Inhibition of Orexin/Hypocretin Neurons Ameliorates Elevated Physical Activity and Energy Expenditure in the A53T Mouse Model of Parkinson’s Disease

Abstract
:1. Introduction
2. Results
2.1. A53T Mice Have Increased Food Intake, Are Leaner, and Have Increased Exploratory Locomotion
2.2. A53T Mice Have Increased SPA and EE
2.3. PD-Associated A53T Pathology Increases Astrogliosis and Inflammation Markers in the LH of A53T Mice in Early Stages of the Disease, and Induces Orexin Neuron Loss in Later Stages of the Disease
2.4. Loss of GAD65 Presynaptic Terminals in LH of the 7-Month-Old A53T Mice
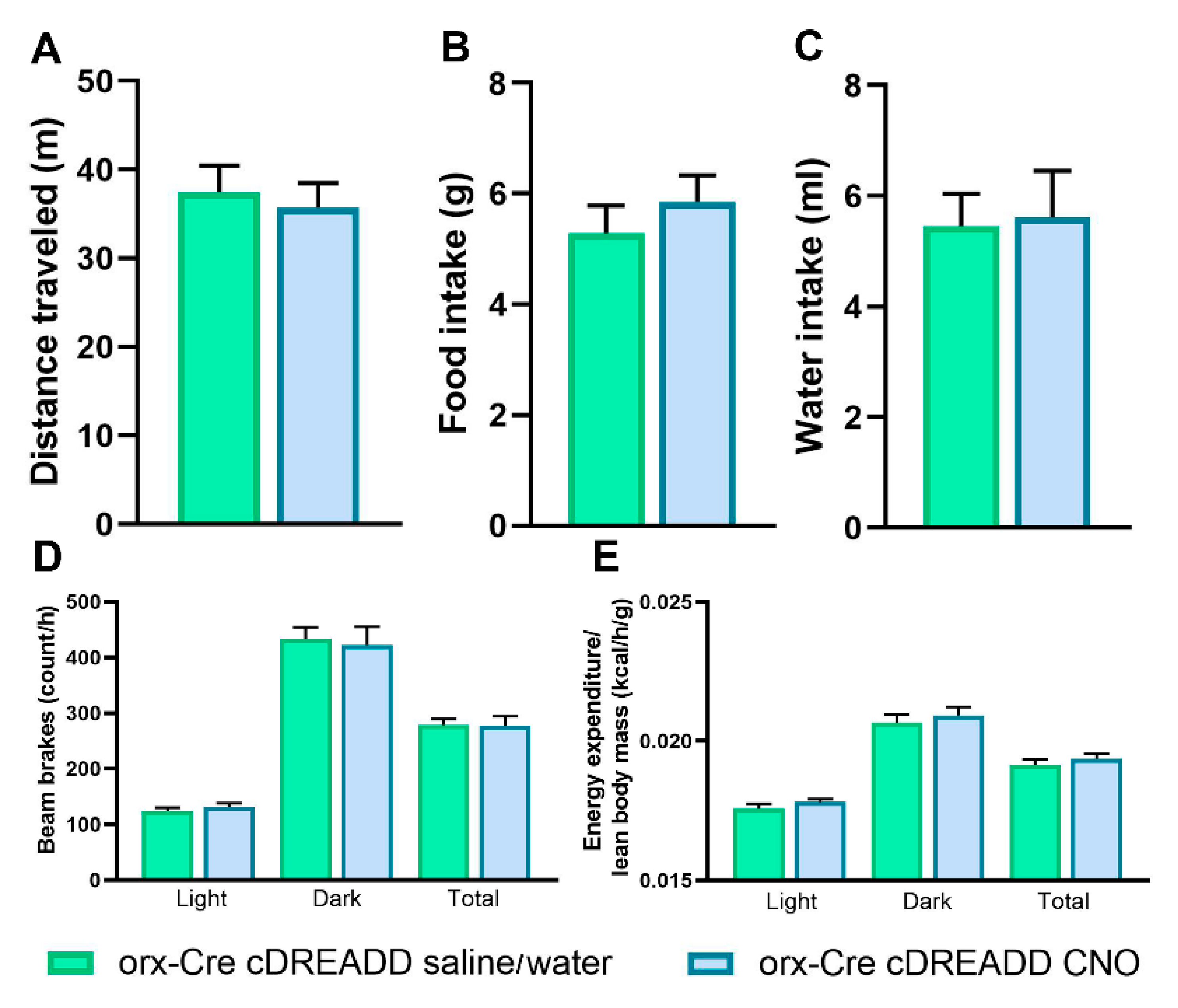
2.5. Clozapine N-Oxide (CNO) Treatment in Mice with an Inactive Designer Receptor Exclusively Activated by Designer Drugs (DREADD) Construct Does Not Affect Food and Water Consumption, Exploratory Locomotion, SPA, or EE
2.6. Chemogenetic Inhibition of Orexin Neurons Ameliorated Increase in Exploratory Locomotion, SPA, and EE
2.7. Confirmation of Injection Placement and DREADD Functionality
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. Food Intake, Body Mass and Composition
4.3. Open Field Test (OFT)
4.4. SPA and EE Analysis
4.5. Viral Injections and CNO Treatment
4.6. Immunohistochemistry
4.7. Immunofluorescence Imaging and Image Analysis
4.8. Unbiased Stereology
4.9. Quantification of Pre-Synaptic Inhibitory Terminals
4.10. Statistical Analyses
4.11. Experimental Design and Exclusion Criteria
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s Disease: Why Is Advancing Age the Biggest Risk Factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primer 2017, 3, 17013. [Google Scholar] [CrossRef]
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Guern, M.L.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson Disease Male-to-Female Ratios Increase with Age: French Nationwide Study and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. [Google Scholar] [CrossRef]
- Tan, L.C.S. Mood Disorders in Parkinson’s Disease. Parkinsonism Relat. Disord. 2012, 18, S74–S76. [Google Scholar] [CrossRef]
- Davis, A.A.; Racette, B. Parkinson Disease and Cognitive Impairment. Neurol. Clin. Pract. 2016, 6, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.G.; Litvan, I. Mild Cognitive Impairment in Parkinson’s Disease. Minerva Med. 2011, 102, 441–459. [Google Scholar] [PubMed]
