Urban Aerosol Particulate Matter Promotes Necrosis and Autophagy via Reactive Oxygen Species-Mediated Cellular Disorders that Are Accompanied by Cell Cycle Arrest in Retinal Pigment Epithelial Cells

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. UPM Preparation
2.2. Cell Culture and Treatment
2.3. Cytotoxicity Assay
2.4. Observation of Cellular and Nuclear Morphology
2.5. Flow Cytometric Analysis for Cell Death and Cell Cycle Distribution
2.6. Analysis of Caspases Activities and Autophagy
2.7. Western Blot Analysis
2.8. Intracellular ROS Detection
2.9. Mitochondrial Membrane Potential (MMP, ∆Ψm) Analysis
2.10. Immunofluorescence
2.11. Statistical Analysis
3. Results
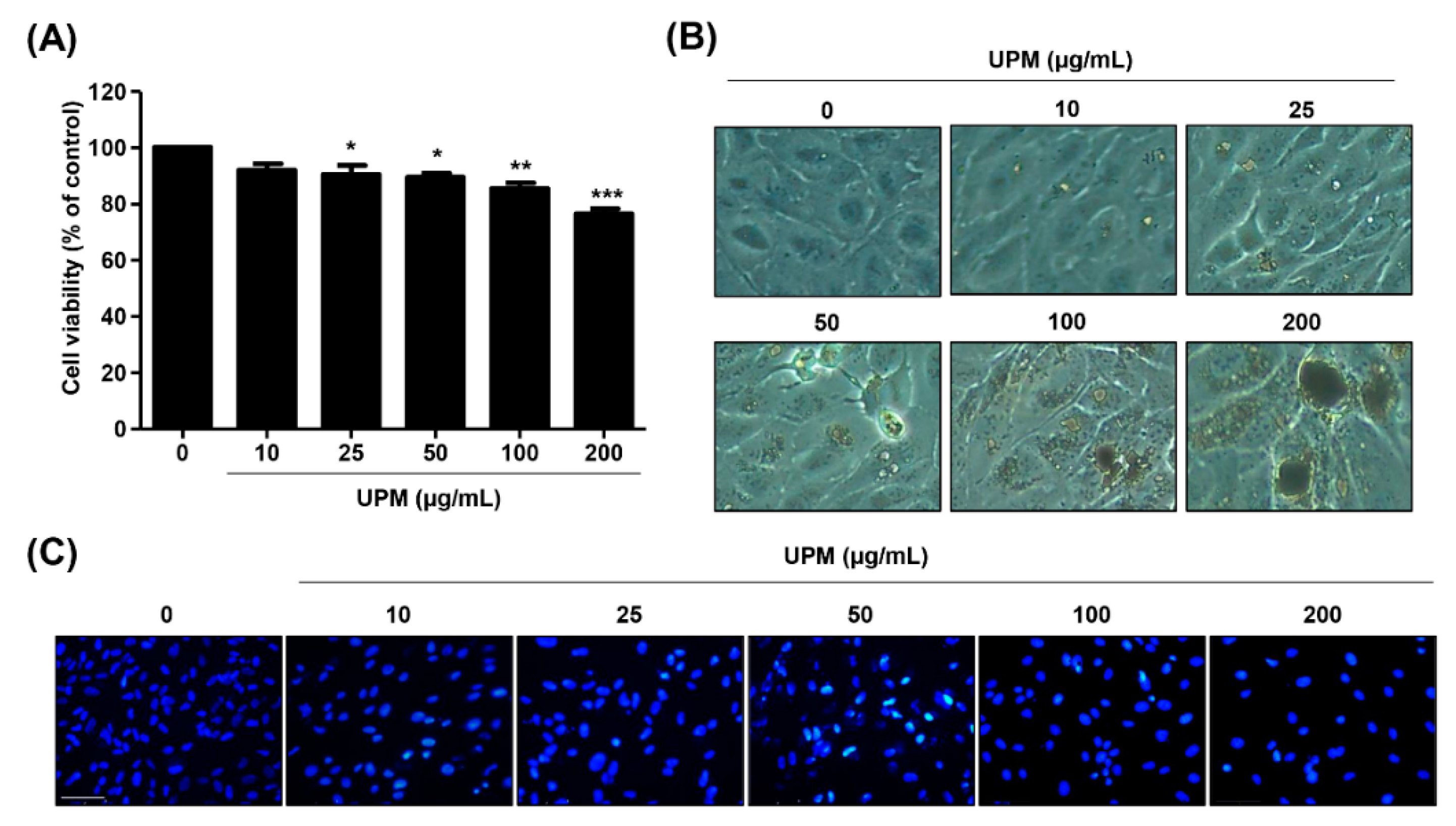
3.1. UPM Has Cytotoxicity in ARPE-19 Cells
