miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia

 , ,
, ,
Abstract
:1. Introduction
2. Results
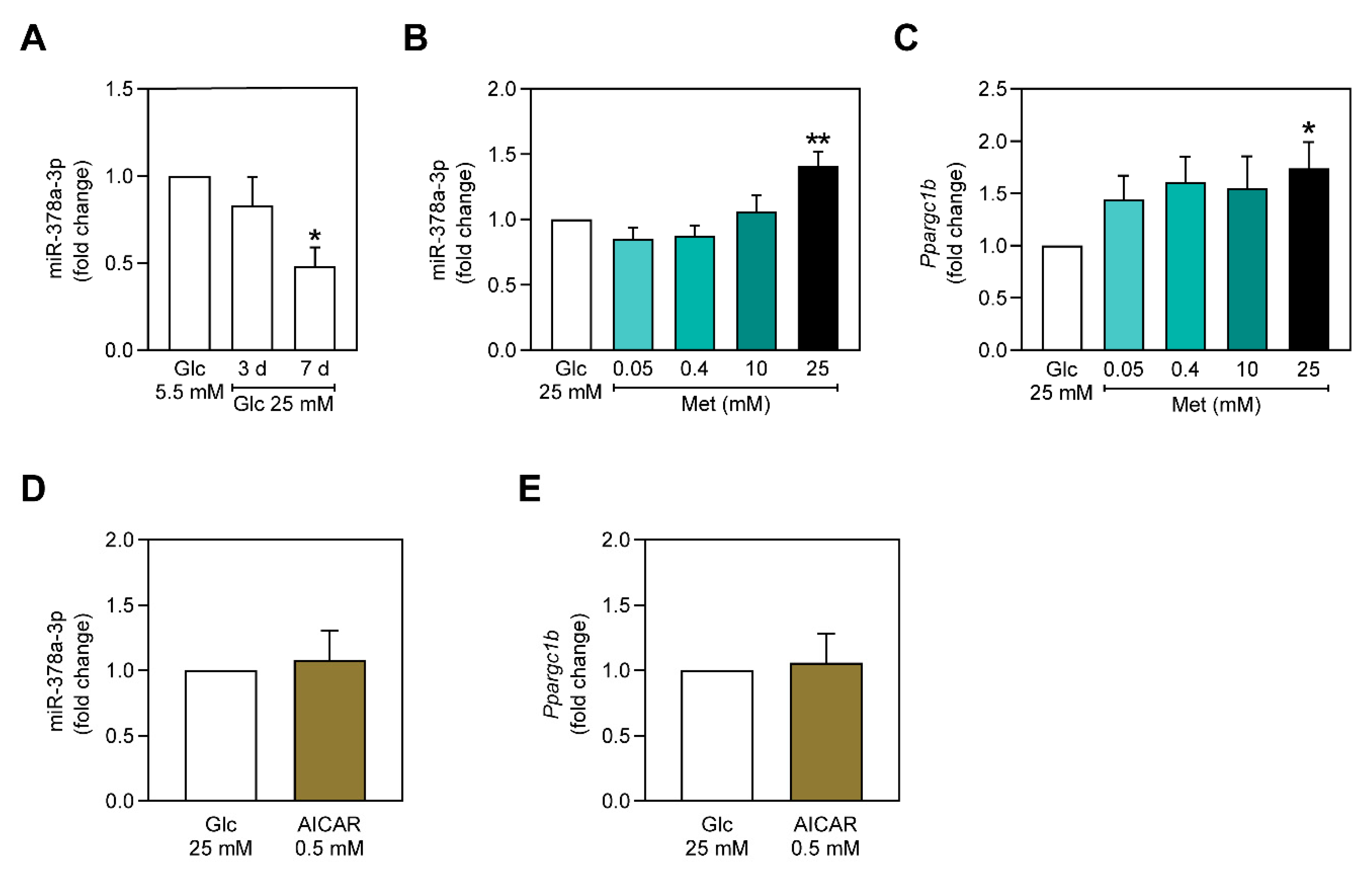
2.1. miR-378a-3p is Downregulated in C2C12 Myoblasts Exposed to Hyperglycemia-Mimicking Conditions
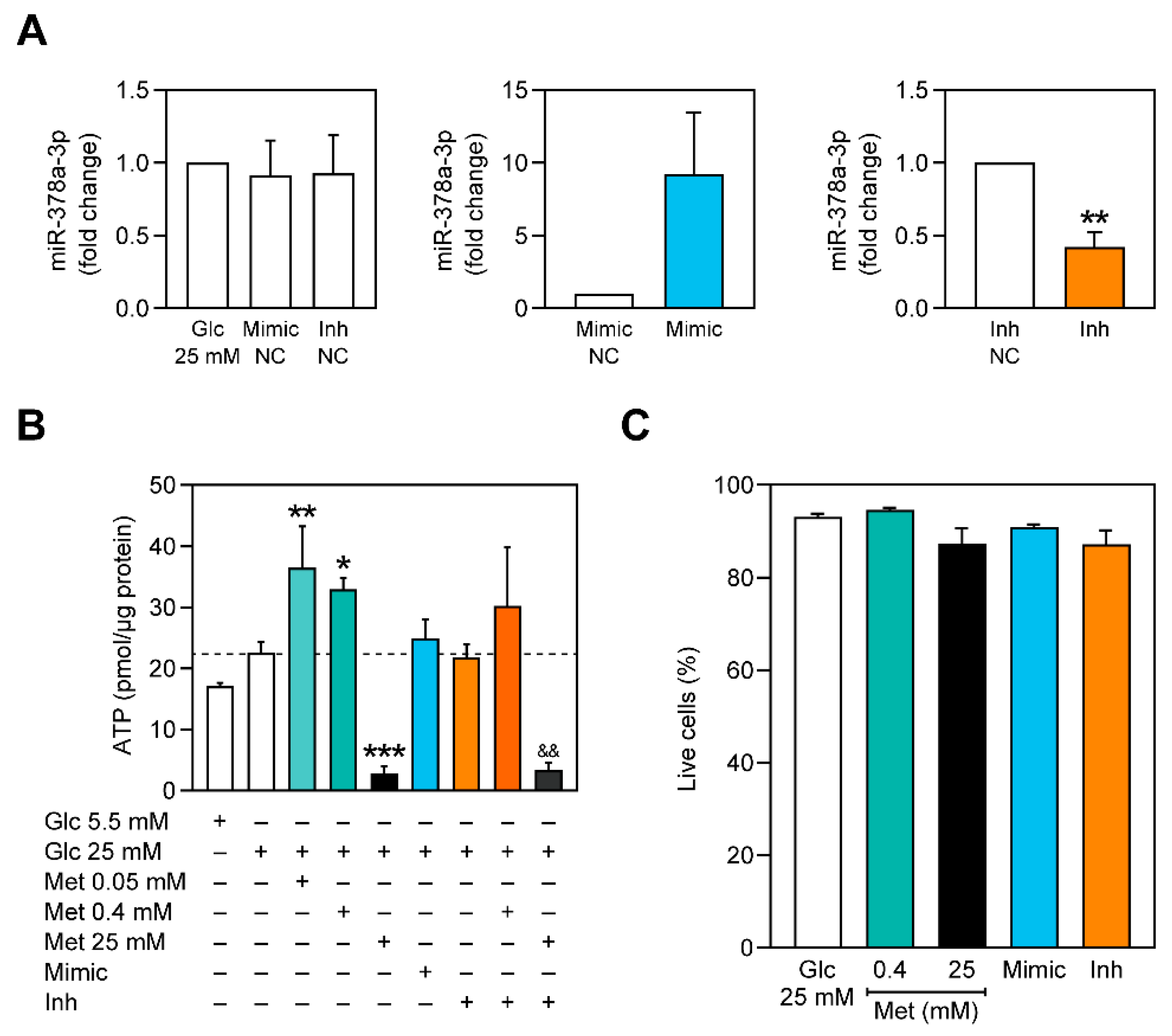
2.2. Metformin Upregulates miR-378a-3p and Ppargc1b Expression Independently of AMPK
2.3. miR-378a-3p Is Required for Metformin-Associated Increase of Cellular ATP Content
2.4. PEPCK Activity Is Decreased with Increasing Concentrations of Metformin and miR-378a-3p Silencing
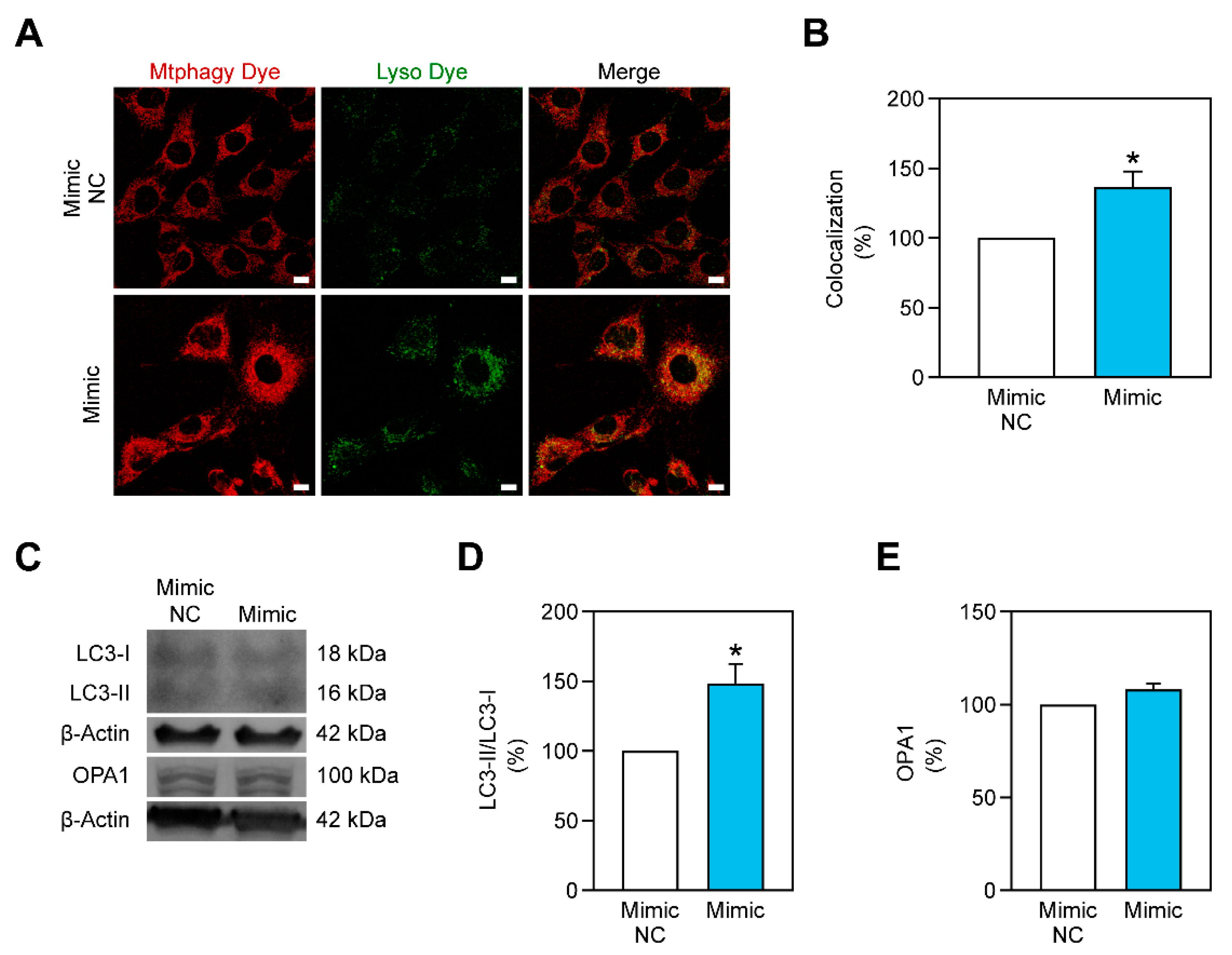
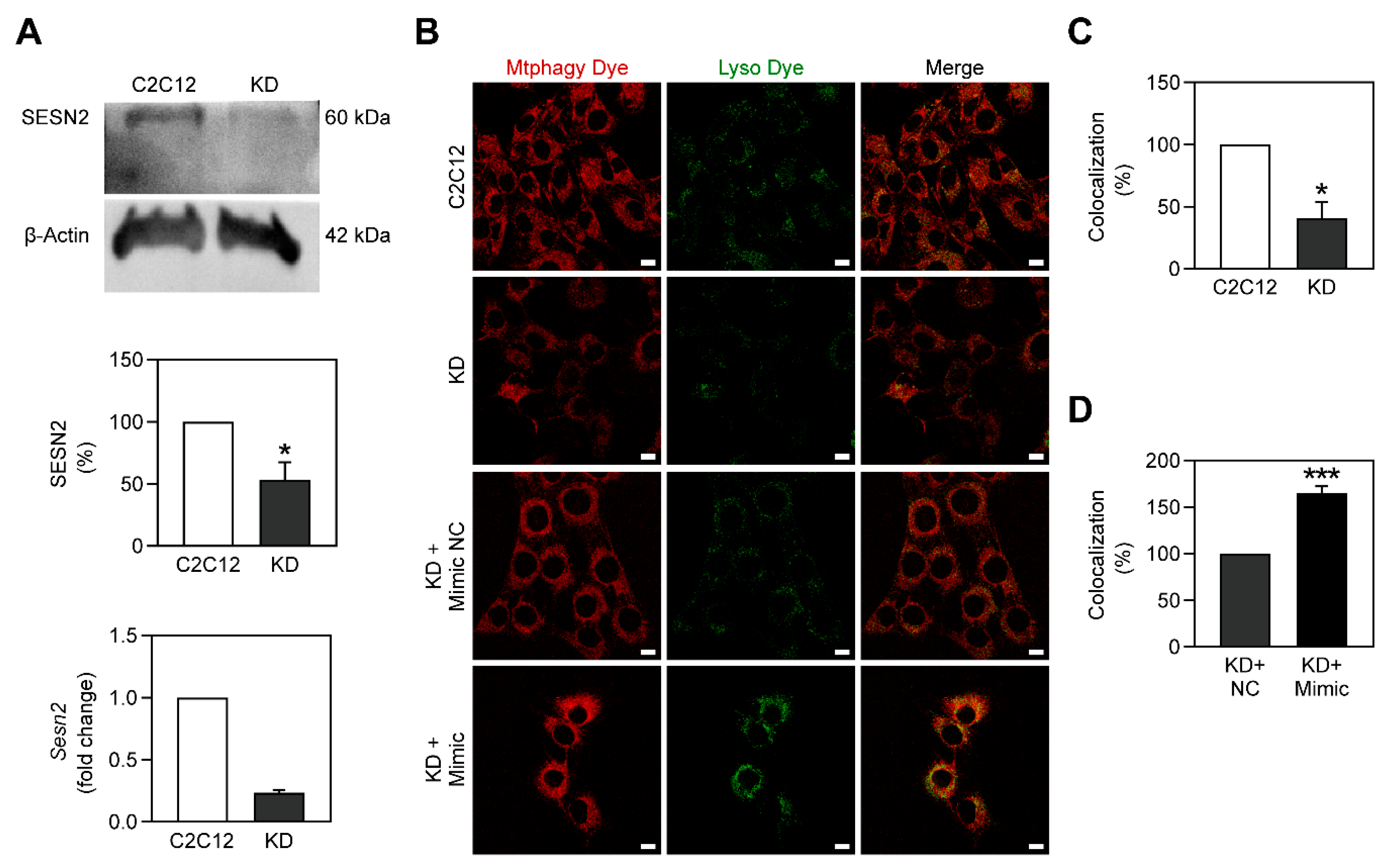
2.5. Mitophagy Is Stimulated by miR-378a-3p
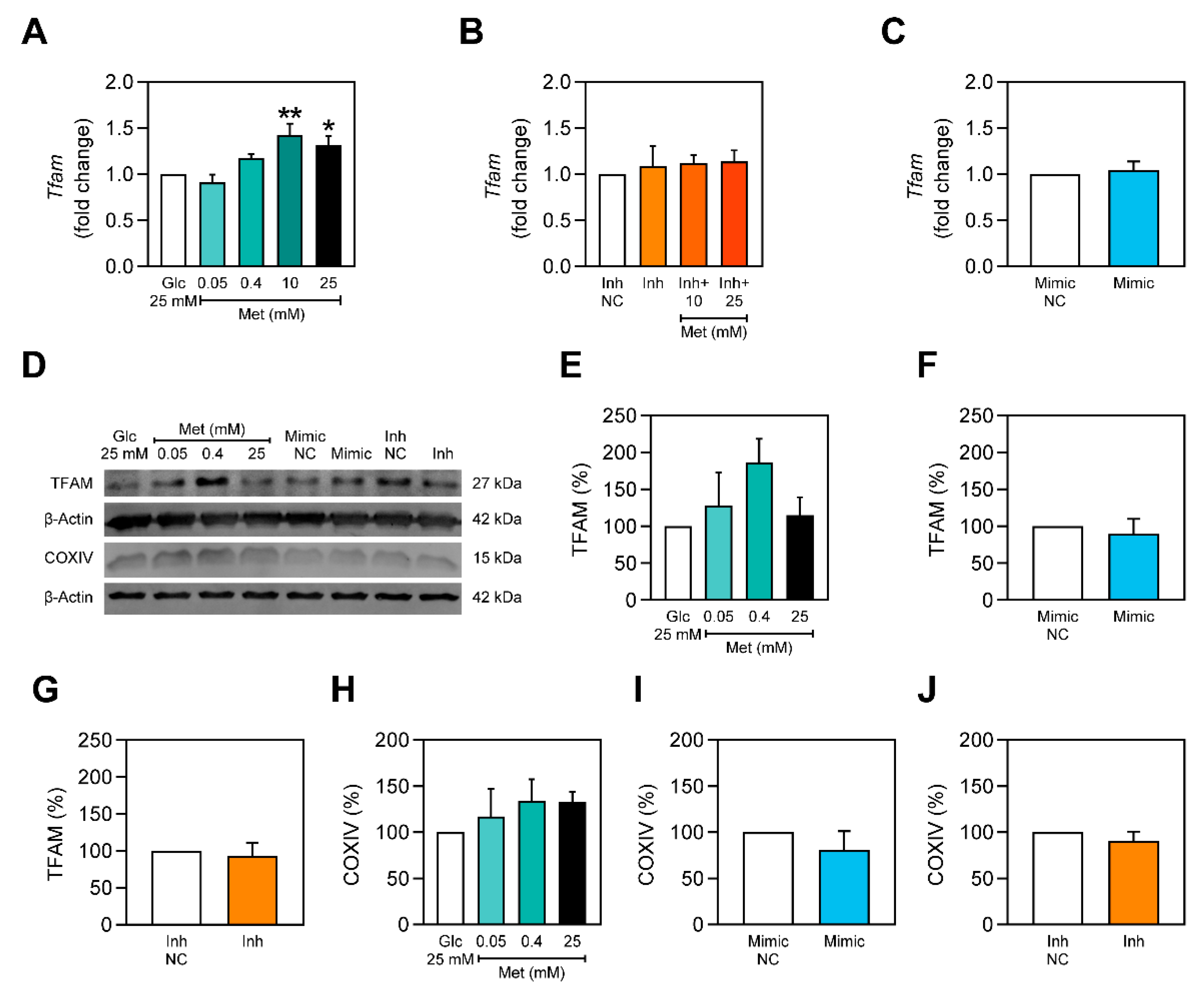
2.6. Metformin Stimulates Tfam Expression only in the Presence of miR-378a-3p
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Transfection with miR-378a-3p Mimics and Inhibitors
4.3. Generation of Sesn2 Knockdown Cells
4.4. Cell Viability Assay
4.5. RT-qPCR
4.6. ATP Content
4.7. PEPCK Activity
4.8. Western Blot
4.9. Mitophagy Detection
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| AMPK | AMP-activated protein kinase |
| BAT | Brown adipose tissue |
| CRAT | Carnitine-O-acetyltransferase |
| HFD | High-fat diet |
| Met | Metformin |
| miRNA | MicroRNA |
| PEP | Phosphoenolpyruvate |
| PEPCK | PEP carboxykinase |
| PGC-1 | PPARγ coactivator-1 |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| ROS | Reactive oxygen species |
