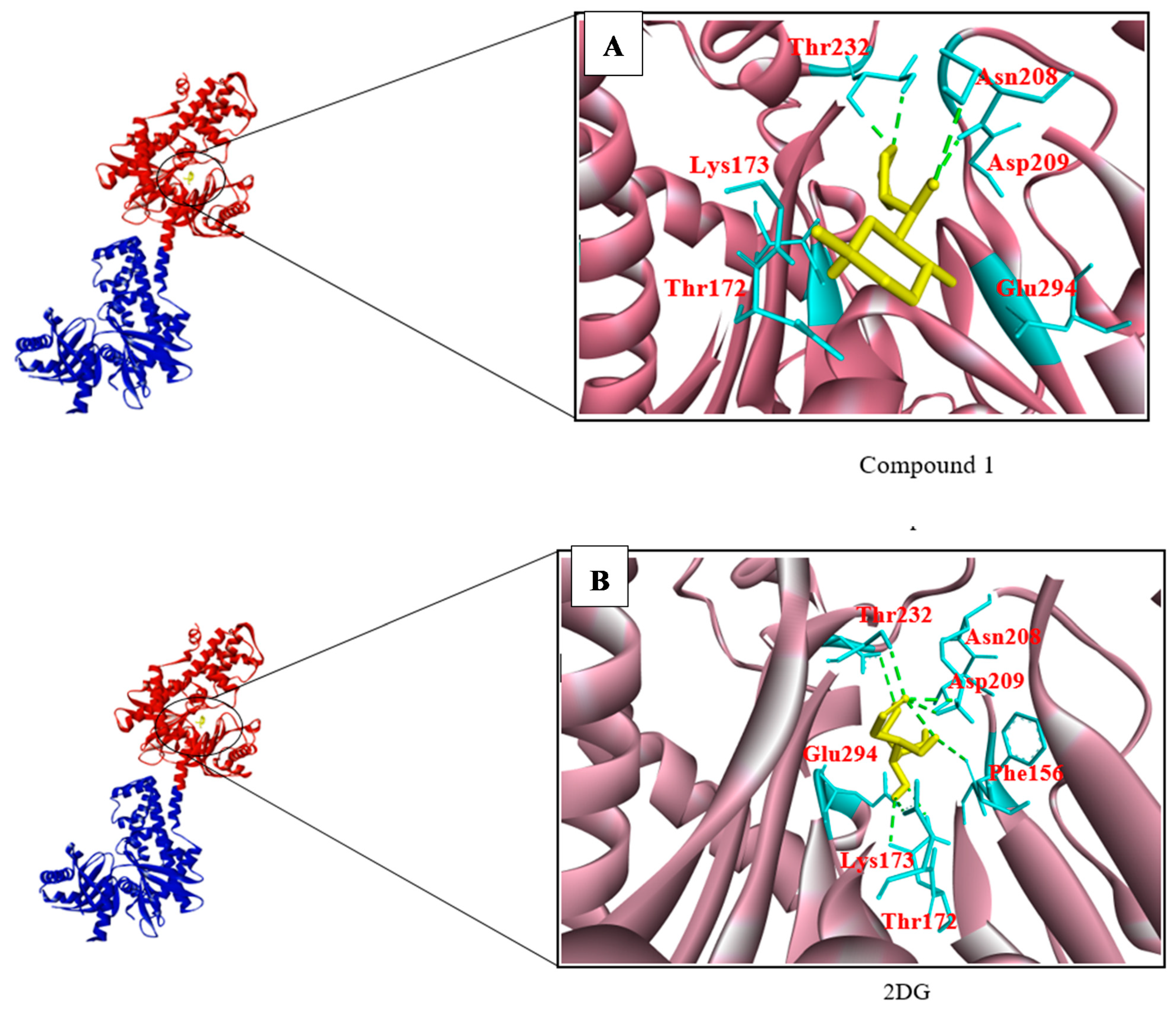
3.2. Structure-Based Screening
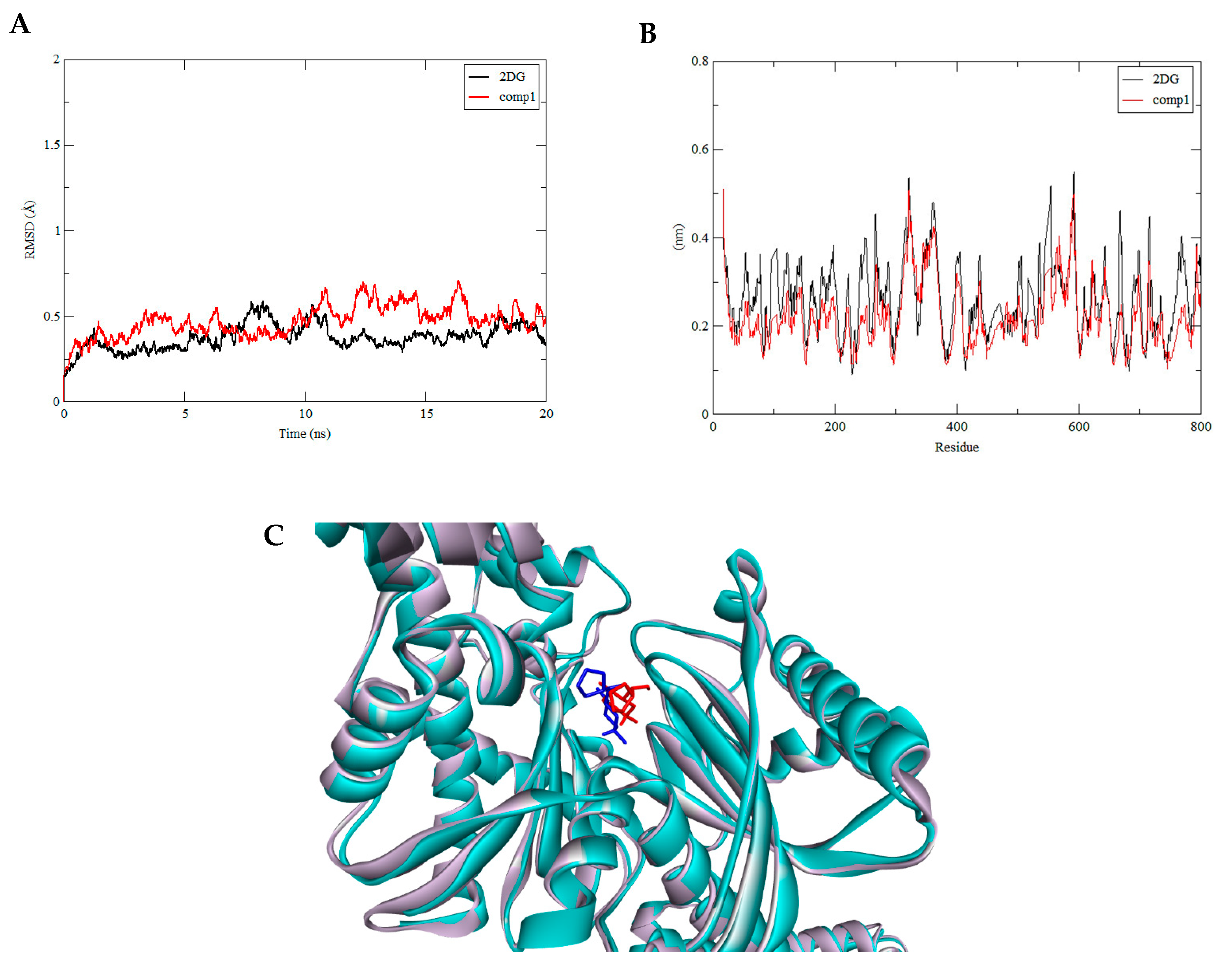
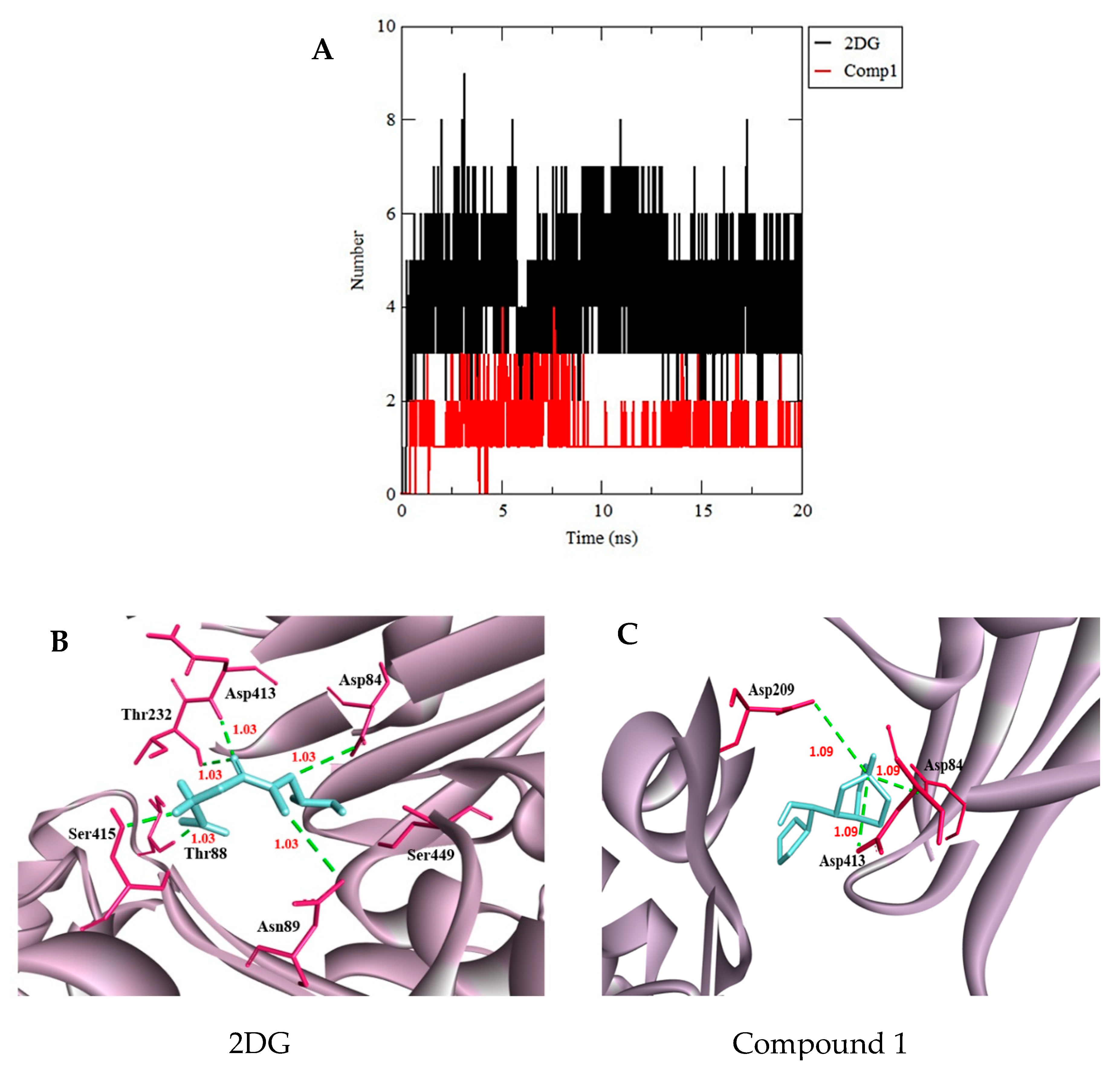
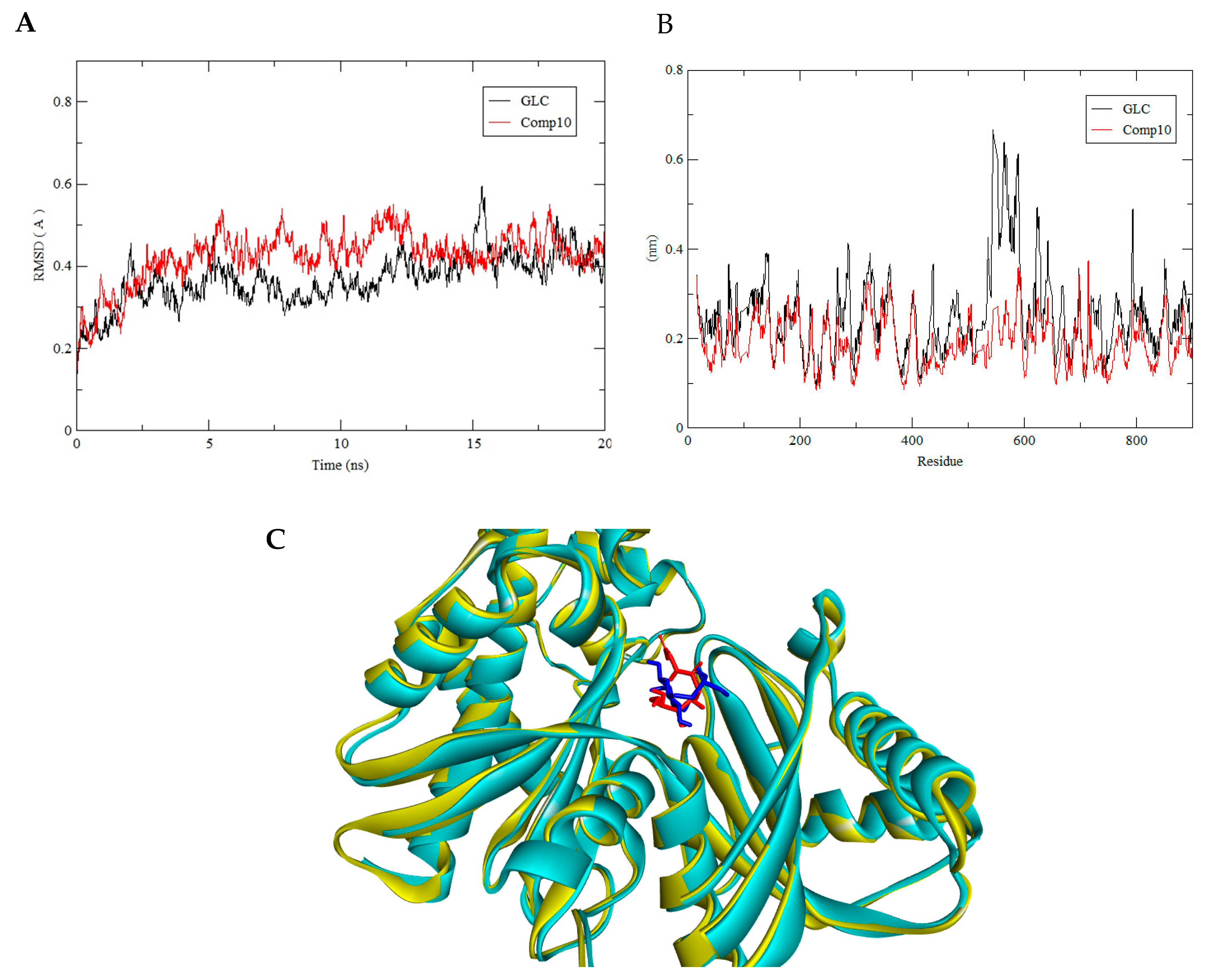
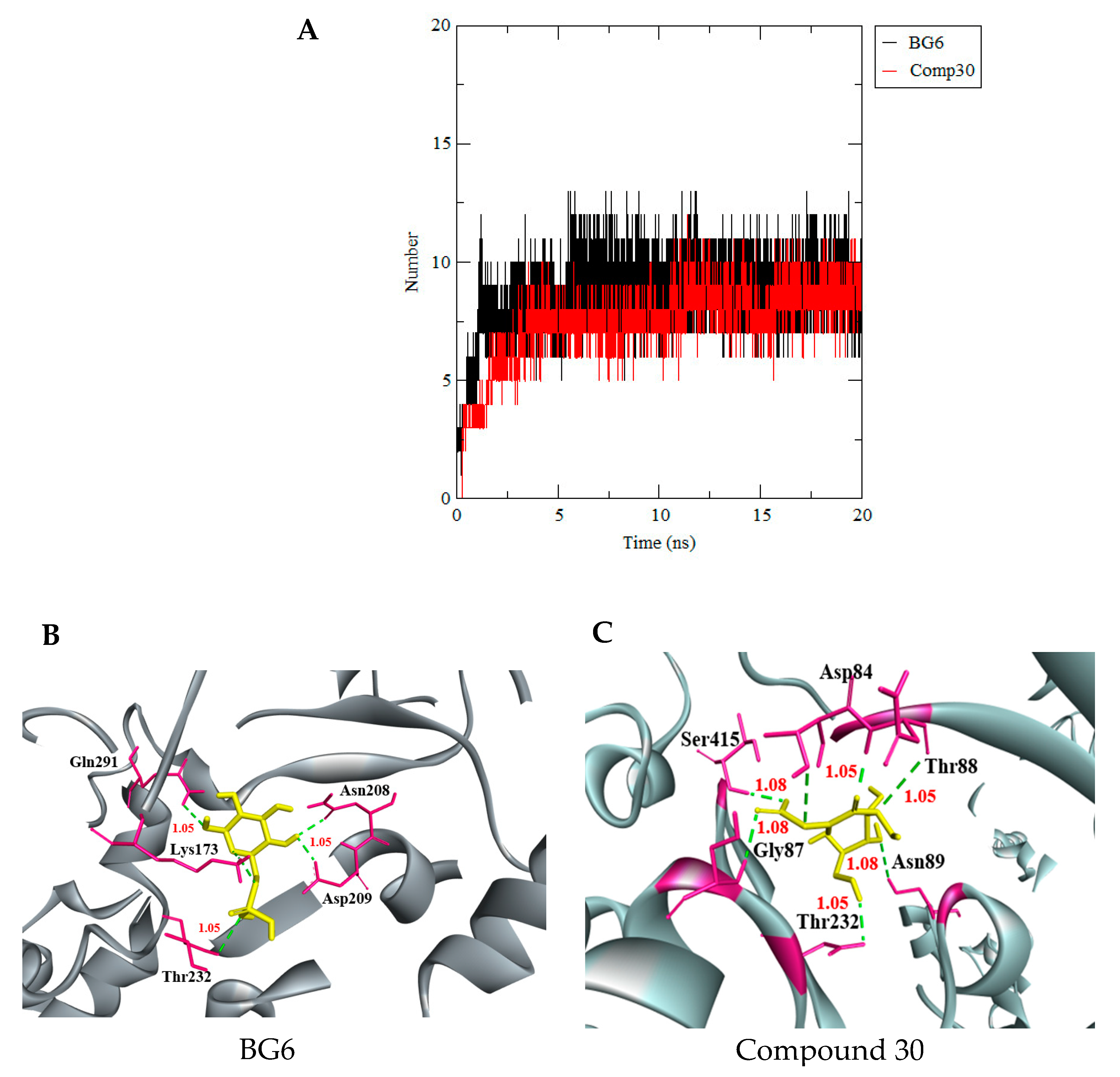
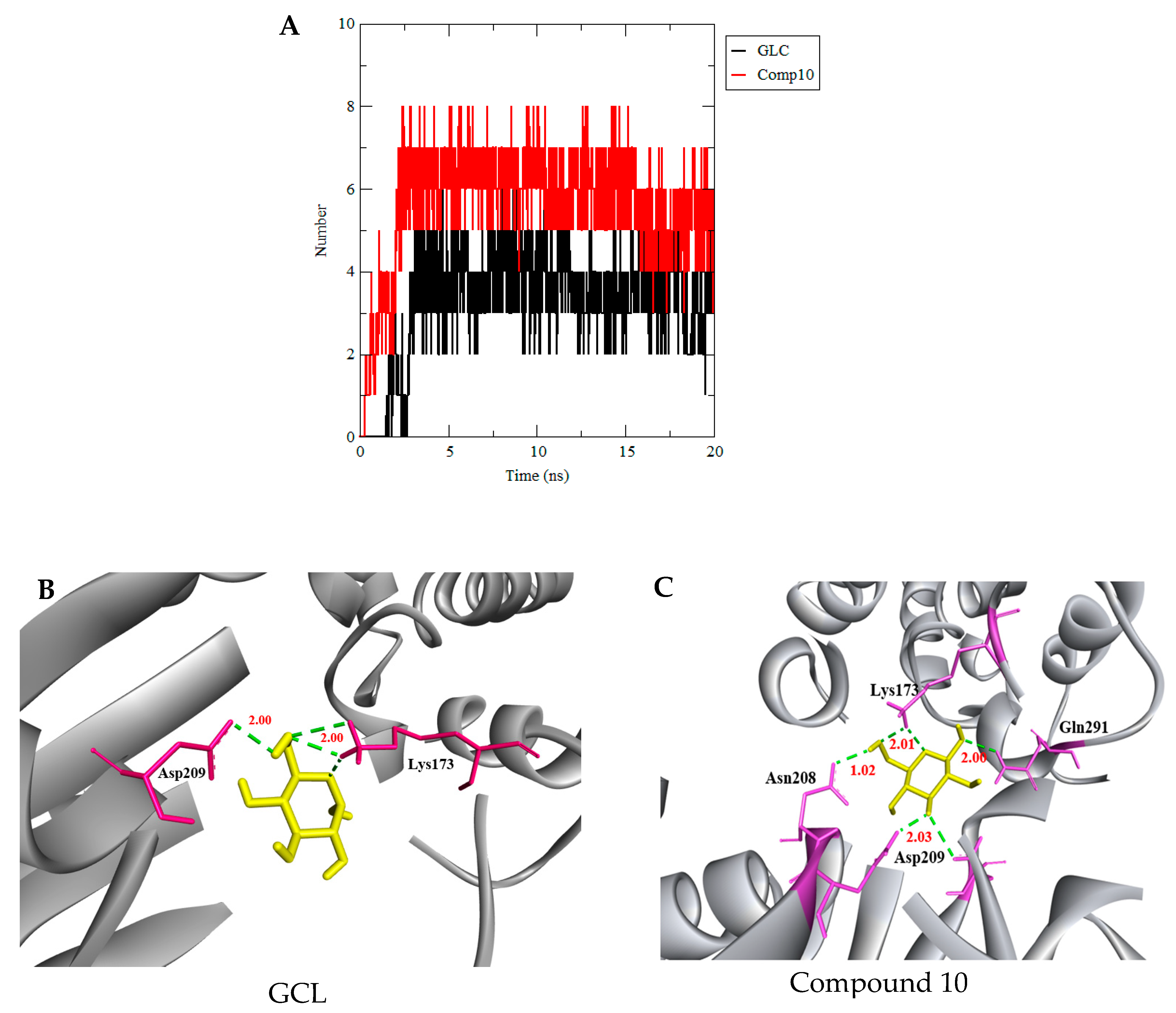
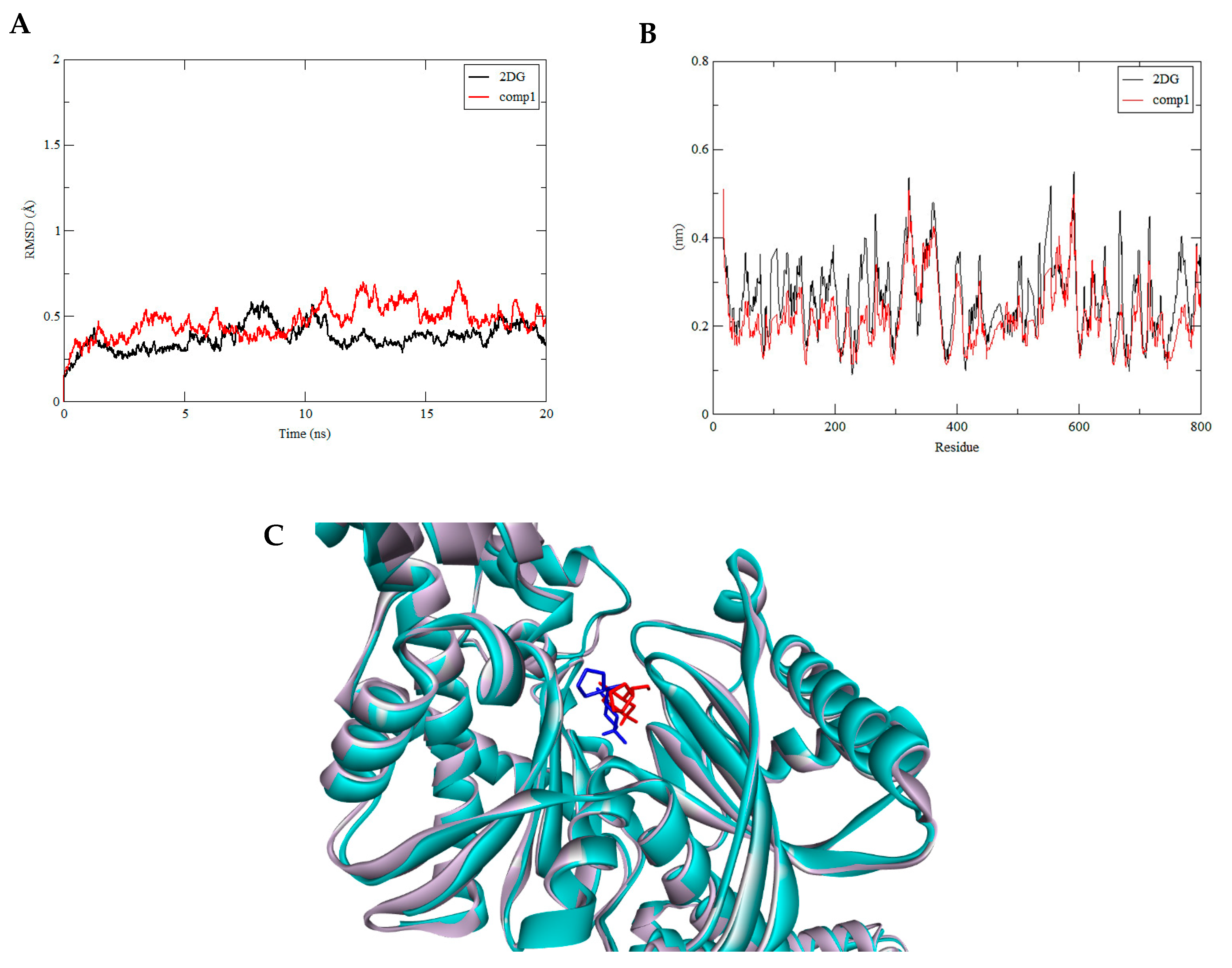