- De Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.-B.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.F.; Gautvik, V.T.; Bartlett, F.S.; et al. The Hypocretins: Hypothalamus-Specific Peptides with Neuroexcitatory Activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.C.; Richardson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and Orexin Receptors: A Family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors That Regulate Feeding Behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Pop, M.G.; Crivii, C.; Opincariu, I. Anatomy and Function of the Hypothalamus. Hypothal. Health Dis. 2018. [Google Scholar] [CrossRef]
- Sakurai, T.; Nagata, R.; Yamanaka, A.; Kawamura, H.; Tsujino, N.; Muraki, Y.; Kageyama, H.; Kunita, S.; Takahashi, S.; Goto, K.; et al. Input of Orexin/Hypocretin Neurons Revealed by a Genetically Encoded Tracer in Mice. Neuron 2005, 46, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; McCormack, S.; España, R.A.; Crocker, A.; Scammell, T.E. Afferents to the Orexin Neurons of the Rat Brain. J. Comp. Neurol. 2006, 494, 845–861. [Google Scholar] [CrossRef] [Green Version]
- Flores, Á.; Valls-Comamala, V.; Costa, G.; Saravia, R.; Maldonado, R.; Berrendero, F. The Hypocretin/Orexin System Mediates the Extinction of Fear Memories. Neuropsychopharmacology 2014, 39, 2732–2741. [Google Scholar] [CrossRef] [Green Version]
- James, M.H.; Campbell, E.J.; Dayas, C.V. Role of the Orexin/Hypocretin System in Stress-Related Psychiatric Disorders. Curr. Top. Behav. Neurosci. 2017, 33, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.L.; Molosh, A.; Fitz, S.D.; Truitt, W.A.; Shekhar, A. Orexin, Stress, and Anxiety/Panic States. Prog. Brain Res. 2012, 198, 133–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavanji, V.; Butterick, T.A.; Duffy, C.M.; Nixon, J.P.; Billington, C.J.; Kotz, C.M. Orexin/Hypocretin Treatment Restores Hippocampal-Dependent Memory in Orexin-Deficient Mice. Neurobiol. Learn. Mem. 2017, 146, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muschamp, J.W.; Hollander, J.A.; Thompson, J.L.; Voren, G.; Hassinger, L.C.; Onvani, S.; Kamenecka, T.M.; Borgland, S.L.; Kenny, P.J.; Carlezon, W.A. Hypocretin (Orexin) Facilitates Reward by Attenuating the Antireward Effects of Its Cotransmitter Dynorphin in Ventral Tegmental Area. Proc. Natl. Acad. Sci. USA 2014, 111, E1648–E1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, B.M.; Hosseinzadeh, H. A Review of the Role of Orexin System in Pain Modulation. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 90, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. Roles of Orexin/Hypocretin in Regulation of Sleep/Wakefulness and Energy Homeostasis. Sleep Med. Rev. 2005, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, J.W.; Campbell, E.J.; James, M.H.; Graham, B.A.; Dayas, C.V. Orexin Antagonists for Neuropsychiatric Disease: Progress and Potential Pitfalls. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Girault, E.M.; Yi, C.-X.; Fliers, E.; Kalsbeek, A. Orexins, Feeding, and Energy Balance. Prog. Brain Res. 2012, 198, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, N.; Sakurai, T. Orexin/Hypocretin: A Neuropeptide at the Interface of Sleep, Energy Homeostasis, and Reward System. Pharmacol. Rev. 2009, 61, 162–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inutsuka, A.; Yamanaka, A. The Physiological Role of Orexin/Hypocretin Neurons in the Regulation of Sleep/Wakefulness and Neuroendocrine Functions. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lecea, L.; Huerta, R. Hypocretin (Orexin) Regulation of Sleep-to-Wake Transitions. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotz, C.M. Integration of Feeding and Spontaneous Physical Activity: Role for Orexin. Physiol. Behav. 2006, 88, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Perez-Leighton, C.E.; Little, M.R.; Grace, M.K.; Billington, C.J.; Kotz, C.M. Orexin Signaling in Rostral Lateral Hypothalamus and Nucleus Accumbens Shell in the Control of Spontaneous Physical Activity in High and Low Activity Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016. [Google Scholar] [CrossRef]
- Blais, A.; Drouin, G.; Chaumontet, C.; Voisin, T.; Couvelard, A.; Even, P.C.; Couvineau, A. Impact of Orexin-A Treatment on Food Intake, Energy Metabolism and Body Weight in Mice. PLoS ONE 2017, 12, e0169908. [Google Scholar] [CrossRef] [Green Version]
- Teske, J.A.; Mavanji, V. Energy Expenditure: Role of Orexin. Vitam. Horm. 2012, 89, 91–109. [Google Scholar] [CrossRef] [Green Version]
- Zink, A.N.; Bunney, P.E.; Holm, A.A.; Billington, C.J.; Kotz, C.M. Neuromodulation of Orexin Neurons Reduces Diet-Induced Adiposity. Int. J. Obes. 2005 2018, 42, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Miyamoto, M.; Miyamoto, T.; Iwanami, M.; Hirata, K. Sleep Disturbances Associated with Parkinson’s Disease. Available online: https://www.hindawi.com/journals/pd/2011/219056/ (accessed on 8 July 2019).