| SESN2 | Sestrin-2 |
| shRNA | Short hairpin RNA |
| T2DM | Type 2 diabetes mellitus |
| TFAM | Mitochondrial transcription factor A |
| ULK1 | Unc-51 like autophagy activating kinase |
References
- Schatz, G. Mitochondria: Beyond oxidative phosphorylation. Biochim. Biophys. Acta Mol. Basis Dis. 1995, 1271, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Natali, A.; Ferrannini, E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: A systematic review. Diabetologia 2006, 49, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1$α$ protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. 2006, 101, 1685–1692. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019, 29, 1511–1523. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vial, G.; Detaille, D.; Guigas, B. Role of mitochondria in the mechanism(s) of action of metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yuan, Y.; Huang, L.; Qiao, M.; Zhang, Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res. Clin. Pract. 2012, 96, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, I.H.; Ertural, D.Y.; Balci, Ş.; Çlnklr, Ü.; Sezer, K.; Tamer, L.; Aras, N. Profiles of circulating miRNAs following metformin treatment in patients with type 2 diabetes. J. Med. Biochem. 2018, 37, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Han, S.; Qian, W.; Gu, Y.; Li, X.; Yang, K. Metformin induces miR-378 to downregulate the CDK1, leading to suppression of cell proliferation in hepatocellular carcinoma. Onco. Targets Ther. 2018, 11, 4451–4459. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.F.; Teodoro, J.S.; Palmeira, C.M.; Rolo, A.P. miR-378a: A new emerging microRNA in metabolism. Cell. Mol. Life Sci. 2020, 77, 1947–1958. [Google Scholar] [CrossRef]
- Eichner, L.J.; Perry, M.-C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378* mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. microRNA-378 promotes autophagy and inhibits apoptosis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, E10849–E10858. [Google Scholar] [CrossRef] [Green Version]
- Carrer, M.; Liu, N.; Grueter, C.E.; Williams, A.H.; Frisard, M.I.; Hulver, M.W.; Bassel-Duby, R.; Olson, E.N. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc. Natl. Acad. Sci. USA 2012, 109, 15330–15335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Mao, C.; Quattrochi, B.; Friedline, R.H.; Zhu, L.J.; Jung, D.Y.; Kim, J.K.; Lewis, B.; Wang, Y.-X. MicroRNA-378 controls classical brown fat expansion to counteract obesity. Nat. Commun. 2014, 5, 4725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, C.; Li, H.; Song, Y.; Zhao, Y.; Zhai, L.; Wang, H.; Zhong, R.; Tang, H.; Zhu, D. MiR-378 activates the pyruvate-PEP futile cycle and enhances lipolysis to ameliorate obesity in mice. EBioMedicine 2016, 5, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1β-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef]