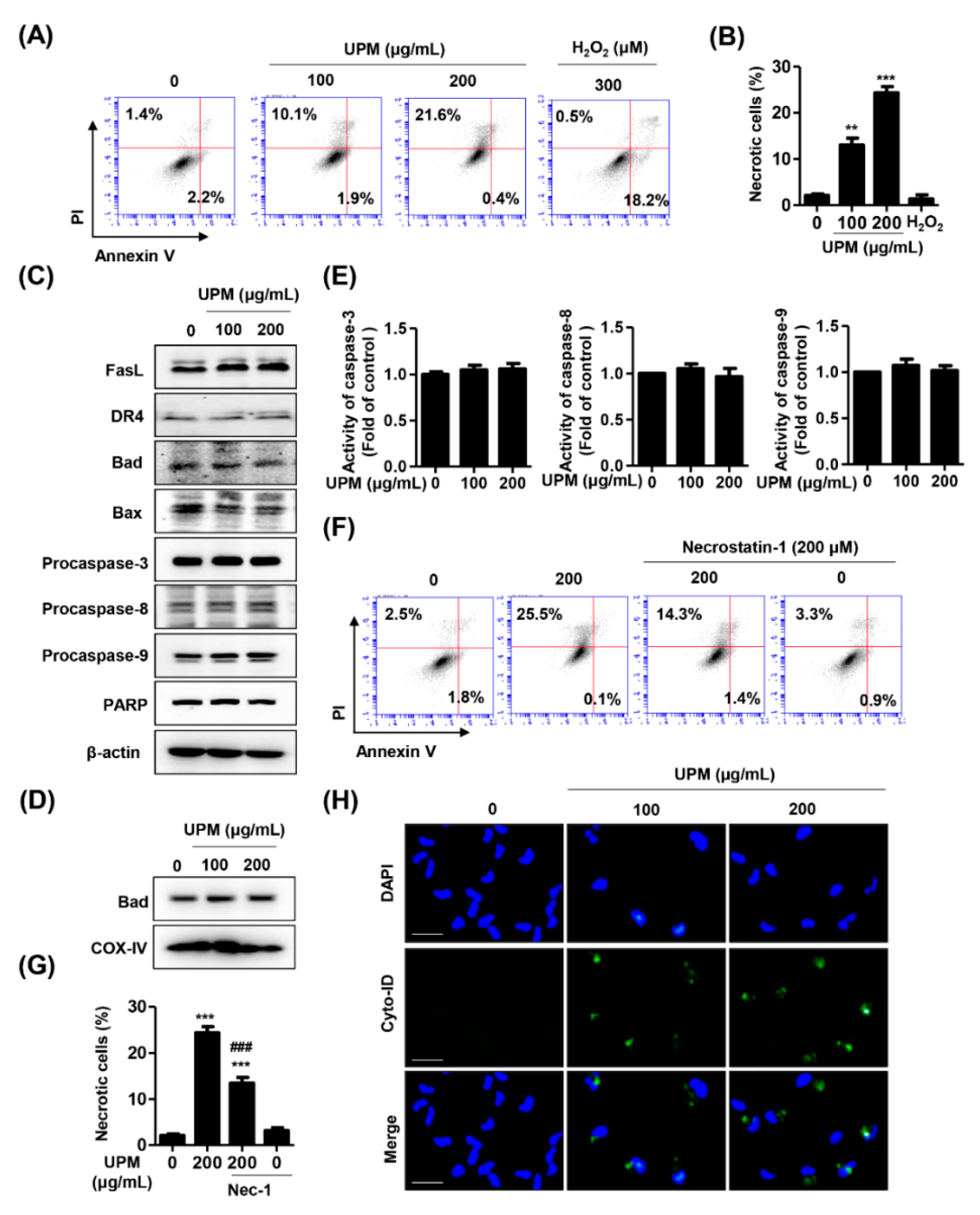
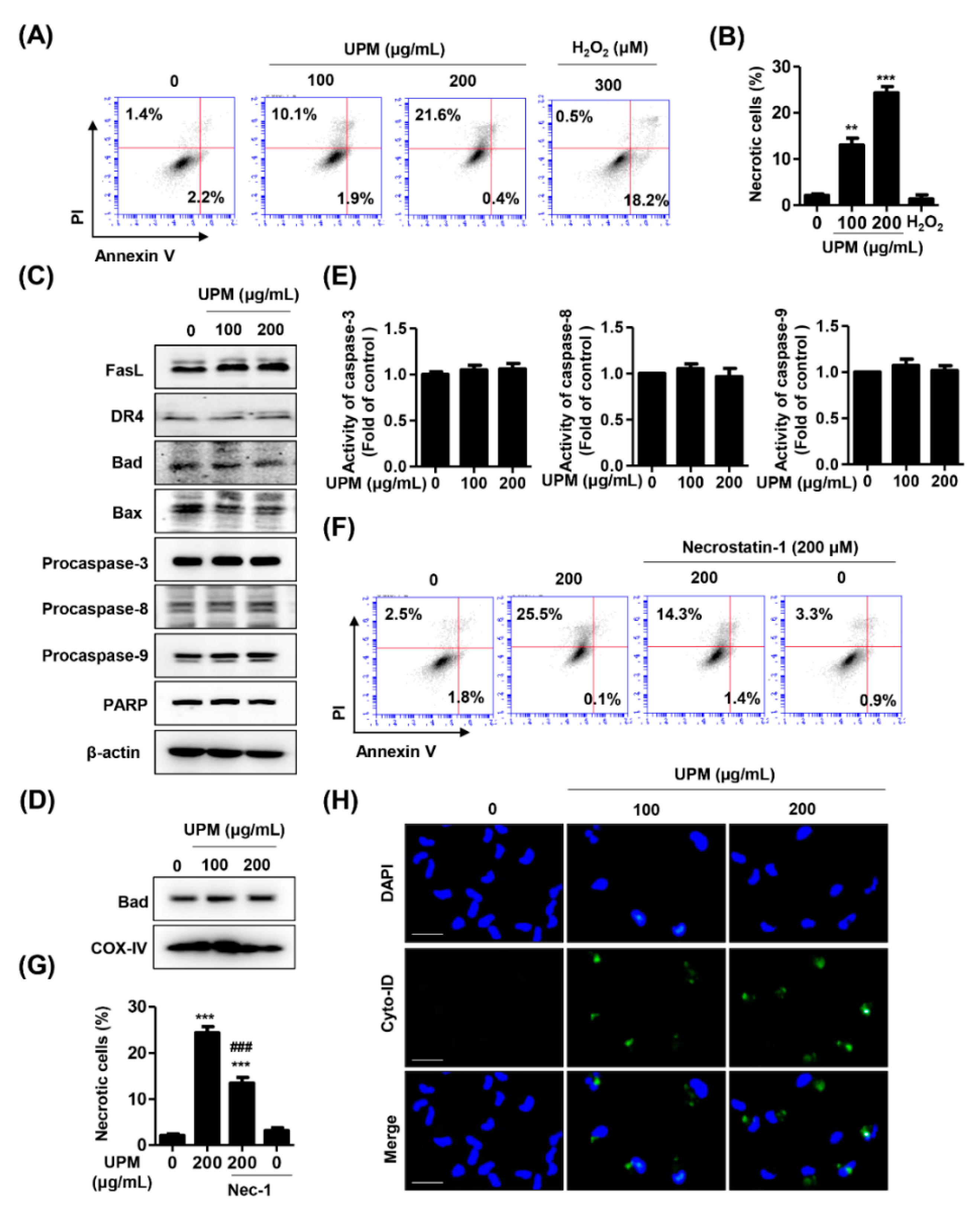
3.2. UPM Induces Necrosis and Autophagy but Not Apoptosis in ARPE-19 Cells
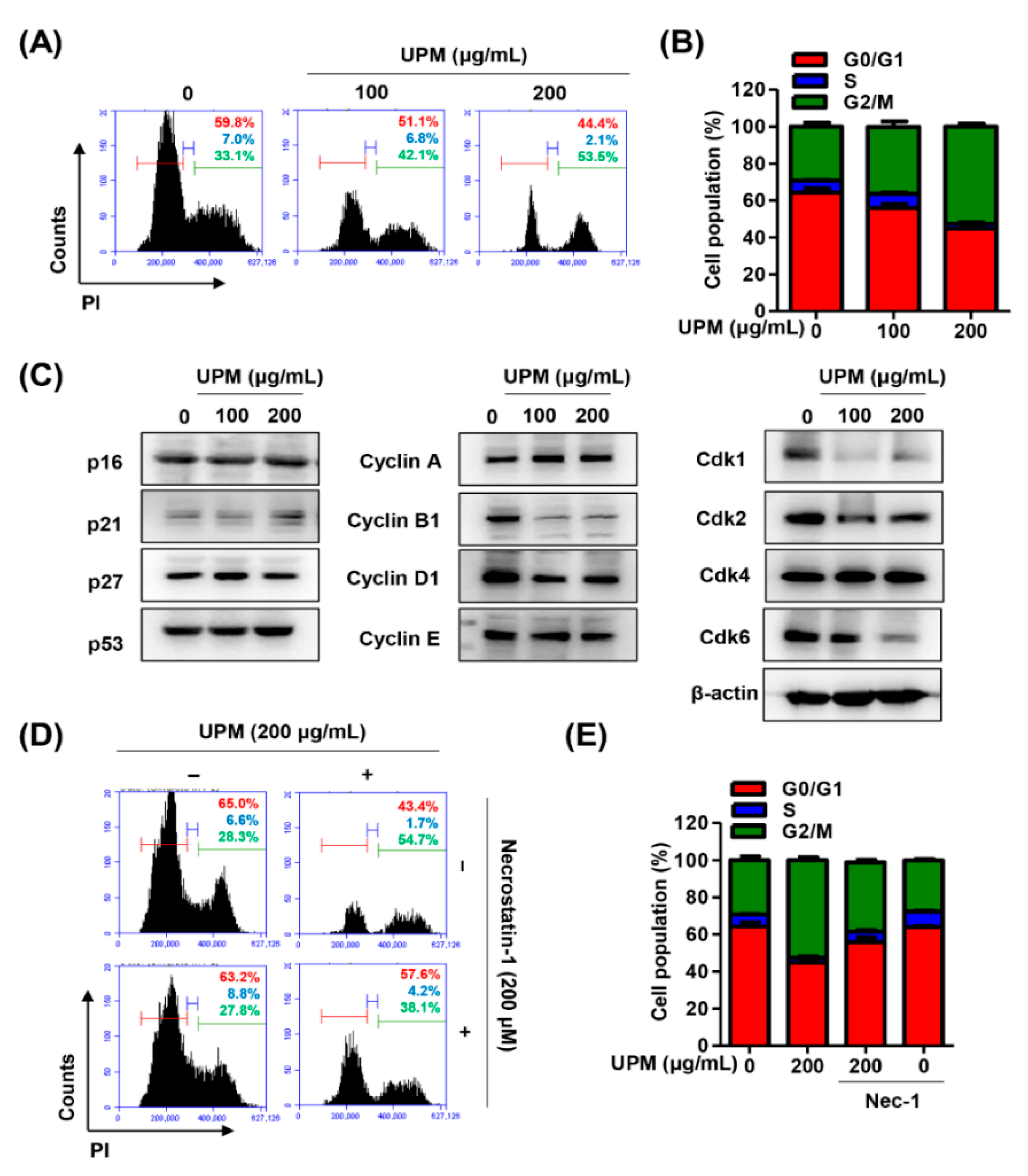