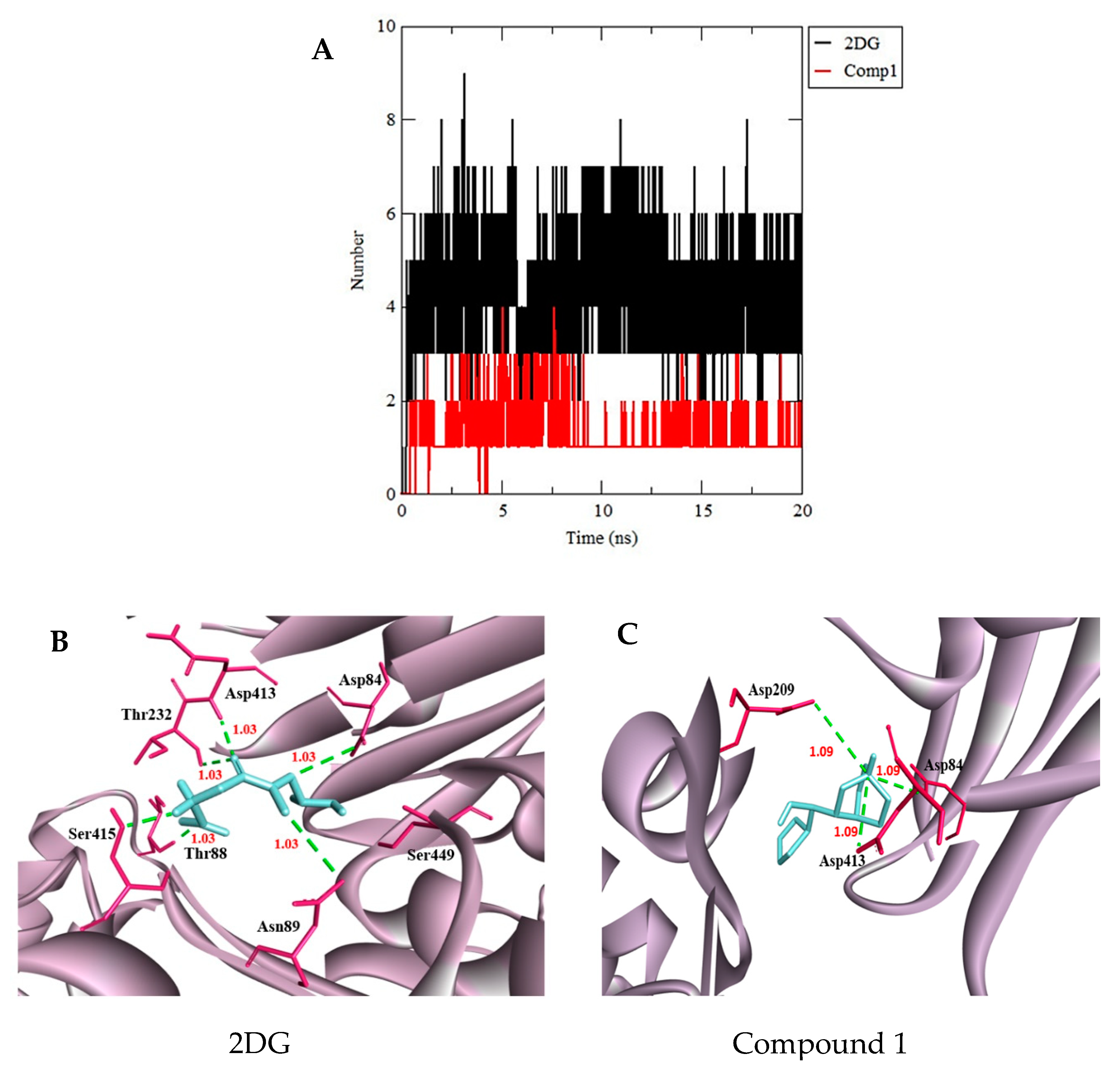
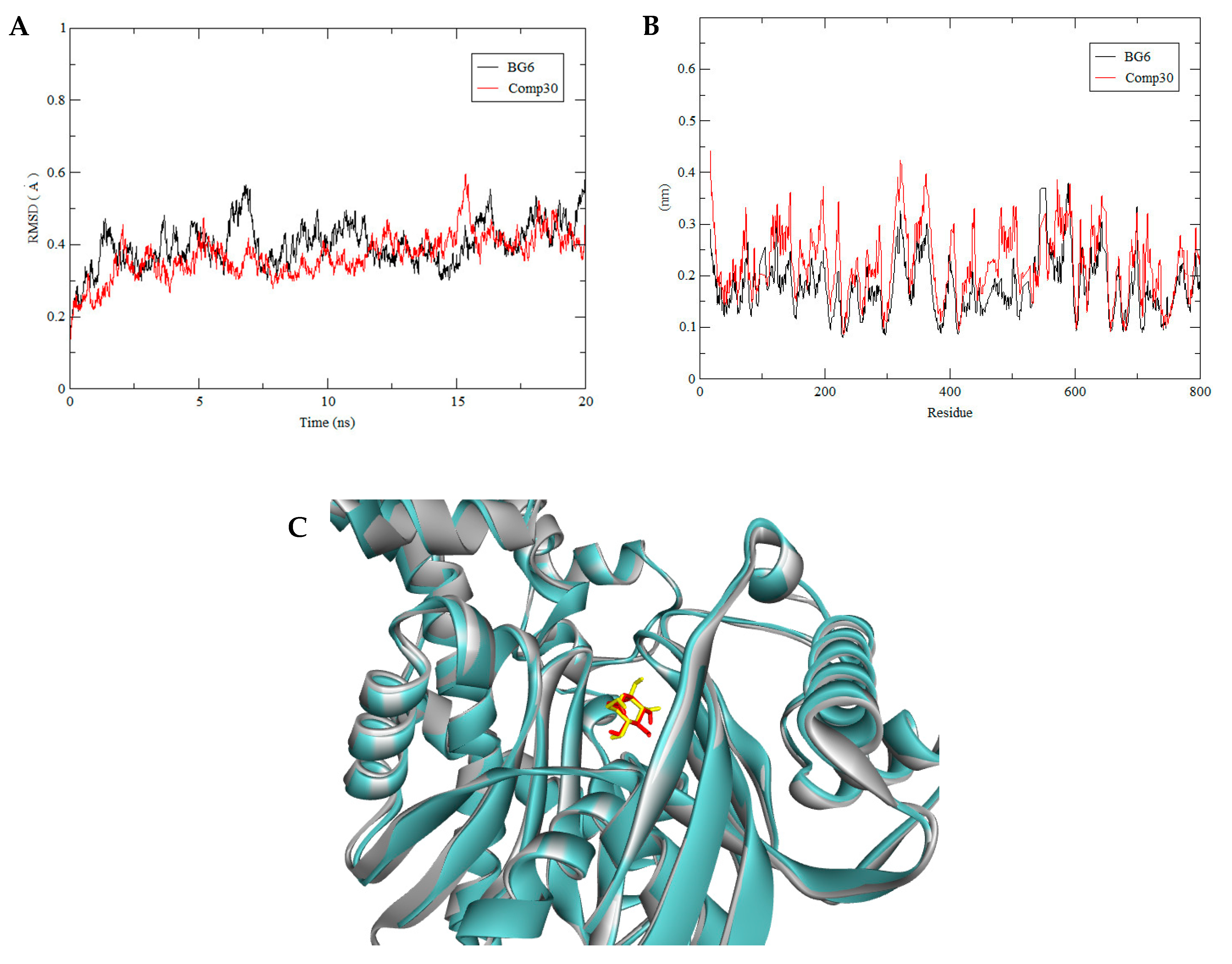
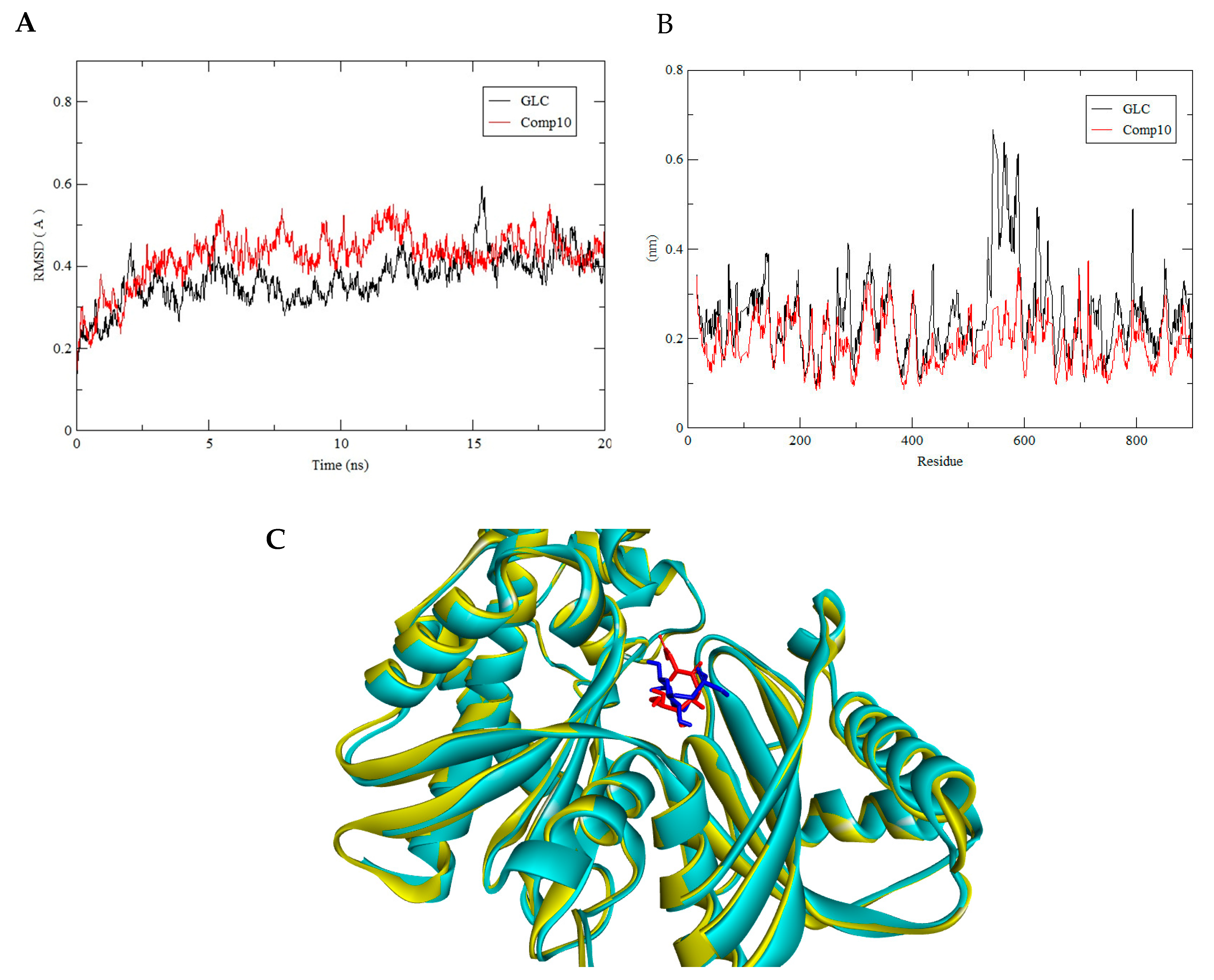
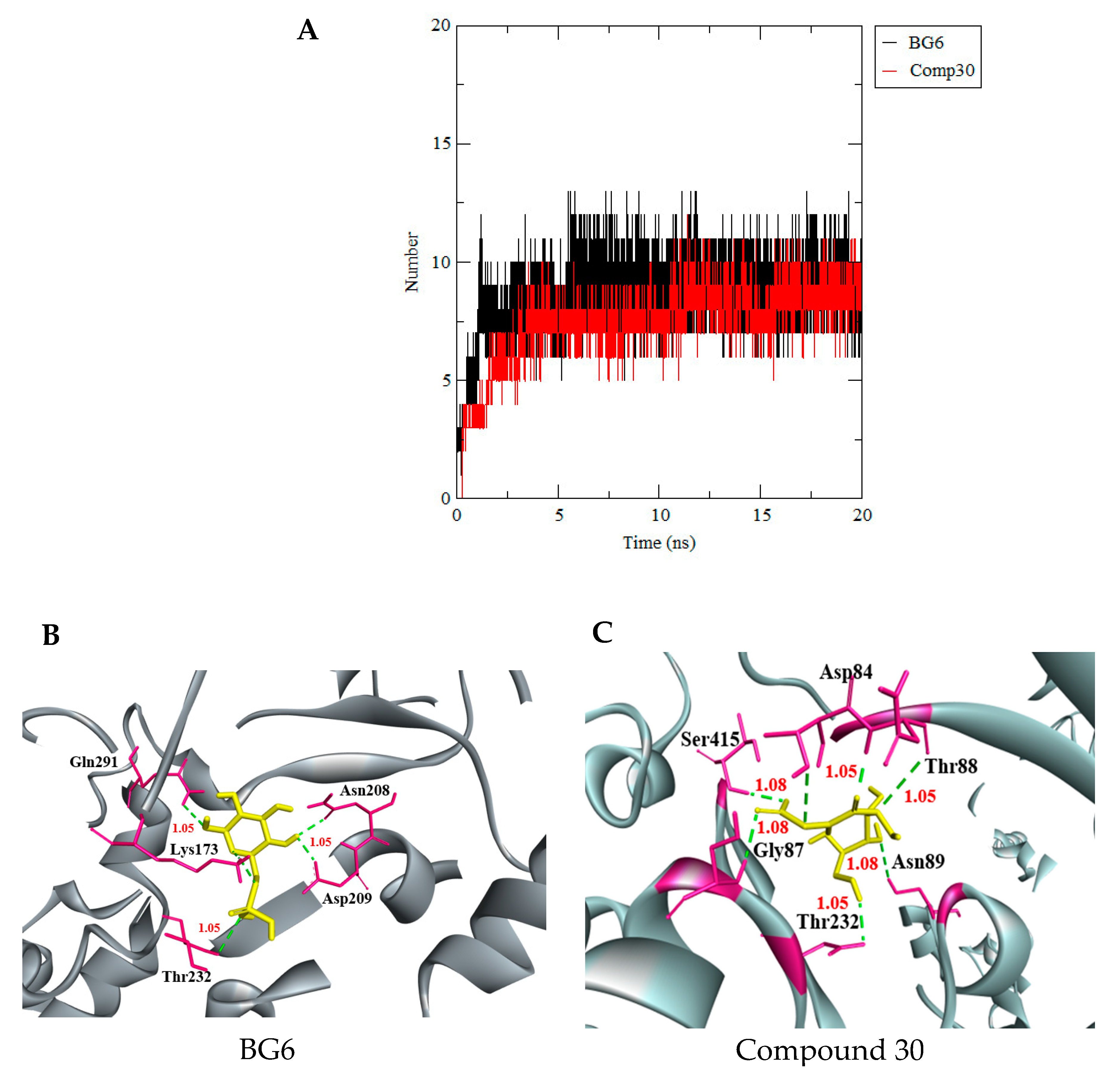
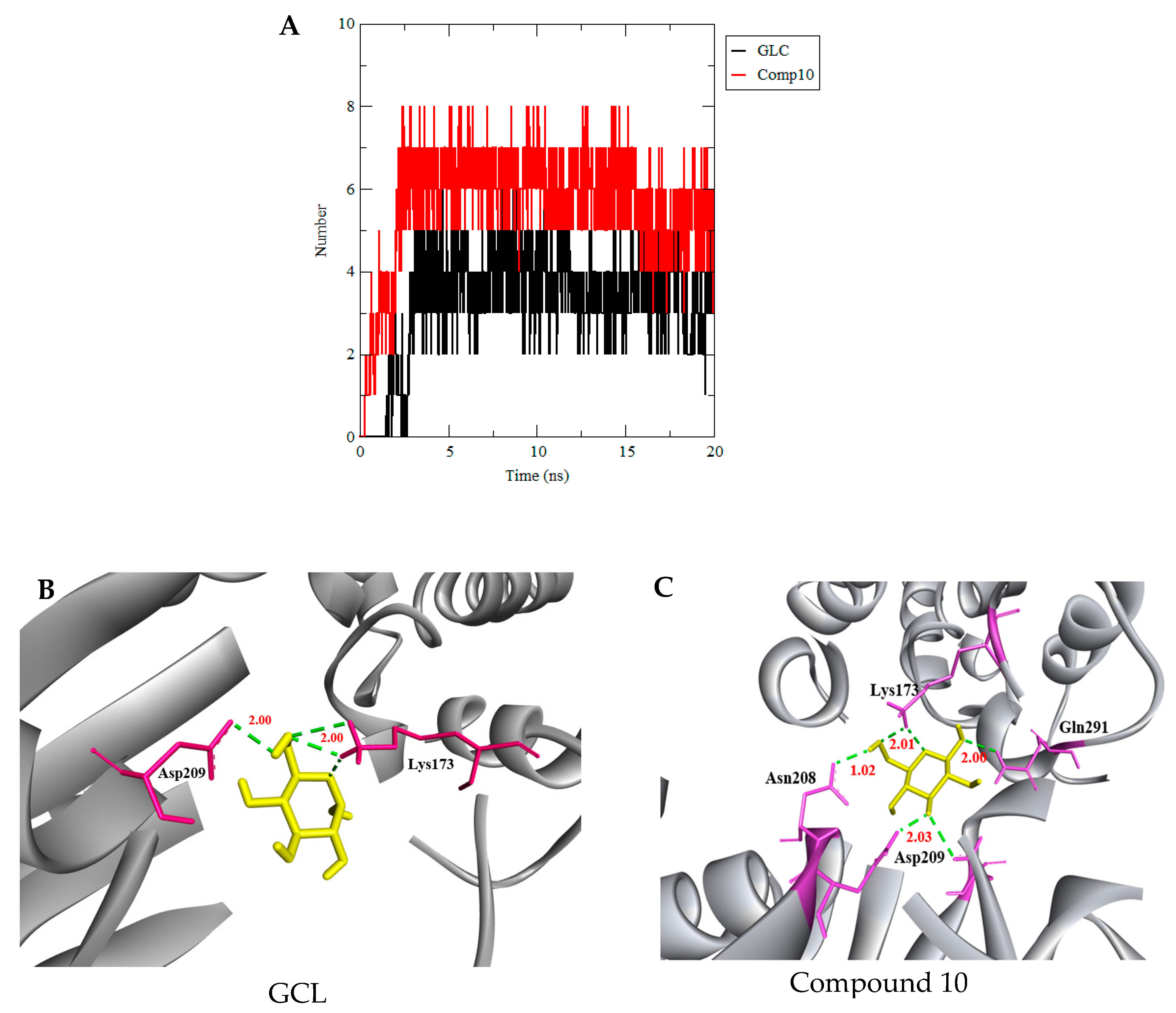
3.3. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henchal, E.A.; Putnak, R.J. The dengue viruses. Am. Soc. Microbiol. 1990, 4, 376–396. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Fernando, S.; Fernanoda, D.J.; Seneviratne, S.L. Dengue viral infections. J. Postgrad. Med. 2004, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Salles, T.S.; Sa-Guimaraes, T.E.; de Alvarenga, E.S.L.; Ribeiro, A.; de Meneses, M.D.F.; Moreire, M.F. History, epidemiology and diagnostics of dengue in the American and Brazillian context: A review. J. BioMed Cent. Parasites Vectors 2018, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Am. Soc. Microbiol. 1998, 11, 480–496. [Google Scholar] [CrossRef] [Green Version]
- Majeed, A.I.; Avabratha, S.K.; Lokesh, R.; Syeda, S. Clinicohaematological profile of dengue in children: A hospital based study. Int. J. Contemp. Pediatr. 2017, 4, 2349–3283. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Immunization, Vaccines and Biologicals. 2019. Available online: http://www.who.int/immunization/diseases/dengue/en/ (accessed on 7 July 2019).