- Ramamoorthy, T.G.; Laverny, G.; Schlagowski, A.-I.; Zoll, J.; Messaddeq, N.; Bornert, J.-M.; Panza, S.; Ferry, A.; Geny, B.; Metzger, D. The transcriptional coregulator PGC-1$β$ controls mitochondrial function and anti-oxidant defence in skeletal muscles. Nat. Commun. 2015, 6, 10210. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.-W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Parmigiani, A.; Budanov, A.V. Sensing the environment through sestrins: Implications for cellular metabolism. In International Review of Cell and Molecular Biology; Jeon, K.W., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 1–32. [Google Scholar]
- Kumar, A.; Shaha, C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 2018, 8, 615. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.-Z.; Li, J.-J.-H.; Xiao, H.-B.; He, Y.; Zhang, L.; Yan, Y.-X. Identification of stress-related microRNA biomarkers in type 2 diabetes mellitus: A systematic review and meta-analysis. J. Diabetes 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Koh, H.W.L.; Zhou, L.; Cheng, H.; Loh, T.P.; Rizi, E.P.; Toh, S.A.; Ronnett, G.V.; Huang, B.E.; Khoo, C.M. Plasma protein and microRNA biomarkers of insulin resistance: A network-based integrative -omics analysis. Front. Physiol. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Greco, M.; Chiefari, E.; Accattato, F.; Corigliano, D.M.; Arcidiacono, B.; Mirabelli, M.; Liguori, R.; Brunetti, F.S.; Pullano, S.A.; Scorcia, V.; et al. Microrna-1281 as a novel circulating biomarker in patients with diabetic retinopathy. Front. Endocrinol. 2020, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; Natoli, A.; Reddy-Luthmoodoo, M.; Heywood, S.E.; Armitage, J.A.; Kingwell, B.A. MicroRNA-194 modulates glucose metabolism and its skeletal muscle expression is reduced in diabetes. PLoS ONE 2016, 11, e0155108. [Google Scholar] [CrossRef] [Green Version]
- Assmann, T.S.; Recamonde-Mendoza, M.; Costa, A.R.; Puñales, M.; Tschiedel, B.; Canani, L.H.; Bauer, A.C.; Crispim, D. Circulating miRNAs in diabetic kidney disease: Case-control study and in silico analyses. Acta Diabetol. 2019, 56, 55–65. [Google Scholar] [CrossRef]
- Jones, A.; Danielson, K.M.; Benton, M.C.; Ziegler, O.; Shah, R.; Stubbs, R.S.; Das, S.; Macartney-Coxson, D. miRNA signatures of insulin resistance in obesity. Obesity 2017, 25, 1734–1744. [Google Scholar] [CrossRef] [Green Version]
- Krist, B.; Florczyk, U.; Pietraszek-Gremplewicz, K.; Józkowicz, A.; Dulak, J. The role of miR-378a in metabolism, angiogenesis, and muscle biology. Int. J. Endocrinol. 2015, 2015, 281756. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Rolo, A.P.; Berthiaume, J.; Bjork, J.A.; Wallace, K.B. Hyperglycemia decreases mitochondrial function: The regulatory role of mitochondrial biogenesis. Toxicol. Appl. Pharmacol. 