- Swick, T.J. Parkinson’s Disease and Sleep/Wake Disturbances. Park. Dis. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Bridoux, A.; Moutereau, S.; Covali-Noroc, A.; Margarit, L.; Palfi, S.; Nguyen, J.-P.; Lefaucheur, J.-P.; Césaro, P.; d’Ortho, M.-P.; Drouot, X. Ventricular Orexin-A (Hypocretin-1) Levels Correlate with Rapid-Eye-Movement Sleep without Atonia in Parkinson’s Disease. Nat. Sci. Sleep 2013, 5, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, C.R.; Scammell, T.E.; Bassetti, C.L. Parkinson’s Disease, Sleepiness and Hypocretin/Orexin. Brain J. Neurol. 2008, 131, e91. [Google Scholar] [CrossRef]
- Fronczek, R.; Overeem, S.; Lee, S.Y.Y.; Hegeman, I.M.; van Pelt, J.; van Duinen, S.G.; Lammers, G.J.; Swaab, D.F. Hypocretin (Orexin) Loss in Parkinson’s Disease. Brain J. Neurol. 2007, 130, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, T.C.; Lai, Y.-Y.; Siegel, J.M. Hypocretin (Orexin) Cell Loss in Parkinson’s Disease. Brain J. Neurol. 2007, 130, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic Animal Models of Parkinson’s Disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Paumier, K.L.; Sukoff Rizzo, S.J.; Berger, Z.; Chen, Y.; Gonzales, C.; Kaftan, E.; Li, L.; Lotarski, S.; Monaghan, M.; Shen, W.; et al. Behavioral Characterization of A53T Mice Reveals Early and Late Stage Deficits Related to Parkinson’s Disease. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-Synuclein-Harboring Familial Parkinson’s Disease-Linked Ala-53 → Thr Mutation Causes Neurodegenerative Disease with α-Synuclein Aggregation in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic Expression of Parkinson’s Disease-Related A53T α-Synuclein Causes Neurodegeneration in Mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Fellner, L.; Jellinger, K.A.; Wenning, G.K.; Stefanova, N. Glial Dysfunction in the Pathogenesis of α-Synucleinopathies: Emerging Concepts. Acta Neuropathol. (Berl.) 2011, 121, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and Inflammation in Parkinson’s Disease. Parkinsonism Relat. Disord. 2012, 18, S207–S209. [Google Scholar] [CrossRef]
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T Human α-Synuclein Overexpression in Transgenic Mice Induces Pervasive Mitochondria Macroautophagy Defects Preceding Dopamine Neuron Degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Oh, S.T.; Jeong, H.J.; Pak, S.C.; Park, H.-J.; Kim, J.; Cho, H.; Jeon, S. MPTP-Induced Vulnerability of Dopamine Neurons in A53T α-Synuclein Overexpressed Mice with the Potential Involvement of DJ-1 Downregulation. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2017, 21, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, R.F. Non-Motor Symptoms in Parkinson’s Disease. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S119–S122. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- Gomez, J.L.; Bonaventura, J.; Lesniak, W.; Mathews, W.B.; Sysa-Shah, P.; Rodriguez, L.A.; Ellis, R.J.; Richie, C.T.; Harvey, B.K.; Dannals, R.F.; et al. Chemogenetics Revealed: DREADD Occupancy and Activation via Converted Clozapine. Science 2017, 357, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected Number of People with Parkinson Disease in the Most Populous Nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Shao, Y.; Le, W. Recent Advances and Perspectives of Metabolomics-Based Investigations in Parkinson’s Disease. Mol. Neurodegener. 