- Pan America Health Organization. Dengue Epidemiology. 2019. Available online: https://www.paho.org/data/index.php/en/mnu-topics/indicadores-dengue-en.html (accessed on 3 March 2019).
- Smith, A.W.; Renhorn, E.K.; Tissera, H.; Bakar, A.S.; Alphey, L.; Kittayapong, P.; Lindsay, S.; Logan, J.; Hatz, C.; Reiter, P.; et al. Dengue tools: Innovative tools and strategies for the surveillance and control of dengue. Glob. Health Action 2014, 4, 256–321. [Google Scholar]
- Cheong, L.Y.; Burkart, K.; Leitao, J.P.; Lakes, T. Assessing weather effects on dengue diseases in Malaysia. Int. J. Environ. Res. Public Health 2013, 10, 6319–6334. [Google Scholar] [CrossRef]
- Perera, R.; Khun, J.R. Structural proteomics of Dengue virus. Natl. Inst. Health 2008, 11, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Wearing, H.J.; Pejman, R. Ecological and immunological determinants of dengue epidemics. Proc. Natl. Acad. Sci. USA 2006, 103, 11802–11807. [Google Scholar] [CrossRef] [Green Version]
- Vinodkumar, C.S.; Kalapannavar, N.K.; Basavarajappa, K.G.; Sanjay, D.; Gowli, C.; Nadig, G.; Prasad, B.S. Episode of coexisting infections with multiple dengue virus serotypes in central Karnataka, India. J. Infect. Public Health 2013, 6, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirusinfection. PLoS Pathog. 2006, 2, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Pierson, T.C. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia virus require glutamine but glucose for efficient replication. J. Virol. 2014, 88, 4366–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyler, H.; Honegger, A.; Bossler, F.; Sponagel, J.; Bulkescher, J.; Lohrey, C. Viral E6/E7 oncogene and cellular hexokinase 2 expressions in HPV-positive cancer cell lines. J. Oncotarget 2017, 8, 6342–6351. [Google Scholar]
- Sanchez, E.; Lagunoff, M. Viral Activation of Cellular Metabolism. J. Virol. 2015, 8, 4321–4334. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 2, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Zagorodna, O.; Martin, M.S.; Rutkowski, T.; Kuwana, T.; Sprite, R.; Kundoson, M. 2-Deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. HHS Public Access 2012, 31, 2738–2749. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [Green Version]
- Landini, M.P. Early enhanced glucose uptake in human cytomegalovirus-infected virus. Virology 1984, 13, 68–77. [Google Scholar]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. Virology 1997, 78, 1527–1531. [Google Scholar] [CrossRef] [Green Version]
- McArdle, J.; Schafer, X.L.; Munger, J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J. Virol. 2011, 85, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLauchlan, J. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta 2009, 1791, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.N.; Durury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of primary metabolic block produced by 2-Doxygulcose. J. Biol. Chem. 1956, 224, 963–969. [Google Scholar]
- Godói, I.P.; Lovato, L.; Lemo, P.; Araújo, V.E.; Bonoto, B.C.; Godman, J.G. CYD-TDV dengue vaccine: Systematic review and meta-analysis of efficacy, immunogenicity and safety. J. Comp. Eff. Res. 2008, 61, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy-Kanniappan, S.; Geschwind, J.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, N.; Lee, S.J.; Choi, T.G.; Bai, K.Y.; Uhm, H.S.; Kim, C.H.; Choi, H.E. Non-thermal plasma with 2-deoxy-D-glucose synergistically induces cell death by targeting glycolysis in blood cancer cells. Sci. Rep. 2015, 5, 8726. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Lin, X.; Bradbury, M.; Kaushal, A.; Li, L.; Spritz, R.; Gius, D. 2-Deoxy-D-Glucose-induced Cytotoxicity and Radiosensitization in Tumor Cells Is Mediated via Disruptions in Thiol Metabolism. Cancer Res. Assoc. 2003, 63, 3413–3417. [Google Scholar]
- Vyas, V.; Jain, A.; Jain, A.V.; Gupta, A. Virtual screening: A fast tool for drug design. Sci. Pharm. 2008, 76, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kim, C.S.; Honzatko, B.; Fromm, H.J. Dual Mechanism for glucose 6-phosphate inhibition of human brain hexokinase. J. Biol. Chem. 1999, 274, 31155–31159. [Google Scholar] [CrossRef] [Green Version]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Lengauer, T.; Lemmen, C.; Rarey, M.; Zimmermann, M. Novel technologies for virtual screening. J. Natl. Libr. Med. 2004, 9, 27–34. [Google Scholar] [CrossRef]
- Lavecchia, A.; Giovannni, C.D. Virtual screening in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 60–71. [Google Scholar] [CrossRef]
- Nawaz, H.M.; Ferreira, J.C.; Nedyalkova, L.; Zhu, H.; Lopez, C.C.; Kirmiziatin, S. The catalytic inactivation of the N-half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. J. Biosci. Rep. 2018, 38, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Printz, R.L.; Osawa, H.; Ardehali, H.; Koch, S.; Granner, D.K. Hexokinase II gene: Structure, regulation and promoter organization. Biochem. Soc. Trans. 1997, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Savic, L.J.; Chapiro, J.; Duwe, G.; Geschwind, J. Targeting glucose metabolism in cancer: A new class of agents for loco-regional and systemic therapy of liver cancer and beyond. Hepatic Oncol. 2016, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles applications and recent advances. Curr. Topics Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminform. 2012, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Tsaioun, K.; Blaaubor, J.B.; Hartung, T. Evidence-Based Absorption, Distribution, Metabolism, Excretion (ADME) and its Interplay with Alternative Toxicity Methods. J. Pharm. Bioallied Sci. 2016, 33, 333–336. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Pezzullo, M.; De Burghgraeve, T.; Kaptein, S.; Pastorino, B.; Dallmeier, K.; Bolognesi, M. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: New prospects for an old drug. J. Antimicrob. Chemother. 2012, 67, 1884–1894. [Google Scholar] [CrossRef] [Green Version]
- Basavannacharya, C.; Vasudevan, S.G. Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem. Biophys. Res. Commun. 2014, 453, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 1998, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckeert, O.A.; Schrey, K.A.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. J. Nucleic Acids Res. 2018, 46, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arome, D.; Chinedu, E. The importance of toxicity testing. J. Pharm. BioSci. 2014, 41, 46–48. [Google Scholar]
- Ruiz, P.; Begluitti, G.; Wheeler, J.; Mumtaz, M. Prediction of acute mammalian toxicity using QSAR method: A case study of sulfur mustard and its breakdown product. J. Mol. 2012, 17, 8982–9001. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.D.; Camacho, J.C. Shape-Based Virtual Screening with Volumetric Aligned Molecular Shapes. J. Natl. Inst. Health 2014, 35, 1824–1834. [Google Scholar] [CrossRef] [Green Version]