2007, 225, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Cao, H.; Ye, C.; Chang, C.; Lu, M.; Jing, Y.; Zhang, D.; Yao, X.; Duan, Z.; Xia, H.; et al. Hepatic miR-378 targets p110$α$ and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat. Commun. 2014, 5, 5684. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Rieusset, J. Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [Green Version]
- Buler, M.; Aatsinki, S.-M.; Izzi, V.; Hakkola, J. Metformin reduces hepatic expression of SIRT3, the mitochondrial deacetylase controlling energy metabolism. PLoS ONE 2012, 7, e49863. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Pavlidou, T.; Rosina, M.; Fuoco, C.; Gerini, G.; Gargioli, C.; Castagnoli, L.; Cesareni, G. Regulation of myoblast differentiation by metabolic perturbations induced by metformin. PLoS ONE 2017, 12, e0182475. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Deng, Z.; Wang, C.; Yang, B.B. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc. Natl. Acad. Sci. USA 2007, 104, 20350–20355. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Gu, W.; Chen, L.; Pan, L.; Chen, J.; Peng, Y. MiR-378 overexpression attenuates high glucose-suppressed osteogenic differentiation through targeting CASP3 and activating PI3K/Akt signaling pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 7249–7261. [Google Scholar]
- Li, W.; Du, M.; Wang, Q.; Ma, X.; Wu, L.; Guo, F.; Ji, H.; Huang, F.; Qin, G. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/parkin pathway. Endocrinology 2017, 158, 2155–2167. [Google Scholar] [CrossRef] [Green Version]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in mitochondrial oxidative stress and mitophagy in subjects with prediabetes and type 2 diabetes mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered mitochondrial function, mitochondrial DNA and reduced metabolic flexibility in patients with diabetic nephropathy. EBioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, D.; Zancan, P.; Coelho, W.S.; Gomez, L.S.; Sola-Penna, M. Metformin reverses hexokinase and 6-phosphofructo-1-kinase inhibition in skeletal muscle, liver and adipose tissues from streptozotocin-induced diabetic mouse. Arch. Biochem. Biophys. 2010, 496, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, X.; Chen, Y.; Zhao, Q. PGC-1 mediates the regulation of metformin in muscle irisin expression and function. Am. J. Transl. Res. 2015, 7, 1850–1859. [Google Scholar]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/miRNA | Primer Sequence (5′ to 3′) | NCBI Nucleotide’s Accession Number | |
|---|---|---|---|
| mmu-miR-378a-3p | RT | TaqMan Assay 002243 | - |
| qPCR | TaqMan Assay 002243 | ||
| 18S rRNA | Forward | GTA ACC CGT TGA ACC CCA TT | NR_046239.1 |
| Reverse | CCA TCC AAT CGG TAG TAG CG | ||
| Ppargc1b | Forward | CTG GAA AGC CCC TGT GAG AG | NM_133249.3 |
| Reverse | CTG GAA AGC CCC TGT GAG AG | ||
| Sesn2 | Forward | TAG CCT GCA GCC TCA CCT AT | NM_144907.1 |