2019, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.R.; Sidhu, A. Mice Expressing the A53T Mutant Form of Human Alpha-Synuclein Exhibit Hyperactivity and Reduced Anxiety-Like Behavior. J. Neurosci. Res. 2010, 88, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanojlovic, M.; Pallais, J.P.; Kotz, C.M. Chemogenetic Modulation of Orexin Neurons Reverses Changes in Anxiety and Locomotor Activity in the A53T Mouse Model of Parkinson’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Rothman, S.M.; Griffioen, K.J.; Fishbein, K.W.; Spencer, R.G.; Makrogiannis, S.; Cong, W.; Martin, B.; Mattson, M.P. Metabolic Abnormalities and Hypoleptinemia in α-Synuclein A53T Mutant Mice. Neurobiol. Aging 2014, 35, 1153–1161. [Google Scholar] [CrossRef] [Green Version]
- Delikanaki-Skaribas, E.; Trail, M.; Wong, W.W.-L.; Lai, E.C. Daily Energy Expenditure, Physical Activity, and Weight Loss in Parkinson’s Disease Patients. Mov. Disord. Off. J. Mov. Disord. Soc. 2009, 24, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Lorefält, B.; Toss, G.; Granérus, A.-K. Weight Loss, Body Fat Mass, and Leptin in Parkinson’s Disease. Mov. Disord. 2009, 24, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Bienfait, K. Sleep Disturbances in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, S117–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and Indifference to Cocaine and Amphetamine in Mice Lacking the Dopamine Transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Uhl, G.R.; Vandenbergh, D.J.; Miner, L.L. Knockout Mice and Dirty Drugs. Drug Addiction. Curr. Biol. CB 1996, 6, 935–936. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, F.; Ray, N.; Strafella, A.P. Impulsivity and Parkinson’s Disease: More than Just Disinhibition. J. Neurol. Sci. 2011, 310, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Mang, G.M.; Dürst, T.; Bürki, H.; Imobersteg, S.; Abramowski, D.; Schuepbach, E.; Hoyer, D.; Fendt, M.; Gee, C.E. The Dual Orexin Receptor Antagonist Almorexant Induces Sleep and Decreases Orexin-Induced Locomotion by Blocking Orexin 2 Receptors. Sleep 2012, 35, 1625–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Qi, R.; Wang, J.; Zhou, W.; Liu, J.; Cai, Y. Evidence for a Role of Orexin/Hypocretin System in Vestibular Lesion-Induced Locomotor Abnormalities in Rats. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoblock, J.R.; Welty, N.; Aluisio, L.; Fraser, I.; Motley, S.T.; Morton, K.; Palmer, J.; Bonaventure, P.; Carruthers, N.I.; Lovenberg, T.W.; et al. Selective Blockade of the Orexin-2 Receptor Attenuates Ethanol Self-Administration, Place Preference, and Reinstatement. Psychopharmacology (Berl.) 2011, 215, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Coborn, J.E.; DePorter, D.P.; Mavanji, V.; Sinton, C.M.; Kotz, C.M.; Billington, C.J.; Teske, J.A. Role of Orexin-A in the Ventrolateral Preoptic Area on Components of Total Energy Expenditure. Int. J. Obes. 2005 2017, 41, 1256–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanojlovic, M.; Pallais Yllescas, J.P.; Vijayakumar, A.; Kotz, C. Early Sociability and Social Memory Impairment in the A53T Mouse Model of Parkinson’s Disease Are Ameliorated by Chemogenetic Modulation of Orexin Neuron Activity. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s Disease α-Synuclein Transgenic Mice Develop Neuronal Mitochondrial Degeneration and Cell Death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Schwach, G.; Ingolic, E.; Adame, A.; Crews, L.; Mante, M.; Pfragner, R.; Schreiner, E.; Windisch, M.; Masliah, E. Lysosomal Pathology Associated with Alpha-Synuclein Accumulation in Transgenic Models Using an EGFP Fusion Protein. J. Neurosci. Res. 2005, 80, 247–259. [Google Scholar] [CrossRef]