- Maia, B.H.E.; Assis, C.L.; Oliveira, T.A.; Silva, M.A.; Taranto, G.A. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. J. Med. Pharm. Chem. 2020, 8, 334–338. [Google Scholar] [CrossRef]
- Lim, S.V.; Basyaruddin, M.; Rahman, A.; Tejo, B.A. Structure-based and ligand-based virtual screening of novel methyltransferase inhibitors. Asia Pac. Bioinform. 2011, 12, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.Y.; Kondreddi, R.R.; Xie, X.; Rao, R.; Nilar, S.; Xu, H.Y.; Lakshminarayana, S.B. A translation inhibitor that suppresses dengue virus in vitro and in vivo. Am. Soc. Microbiol. 2011, 55, 4072–4080. [Google Scholar]
- Sing, J.; Kumar, M.; Mansuri, R.; Sahoo, C.G.; Deep, A. Inhibitor desingning, virtual screening and docking studies for methyltransferase: A potential target against dengue virus. J. Pharm. Bioallied Sci. 2015, 34, 2–8. [Google Scholar]
- Kouretova, J.; Hammamy, Z.M.; Epp, A.; Hardes, K.; Kallis, S.; Zhang, L.; Steinmetzer, T. Effects of NS2B-NS3 protease and furin inhibition on West Nile and Dengue virus replication. J. Enzym. Inhib. Med. Chem. 2017, 32, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Halim, S.A.; Khan, H.; Khan, A.; Wadood, A.; Mabood, F.; Hussain, J.; Al-Harrasi, A. Targeting dengue virus NS-3 helicase by ligand based pharmacophore modeling and structural based virtual screening. Front. Chem. 2017, 5, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, C.M.; Dai, D.; Grosenbach, D.W.; Berhan, A.; Jones, K.F.; Cardwell, K.B.; Jordan, R. A novel inhibitor of dengue virus replication that targets the capsid protein. Am. Soc. Microbiol. J. 2018, 57, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitby, K.; Pierson, T.C.; Geiss, B.; Lane, K.; Engle, M.; Zhou, Y.; Diamond, M.S. Castanospermine a potent inhibitor of dengue virus infection In vitro and In vivo. Am. Soc. Microbiol. 2005, 79, 8698–8706. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.C.; Hu, H.S.; Wu, R.H.; Wu, S.H.; Lee, S.J.; Jiaang, W.T.; Yueh, A. A novel dengue virus inhibitors BP13944, discovered by High-throughput screening with dengue virus replicon cells selectes for resistance in the viral NS2B/NS3 protease. Am. Soc. Microbiol. 2013, 58, 110–119. [Google Scholar]
- Ismail, A.N.; Jusoh, A.S. Molecular Docking and Molecular Dynamics Simulation Studies to Predict Flavonoid Binding on the Surface of DENV2 E Protein. J. Comput. Life Sci. 2017, 9, 499–511. [Google Scholar] [CrossRef]
- Shanmugam, A.; Velmurugan, D.; Gromiha, M. Identification of Dengue Viral RNA Dependent RNA Polymerase Inhibitor using Computational Fragment Based Approaches and Molecular Dynamics Study. J. Biomol. Struct. Dyn. 2015, 2, 1–10. [Google Scholar]
- Shanmugam, A.; Gromiha, M.M. Quercetin derivatives as non-nucleoside inhibitors for dengue polymerase: Molecular docking, molecular dynamics simulation and binding free energy calculation. J. Biomol. Struct. Dyn. 2016, 5, 2895–2909. [Google Scholar]
- Tambunan, S.U.; Zahroar, H.; Utomo, B.B.; Parikesit, A.A. Screening of commercial cyclic peptide as inhibitor NS5 methyltransferase of Dengue virus through Molecular Docking and Molecular Dynamics Simulation. J. Bioinform. 2014, 10, 23–27. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, H.; Bastos, M.D.I.; Ribeiro, M.B.; Maigret, B.; Santana, M.J. New Binding Site Conformations of the Dengue Virus NS3 Protease. Accessed by Molecular Dynamics Simulation. PLoS ONE 2013, 8, e72402. [Google Scholar] [CrossRef] [Green Version]
- Tambunan, S.U.; Nasution, F.A.; Azhima, F.; Parikesti, A.A.; Toepak, P.E.; Idrus, S. Modification of S-Adenosyl-l-Homocysteine as Inhibitor of Nonstructural Protein 5 Methyltransferase Dengue Virus Through Molecular Docking and Molecular Dynamics Simulation. J. Bioinform. 2017, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnam, M.A.M.; Nitsche, C.; Vechi, M.S.; Klein, D.C. C-terminal residue optimization and fragment merging discovery of a potent peptide-hybrid inhibitior of dengue protease. Am. Chem. Soc. 2014, 5, 1037–1042. [Google Scholar]
- Mirza, S.B.; Salmas, R.K.; Fatmi, M.Q.; Durdagi, S. Virtual screening of eighteen million compounds against dengue virus: Combined molecular docking and molecular dynamics simulation study. J. Mol. Graph. Model. 2016, 66, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Luzhkov, V.; Decroly, E.; Canarda, B.; Selisko, B.; Aqvist, J. Evaluation of Adamantane Derivatives as Inhibitors of Dengue Virus mRNA Cap Methyltransferase by Dockingand Molecular Dynamics Simulations. J. Mol. Inform. 2013, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | ZINC ID | 2D Structure | Similarity Score | Number of HBD/HBA | Molecular Weight (g/mol) | MlogP | Drug-Likeness |
|---|---|---|---|---|---|---|---|
| GLC | 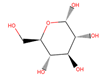 | 5/6 | 180.156 | −3.21 | 0.01 | ||
| 10 | 3,956,760 |  | 0.946 | 4/5 | 182.147 | −2.15 | 0.14 |
| 26 | 16,159,409 | 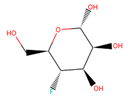 | 0.933 | 4/5 | 182.147 | −2.15 | 0.09 |
| 58 | 3,809,846 |  | 0.913 | 4/5 | 182.147 | −1.93 | −0.01 |
| Compound Number | ZINC ID | 2D Structure | Similarity Score | Number of HBD/HBA | Molecular Weight (g/mol) | MlogP | Drug-Likeness |
|---|---|---|---|---|---|---|---|
| BG6 | 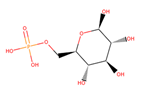 | 6/9 | 260.135 | −3.64 | −0.20 | ||
| 30 | 4,403,145 |  | 0.817 | 6/8 | 238.192 | −3.84 | 0.89 |
| 36 | 4,530,268 | 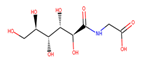 | 0.812 | 6/9 | 252.199 | −4.02 | 0.72 |
| 38 | 1,576,959 | 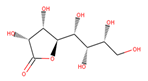 | 0.812 | 6/8 | 238.192 | −3.84 | −1.08 |
| Compound Number | ZINC ID | 2D Structure | Similarity Score | Number of HBD/HBA | Molecular Weight (g/mol) | MlogP | Drug-Likeness |
|---|---|---|---|---|---|---|---|
| 2DG | 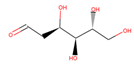 | 4/5 | 164.157 | −1.95 | −1.33 | ||
| 1 | 86,652,948 | 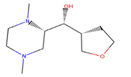 | 0.806 | 2/4 | 215.317 | 0.06 | −0.64 |
| 4 | 86,991,606 | 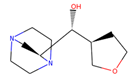 | 0.796 | 2/4 | 213.301 | −0.08 | −1.16 |
| 31 | 86,991,603 | 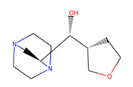 | 0.751 | 2/4 | 213.301 | −0.08 | −1.16 |
| Properties | Model Name | Units | GLC | ZINC 3956760 Compound 10 | ZINC16159409 Compound 26 | ZINC3809846 Compound 58 |
|---|---|---|---|---|---|---|
| Absorption | Water solubility | log mol/L | −1.377 | −1.08 | −1.005 | −1.581 |
| Caco2 permeability | Log 10−6 cm/s | −0.249 | 0.251 | 0.315 | 0.214 | |
| Intestinal absorption (human) | %(Absorbed) | 21.51 | 59.553 | 59.79 | 59.316 | |
| Skin Permeability | logKp | −3.041 | −3.044 | −3.107 | −2.939 | |
| Distribution | Fraction unbound (human) | logL/Kg | 0.82 | 0.841 | 0.84 | 0.853 |
| BBB permeability | logBB | −0.943 | −0.786 | −0.9 | −0.872 | |
| CNS permeability | logPS | −3.636 | −3.552 | −3.552 | −3.192 | |
| Metabolism | CYP2D6 | Yes/No | No | No | No | No |
| CYP1A2 | Yes/No | No | No | No | No | |