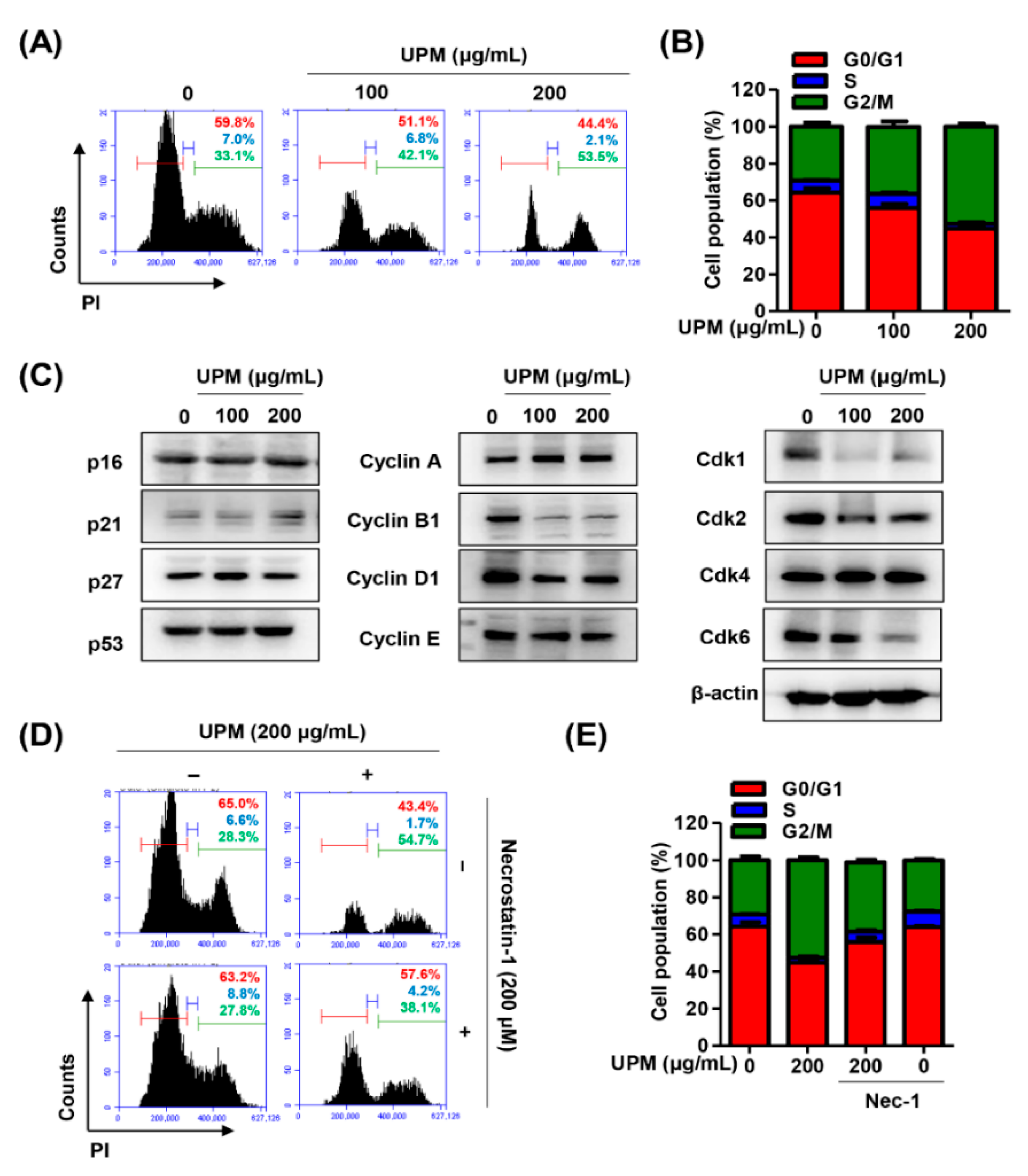
3.3. UPM Promotes Cell Cycle Arrest at the G2/M Phase in ARPE-19 Cells
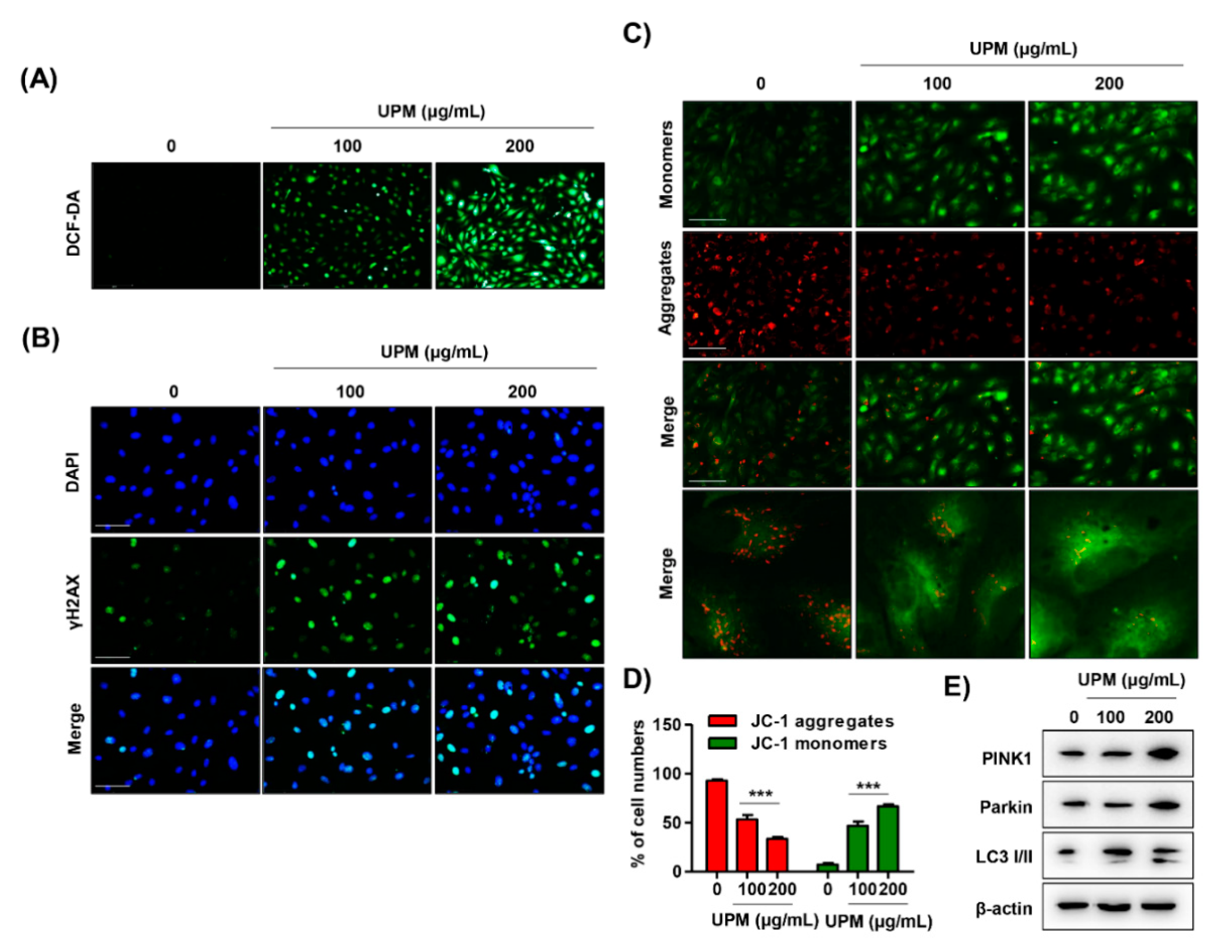
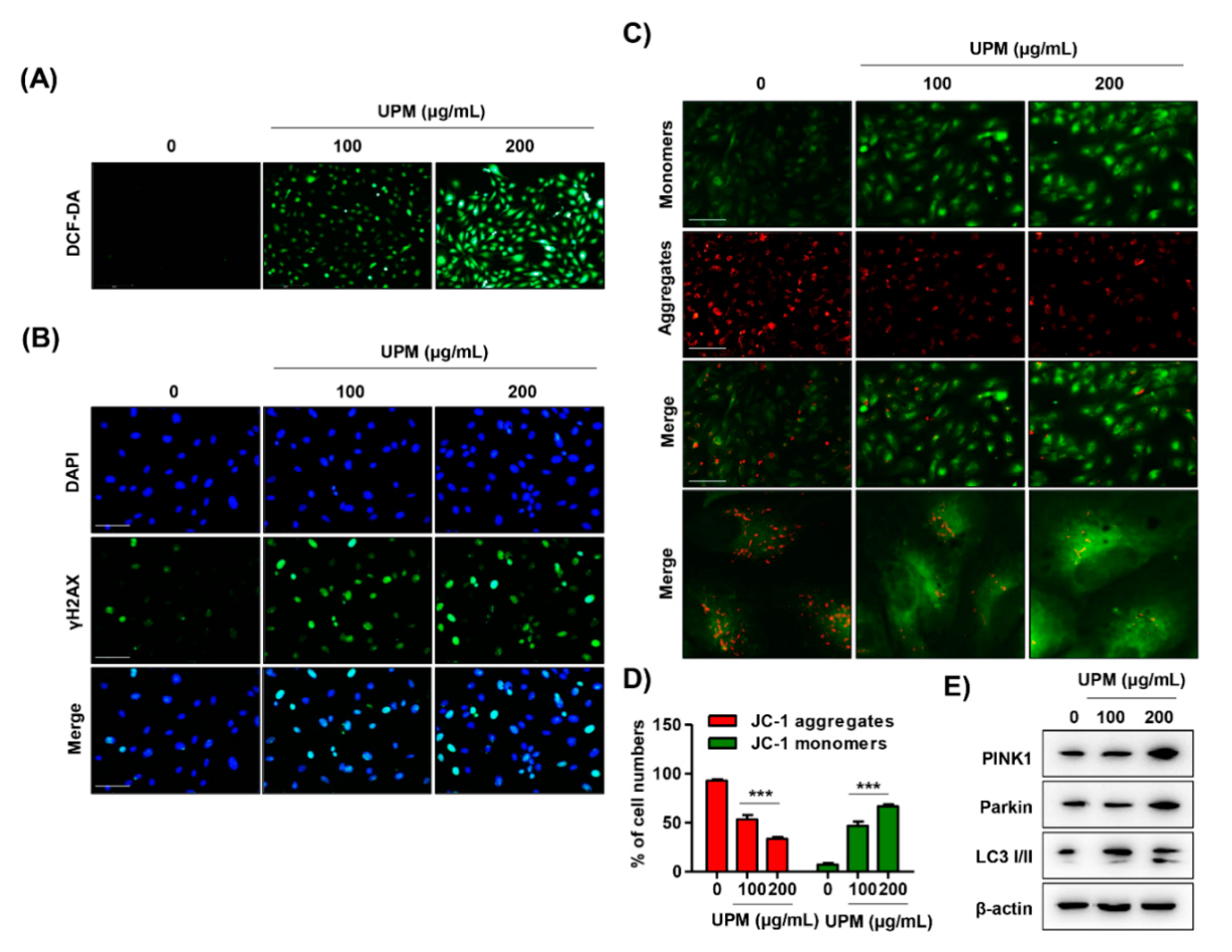