- Kremer, H.P.; Bots, G.T. Lewy Bodies in the Lateral Hypothalamus: Do They Imply Neuronal Loss? Mov. Disord. Off. J. Mov. Disord. Soc. 1993, 8, 315–320. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Katsuki, H.; Michinaga, S. Anti-Parkinson Drugs and Orexin Neurons. Vitam. Horm. 2012, 89, 279–290. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-Synuclein Oligomers: A New Hope. Acta Neuropathol. (Berl.) 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms Underlying Extensive Ser129-Phosphorylation in α-Synuclein Aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chee, M.J.; Hebert, A.J.; Briançon, N.; Flaherty, S.E.; Pissios, P.; Maratos-Flier, E. Conditional Deletion of Melanin-Concentrating Hormone Receptor 1 from GABAergic Neurons Increases Locomotor Activity. Mol. Metab. 2019, 29, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Bubser, M.; Fadel, J.R.; Jackson, L.L.; Meador-Woodruff, J.H.; Jing, D.; Deutch, A.Y. Dopaminergic Regulation of Orexin Neurons. Eur. J. Neurosci. 2005, 21, 2993–3001. [Google Scholar] [CrossRef] [Green Version]
- di Michele, F.; Luchetti, S.; Bernardi, G.; Romeo, E.; Longone, P. Neurosteroid and Neurotransmitter Alterations in Parkinson’s Disease. Front. Neuroendocrinol. 2013, 34, 132–142. [Google Scholar] [CrossRef]
- O’Gorman Tuura, R.L.; Baumann, C.R.; Baumann-Vogel, H. Beyond Dopamine: GABA, Glutamate, and the Axial Symptoms of Parkinson Disease. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Błaszczyk, J.W. Parkinson’s Disease and Neurodegeneration: GABA-Collapse Hypothesis. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Borgkvist, A.; Avegno, E.M.; Wong, M.Y.; Kheirbek, M.A.; Sonders, M.S.; Hen, R.; Sulzer, D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron 2015, 87, 976–988. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, G.; Cerri, S.; Blandini, F. A Further Update on the Role of Excitotoxicity in the Pathogenesis of Parkinson’s Disease. J. Neural Transm. Vienna Austria 1996 2014, 121, 849–859. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Qin, Z. Molecular Mechanisms of Excitotoxicity and Their Relevance to Pathogenesis of Neurodegenerative Diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The Nasal and Gut Microbiome in Parkinson’s Disease and Idiopathic Rapid Eye Movement Sleep Behavior Disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inutsuka, A.; Yamanaka, A. The Regulation of Sleep and Wakefulness by the Hypothalamic Neuropeptide Orexin/Hypocretin. Nagoya J. Med. Sci. 2013, 75, 29–36. [Google Scholar]
- Feng, H.; Wen, S.-Y.; Qiao, Q.-C.; Pang, Y.-J.; Wang, S.-Y.; Li, H.-Y.; Cai, J.; Zhang, K.-X.; Chen, J.; Hu, Z.-A.; et al. Orexin Signaling Modulates Synchronized Excitation in the Sublaterodorsal Tegmental Nucleus to Stabilize REM Sleep. Nat. Commun. 2020, 11, 3661. [Google Scholar] [CrossRef]
- Mehta, R.; Khanday, M.A.; Mallick, B.N. REM Sleep Loss Associated Changes in Orexin-A Levels in Discrete Brain Areas in Rats. Neurosci. Lett. 2015, 590, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Branch, A.F.; Navidi, W.; Tabuchi, S.; Terao, A.; Yamanaka, A.; Scammell, T.E.; Diniz Behn, C. Progressive Loss of the Orexin Neurons Reveals Dual Effects on Wakefulness. Sleep 2016, 39, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, I.Z.; Naidu, Y.; Reddy, P.; Chaudhuri, K.R. Narcolepsy in Parkinson’s Disease. Expert Rev. Neurother. 2010, 10, 879–884. [Google Scholar] [CrossRef]