| Reverse | GAT TTT GAG GTT CCG TTC CA | ||
| Tfam | Forward | CTT TGA GCC TTG ACA GAA G | NM_009360.4 |
| Reverse | ATA TGT AAC GGT CAT CAG TG | ||
| Antibody | MW (kDa) | Dilution | Supplier | Reference Number |
|---|---|---|---|---|
| Primary antibodies | ||||
| β-actin | 42 | 1:2500 | Sigma | A5441 |
| COXIV | 15 | 1:1000 | Invitrogen | PA5-19471 |
| LC3B | 18, 16 | 1:1000 | Sigma-Aldrich | L7543 |
| OPA1 | 100 | 1:1000 | BD Biosciences | 612607 |
| SESN2 | 60 | 1:100 | Santa Cruz | sc-393195 |
| TFAM | 27 | 1:1000 | Aviva Systems Biology | ARP36993 |
| Secondary antibodies | ||||
| Biotin-XX goat anti-mouse IgG | - | 1:2500 | Invitrogen | W10132 |
| Biotin-XX goat anti-rabbit IgG | - | 1:2500 | Invitrogen | W10142 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, I.F.; Teodoro, J.S.; Castela, A.C.; Palmeira, C.M.; Rolo, A.P. miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 541. https://doi.org/10.3390/ijms22020541
Machado IF, Teodoro JS, Castela AC, Palmeira CM, Rolo AP. miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia. International Journal of Molecular Sciences. 2021; 22(2):541. https://doi.org/10.3390/ijms22020541
Chicago/Turabian StyleMachado, Ivo F., João S. Teodoro, Ana C. Castela, Carlos M. Palmeira, and Anabela P. Rolo. 2021. "miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia" International Journal of Molecular Sciences 22, no. 2: 541. https://doi.org/10.3390/ijms22020541