3.4. UPM Disrupts DNA and Mitochondria in ARPE-19 Cells
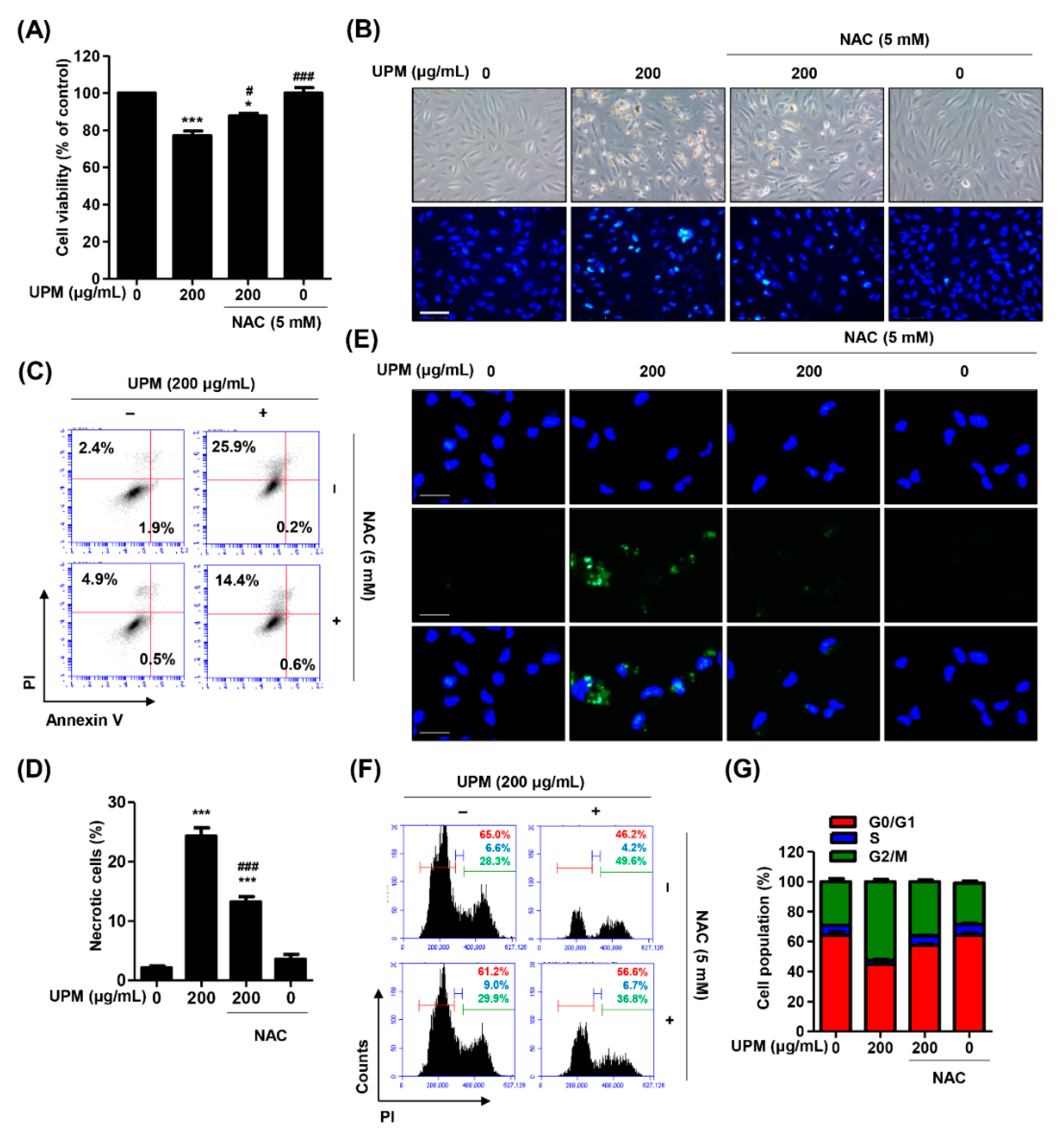
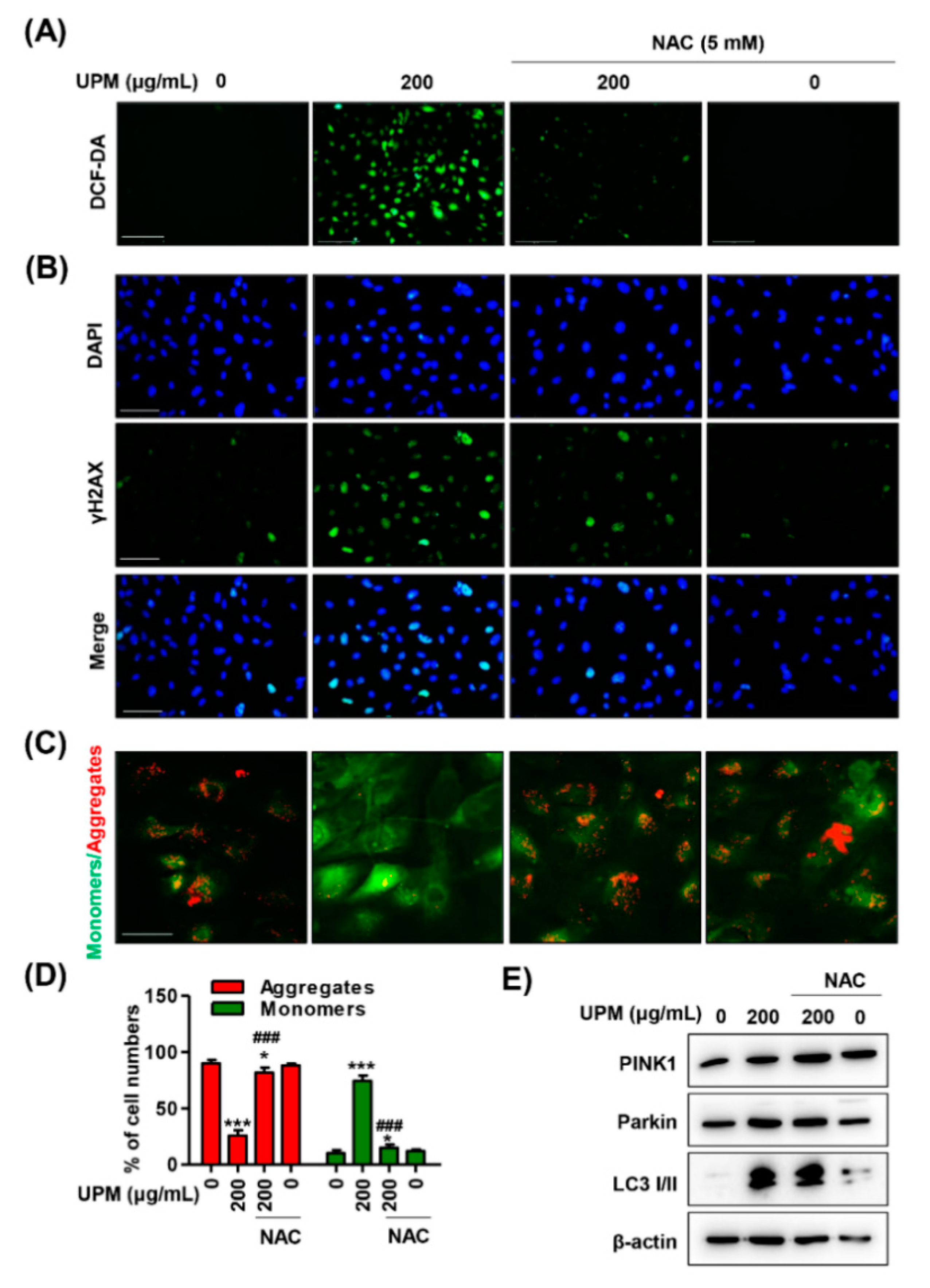
3.5. The Excess ROS Induced by UPM Triggers Necrosis, Autophagy, and Cell Cycle Arrest in ARPE-19 Cells