- Mahoney, C.E.; Cogswell, A.; Koralnik, I.J.; Scammell, T.E. The Neurobiological Basis of Narcolepsy. Nat. Rev. Neurosci. 2019, 20, 83–93. [Google Scholar] [CrossRef]
- Kantor, S.; Mochizuki, T.; Janisiewicz, A.M.; Clark, E.; Nishino, S.; Scammell, T.E. Orexin Neurons Are Necessary for the Circadian Control of REM Sleep. Sleep 2009, 32, 1127–1134. [Google Scholar] [CrossRef]
- Azeez, I.A.; Del Gallo, F.; Cristino, L.; Bentivoglio, M. Daily Fluctuation of Orexin Neuron Activity and Wiring: The Challenge of “Chronoconnectivity”. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Matsuki, T.; Nomiyama, M.; Takahira, H.; Hirashima, N.; Kunita, S.; Takahashi, S.; Yagami, K.; Kilduff, T.S.; Bettler, B.; Yanagisawa, M.; et al. Selective Loss of GABA(B) Receptors in Orexin-Producing Neurons Results in Disrupted Sleep/Wakefulness Architecture. Proc. Natl. Acad. Sci. USA 2009, 106, 4459–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, K. The Mouse Brain in Stereotaxic Coordinates; Acad. Press: Amsterdam, The Netherland, 2008; ISBN 978-0-12-369460-7. [Google Scholar]
- Stanojlovic, M.; Pallais Yllescas, J.P.; Mavanji, V.; Kotz, C. Chemogenetic Activation of Orexin/Hypocretin Neurons Ameliorates Aging-Induced Changes in Behavior and Energy Expenditure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R571–R583. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin Is Required for Normal Excitatory Synapse Development in Cortical and Striatal Circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, D.M.; Eroglu, C. Quantifying Synapses: An Immunocytochemistry-Based Assay to Quantify Synapse Number. J. Vis. Exp. JoVE 2010. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Composition | Exploratory Locomotion | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | Age (Months) | Food Intake (g) | Body Mass (g) | Lean Mass (g) | Fat Mass (g) | Fat to Lean Mass Ratio | Distance Traveled (m) |
| WT | 3 | 3.05 ± 0.17 @#$& | 28.30 ± 0.57 #$ | 25.94 ± 0.37 | 1.07 ± 0.14 @$ | 0.0413 ± 0.0058 @#$ | 43.38 ± 1.56 $ |
| 5 | 3.52 ± 0.12 $ | 29.72 ± 0.24 $ | 26.61 ± 0.36 | 1.78 ± 0.13 $ | 0.0670 ± 0.0055 #$ | 38.26 ± 1.73 & | |
| 7 | 3.80 ± 0.14 *& | 30.72 ± 0.70 $ | 26.93 ± 0.30 | 3.07 ± 0.37 *$ | 0.1142 ± 0.0146 *$ | 34.06 ± 1.45 & | |
| 9 | 4.04 ± 0.13 *& | 31.90 ± 0.80 * | 27.47 ± 0.46 | 4.25 ± 0.42 $ | 0.1552 ± 0.0162 *!$& | 31.26 ± 2.15 & | |
| 11 | 4.32 ± 0.18 *! | 34.47 ± 0.85 *!@ | 27.45 ± 0.19 | 6.02 ± 0.67 *!@#& | 0.2192 ± 0.0242 *!@#& | 27.08 ± 1.54 *& | |
| A53T | 3 | 3.86 ± 0.14 #& | 28.02 ± 0.18 #$ | 26.22 ± 0.36 | 1.03 ± 0.16 #$ | 0.0392 ± 0.0060 #$ | 52.63 ± 1.96 @# |
| 5 | 4.14 ± 0.13 | 29.60 ± 0.35 | 26.46 ± 0.53 | 1.51 ± 0.19 | 0.0576 ± 0.0078 | 61.87 ± 1.84 @#& | |
| 7 | 4.50 ± 0.14 & | 30.72 ± 0.35 | 26.63 ± 0.54 | 2.01 ± 0.28 | 0.0768 ± 0.0122 | 81.34 ± 4.37 *!$& | |
| 9 | 4.74 ± 0.09 *$& | 31.32 ± 0.62 * | 27.41 ± 0.28 | 2.75 ± 0.27 * | 0.1011 ± 0.0096 *& | 89.70 ± 5.02 *!$& | |
| 11 | 3.90 ± 0.21 # | 31.82 ± 1.10 * | 26.98 ± 0.44 | 2.81 ± 0.26 *& | 0.1039 ± 0.0089 *& | 65.40 ± 3.91 @#& | |
| Spontaneous Physical Activity (SPA) | Energy Expenditure (EE) | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | Age (months) | Beam Breaks—Light Phase (count/h) | Beam Breaks—Dark Phase (count/h) | Beam Breaks—Total (count/h) | Energy Expenditure/Lean Body Mass—Light Phase (kcal/h/g) | Energy Expenditure/Lean Body Mass—Dark Phase (kcal/h/g) | Energy Expenditure/Lean Body Mass—Total (kcal/h/g) |