| Excretion | Oral Rat Acute Toxicity (LD50) | mol/kg | 0.626 | 0.535 | 0.535 | 0.534 |
| Renal OCT2 substrate | Yes/No | No | No | No | No |
| Properties | Model Name | Units | BG6 | ZINC 4403145 Compound 30 | ZINC 4530268 Compound 36 | ZINC1576959 Compound 38 |
|---|---|---|---|---|---|---|
| Absorption | Water solubility | log mol/L | −0.811 | −1.753 | −1.995 | −1.753 |
| Caco2 permeability | Log 10−6 cm/s | −0.341 | −0.104 | −0.371 | −0.104 | |
| Intestinal absorption (human) | %(Absorbed) | 35.51 | 26.376 | 0 | 26.376 | |
| Skin Permeability | logKp | −2.82 | −2.744 | −2.735 | −2.744 | |
| Distribution | Fraction unbound (human) | logL/Kg | 0.716 | 0.79 | 0.886 | 0.232 |
| BBB permeability | logBB | −1.414 | −1.032 | −0.928 | −1.032 | |
| CNS permeability | logPS | −4.211 | −3.46 | −3.604 | −3.46 | |
| Metabolism | CYP2D6 | Yes/No | No | No | No | No |
| CYP1A2 | Yes/No | No | No | No | No | |
| Excretion | Oral Rat Acute Toxicity (LD50) | mol/kg | 0.414 | 0.787 | 1.056 | 0.787 |
| Renal OCT2 substrate | Yes/No | No | No | No | No |
| Properties | Model Name | Units | 2DG | ZINC86652948 Compound 1 | ZINC 86991606 Compound 4 | ZINC86991603 Compound 31 |
|---|---|---|---|---|---|---|
| Absorption | Water solubility | log mol/L | −0.512 | −1.987 | −1.654 | −1.376 |
| Caco2 permeability | Log 10−6 cm/s | 1.559 | 1.985 | 1.654 | 1.098 | |
| Intestinal absorption (human) | %(Absorbed) | 86.433 | 86.983 | 87.984 | 86.764 | |
| Skin Permeability | logKp | −3.41 | −3.98 | −2.34 | −2.75 | |
| Distribution | Fraction unbound (human) | logL/Kg | 0.916 | 0.914 | 0.158 | 0.985 |
| BBB permeability | logBB | −0.043 | −0.025 | −0.265 | −0.265 | |
| CNS permeability | logPS | −3.434 | −3.254 | −3.214 | −3.147 | |
| Metabolism | CYP2D6 | Yes/No | No | No | No | No |
| CYP1A2 | Yes/No | No | No | No | No | |
| Excretion | Oral Rat Acute Toxicity (LD50) | mol/kg | 1.153 | 1.258 | 1.245 | 1.452 |
| Renal OCT2 substrate | Yes/No | No | No | No | No |
| ZINC ID | Classification | |||||
|---|---|---|---|---|---|---|
| Acute Toxicity | Toxicity End Point | Organ Toxicity | ||||
| Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||
| GLC | I. Toxicity Class: 6 II. LD50:2300 mg/kg III. accuracy: 70.97% | Inactive Ps: 0.82 | Inactive Ps: 0.99 | Inactive Ps: 0.87 | Inactive Ps: 0.81 | Inactive Ps: 0.98 |
| 3956760 Compound 10 | I. Toxicity Class: 6 II. LD50:14,388 mg/kg III. accuracy: 67.38% | Inactive Ps: 0.67 | Inactive Ps: 0.99 | Inactive Ps: 0.68 | Inactive Ps: 0.68 | Inactive Ps: 0.98 |
| 16159409 Compound 26 | I. Toxicity Class: 6 II. LD50:2300 mg/kg III. accuracy: 68.07% | Inactive Ps: 0.73 | Inactive Ps: 0.98 | Inactive Ps: 0.68 | Inactive Ps: 0.64 | Inactive Ps: 0.89 |
| 3809846 Compound 58 | I. Toxicity Class: 6 II. LD50:14,388 mg/kg III. accuracy: 67.38% | Inactive Ps: 0.73 | Inactive Ps: 0.98 | Inactive Ps: 0.68 | Inactive Ps: 0.64 | Inactive Ps: 0.89 |
| ZINC ID | Classification | |||||
|---|---|---|---|---|---|---|
| Acute Toxicity | Toxicity End Point | Organ Toxicity | ||||
| Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||
| BG6 | I. Toxicity Class: 4 II. LD50:1500 mg/kg III. accuracy: 67.38% | Inactive Ps: 0.74 | Inactive Ps: 0.99 | Inactive Ps:0.74 | Inactive Ps:0.81 | Inactive Ps:0.94 |
| 4403145 Compound 30 | I. Toxicity Class: 6 II. LD50:15,900 mg/kg III. accuracy: 70.97% | Inactive Ps: 0.89 | Inactive Ps: 0.99 | Inactive Ps: 0.85 | Inactive Ps: 0.68 | Inactive Ps: 0.94 |
| 4530268 Compound 36 | I. Toxicity Class: 6 II. LD50:5500 mg/kg III. accuracy: 69.26% | Inactive Ps: 0.69 | Inactive Ps: 0.99 | Inactive Ps: 0.73 | Inactive Ps: 0.68 | Inactive Ps: 0.93 |
| 1576959 Compound 38 | I. Toxicity Class: 6 II. LD50:15,900 mg/kg III. accuracy: 70.97% | Inactive Ps: 0.89 | Inactive Ps: 0.99 | Inactive Ps: 0.85 | Inactive Ps: 0.68 | Inactive Ps: 0.94 |
| ZINC ID | Classification | |||||
|---|---|---|---|---|---|---|
| Acute Toxicity | Toxicity End Point | Organ Toxicity | ||||
| Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||
| 2DG | I. Toxicity Class: 3 II. LD50:1500 mg/kg III. accuracy: 67.38% | Inactive Ps: 0.74 | Inactive Ps: 0.99 | Inactive Ps:0.74 | Inactive Ps:0.81 | Inactive Ps:0.94 |
| 86652948 Compound 1 | I. Toxicity Class: 6 II. LD50:15,900 mg/kg III. accuracy: 70.97% | Inactive Ps: 0.89 | Inactive Ps: 0.99 | Inactive Ps: 0.85 | Inactive Ps: 0.68 | Inactive Ps: 0.94 |
| 86991606 Compound 4 | I. Toxicity Class: 6 II. LD50:5500 mg/kg III. accuracy: 69.26% | Inactive Ps: 0.69 | Inactive Ps: 0.99 | Inactive Ps: 0.73 | Inactive Ps: 0.68 | Inactive Ps: 0.93 |
| 86991603 Compound 31 | I. Toxicity Class: 6 II. LD50:15,900 mg/kg III. accuracy: 70.97% | Inactive Ps: 0.89 | Inactive Ps: 0.99 | Inactive Ps: 0.85 | Inactive Ps: 0.68 | Inactive Ps: 0.94 |
| ZINC ID | Binding Energy (Kcal/mol) Chain A Terminal N | Hydrogen Bond Number Chain A Terminal N | Catalytic Residue |
|---|---|---|---|
| GLC | (−7.2) | 6 | Glu260, Lys173, Asp209, Asn235 |
| 3956760 Compound 10 | (−7.2) | 7 | Glu260, Gln291, Phe156, Asn208, Lys173, Asp209 |
| 16159409 Compound 26 | (−7.0) | 5 | Ser155, Asp209, Asn208, Lys173, Glu294 |
| 3809846 Compound 58 | (−6.10) | 2 | Asp209, Asn209, Ser155 |
| ZINC ID | Binding Energy (Kcal/mol) Chain A, Terminal N | Hydrogen Bond Number Chain A Terminal N | Catalytic Residue |
|---|---|---|---|
| BG6 | (−7.9) | 6 | Gln291, Pro157, Lys173, Thr232, Gly87, Asp209, Asn208 |
| 4403145 Compound 30 | (−7.8) | 11 | Gly87, Asn89, Thr232, Arg91, Asp84, Lys173 |
| 4530268 Compound 36 | (−7.4) | 9 | Asp84, Thr88, Thr323, Asn89 |
| 1576959 Compound 38 | (−7.0) | 5 | Asn89, Thr88, Ser449 |
| ZINC ID | Binding Energy (Kcal/mol) Chain A Terminal N | Hydrogen Bond Number Chain A Terminal N | Catalytic Residue |
|---|---|---|---|
| 2DG | (−6.0) | 8 | Asn208, Asp209, Thr172, Lys173, Glu294, Glu260, Phe156 |
| 86652948 Compound 1 | (−6.8) | 4 | Asp209, Asn208, Glu294, Thr172, Thr232, Lys173 |
| 86991606 Compound 4 | (−6.3) | 3 | Asp209, Asp84, Gly87, Thr88, Asp413, Thr232 |
| 86991603 Compound 31 | (−6.3) | 1 | Asp413, Asp84, Asp209 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanbin, S.; Ahmad Fuad, F.A.; Abdul Hamid, A.A. Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics. BioTech 2021, 10, 1. https://doi.org/10.3390/biotech10010001
Tanbin S, Ahmad Fuad FA, Abdul Hamid AA. Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics. BioTech. 2021; 10(1):1. https://doi.org/10.3390/biotech10010001
Chicago/Turabian StyleTanbin, Suriyea, Fazia Adyani Ahmad Fuad, and Azzmer Azzar Abdul Hamid. 2021. "Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics" BioTech 10, no. 1: 1. https://doi.org/10.3390/biotech10010001
APA StyleTanbin, S., Ahmad Fuad, F. A., & Abdul Hamid, A. A. (2021). Virtual Screening for Potential Inhibitors of Human Hexokinase II for the Development of Anti-Dengue Therapeutics. BioTech, 10(1), 1. https://doi.org/10.3390/biotech10010001