| WT | 3 | 121.62 ± 21.60 | 522.03 ± 96.71 & | 321.82 ± 57.91 & | 0.01842 ± 0.00027 | 0.02277 ± 0.00061 | 0.02060 ± 0.00042 |
| 5 | 134.39 ± 20.41 & | 532.01 ± 105.72 & | 333.20 ± 62.34 & | 0.01818 ± 0.00031 | 0.02148 ± 0.00039 | 0.01983 ± 0.00032 | |
| 7 | 136.04 ± 31.71 & | 462.14 ± 78.21 & | 299.09 ± 51.41 & | 0.01760 ± 0.00016 & | 0.02067 ± 0.00028 & | 0.01913 ± 0.00022 & | |
| 9 | 101.11 ± 9.83 & | 332.15 ± 31.23 & | 216.63 ± 18.07 & | 0.01768 ± 0.00029 & | 0.02041 ± 0.00032 & | 0.01905 ± 0.00029 & | |
| 11 | 96.27 ± 9.68 | 284.67 ± 28.33 | 190.47 ± 16.65 | 0.01748 ± 0.00024 & | 0.02043 ± 0.00042 | 0.01896 ± 0.00029 | |
| A53T | 3 | 291.06 ± 64.15 @# | 1421.47 ± 237.51 @$& | 856.27 ± 148.58 @#$& | 0.1918 ± 0.0027 | 0.02383 ± 0.00045 | 0.02151 ± 0.00032 |
| 5 | 485.76 ± 42.57 @$& | 1700.42 ± 85.64 $& | 1093.09 ± 57.36 $& | 0.01949 ± 0.0033 | 0.02375 ± 0.00044 | 0.02162 ± 0.00032 | |
| 7 | 756.28 ± 85.30 *!$& | 2087.91 ± 152.22 *$& | 1422.09 ± 107.65 *$& | 0.01986 ± 0.0040 & | 0.02359 ± 0.00030 & | 0.02173 ± 0.00029 & | |
| 9 | 595.45 ± 31.56 *$& | 1905.05 ± 157.43 *$& | 1250.25 ± 92.00 *$& | 0.01963 ± 0.0035 & | 0.02357 ± 0.00021 $& | 0.02160 ± 0.00026 & | |
| 11 | 204.73 ± 41.09 !@# | 755.40 ± 143.23 *!@# | 480.07 ± 89.59 *!@# | 0.01944 ± 0.0045 & | 0.02163 ± 0.00052 # | 0.02053 ± 0.00044 | |
| Exploratory Locomotion | Spontaneous Physical Activity (SPA) | |||
|---|---|---|---|---|
| Genotype | Distance traveled (m) | Beam breaks—light phase (count) | Beam breaks—dark phase (count) | Beam breaks—total (count) |
| Orx-Cre cDREADD CNO | 38.37 ± 2.30 !@ | 136.69 ± 24.12 !@ | 480.69 ± 55.24 !@ | 308.69 ± 27.26 !@ |
| Orx/Cre;A53T cDREADD CNO | 92.11 ± 4.62 *@ | 837.18 ± 62.52 *@ | 2035.29 ± 207.31 *@ | 1436.24 ± 130.06 *@ |
| Orx/Cre;A53T iDREADD CNO | 58.78 ± 3.80 *! | 582.85 ± 62.97 *! | 1113.04 ± 64.17 *! | 847.94 ± 44.36 *! |
| Energy expenditure (EE) | ||||
| Genotype | Energy expenditure/lean body mass—light phase (kcal/h/g) | Energy expenditure/lean body mass—dark phase (kcal/h/g) | Energy expenditure/lean body mass—totlal (kcal/h/g) | Food intake (g) |
| Orx-Cre cDREADD CNO | 0.01809 ± 0.00020 ! | 0.02113 ± 0.00024 ! | 0.01932 ± 0.00026 ! | 5.05 ± 0.40 |
| Orx/Cre;A53T cDREADD CNO | 0.01919 ± 0.00040 * | 0.02356 ± 0.00042 *@ | 0.02136 ± 0.00022 *@ | 6.48 ± 0.63 |
| Orx/Cre;A53T iDREADD CNO | 0.01824 ± 0.00040 | 0.02164 ± 0.0041 ! | 0.01994 ± 0.00038 ! | 5.50 ± 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanojlovic, M.; Pallais, J.P.; Kotz, C.M. Inhibition of Orexin/Hypocretin Neurons Ameliorates Elevated Physical Activity and Energy Expenditure in the A53T Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 795. https://doi.org/10.3390/ijms22020795
Stanojlovic M, Pallais JP, Kotz CM. Inhibition of Orexin/Hypocretin Neurons Ameliorates Elevated Physical Activity and Energy Expenditure in the A53T Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(2):795. https://doi.org/10.3390/ijms22020795
Chicago/Turabian StyleStanojlovic, Milos, Jean Pierre Pallais, and Catherine M. Kotz. 2021. "Inhibition of Orexin/Hypocretin Neurons Ameliorates Elevated Physical Activity and Energy Expenditure in the A53T Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 2: 795. https://doi.org/10.3390/ijms22020795