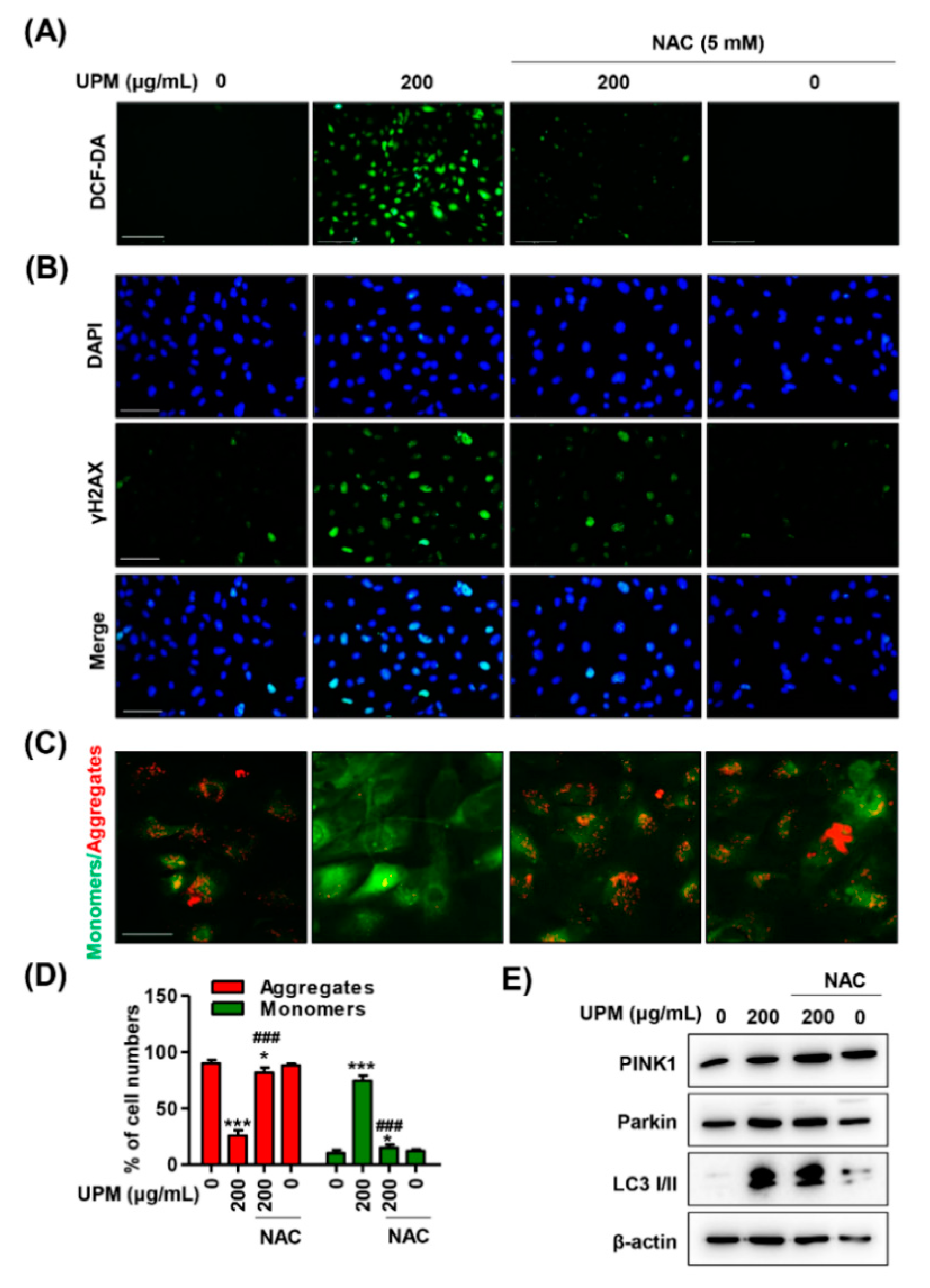
3.6. ROS Directly Regulate UPM-Induced DNA and Mitochondrial Damage in ARPE-19 Cells
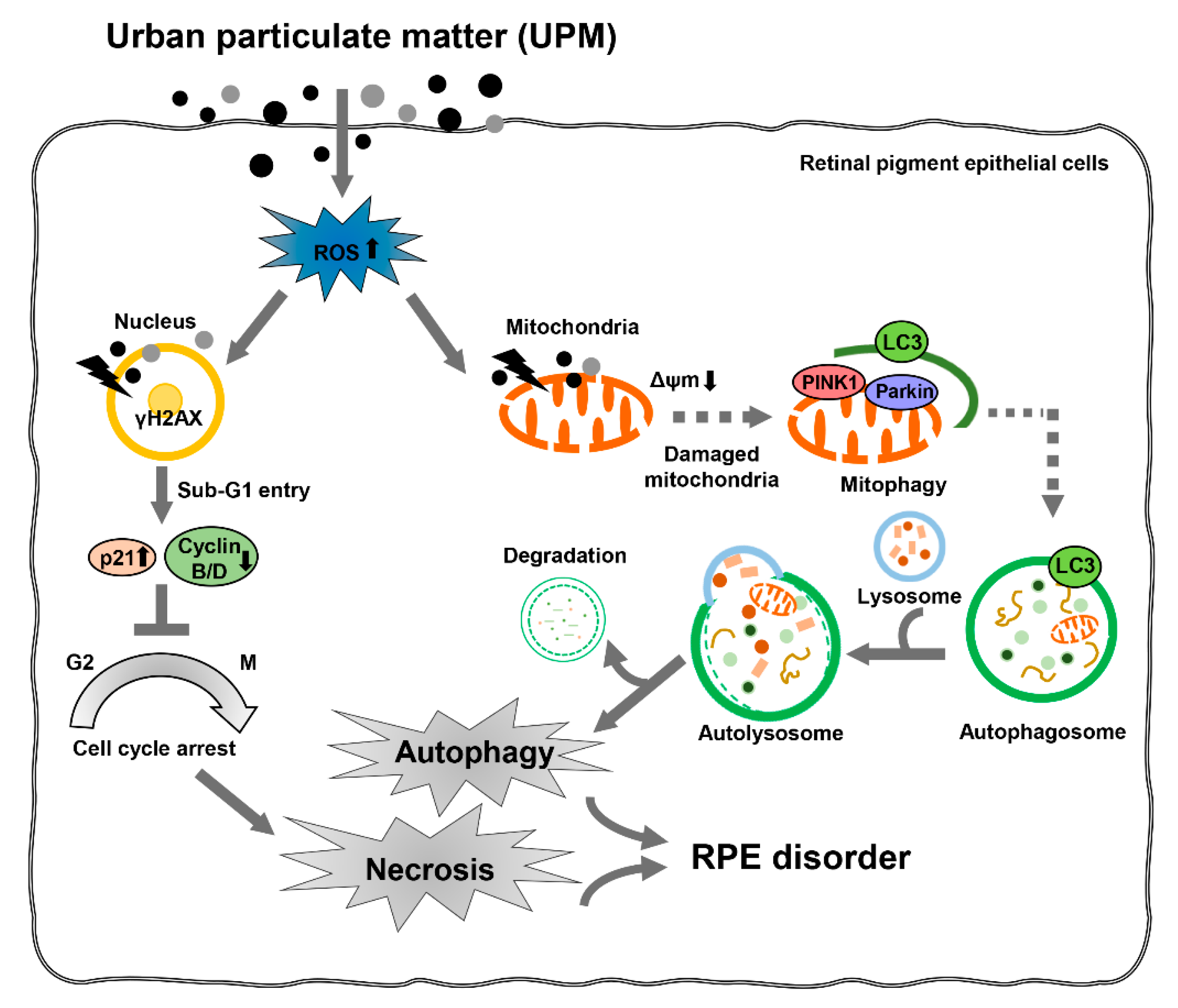
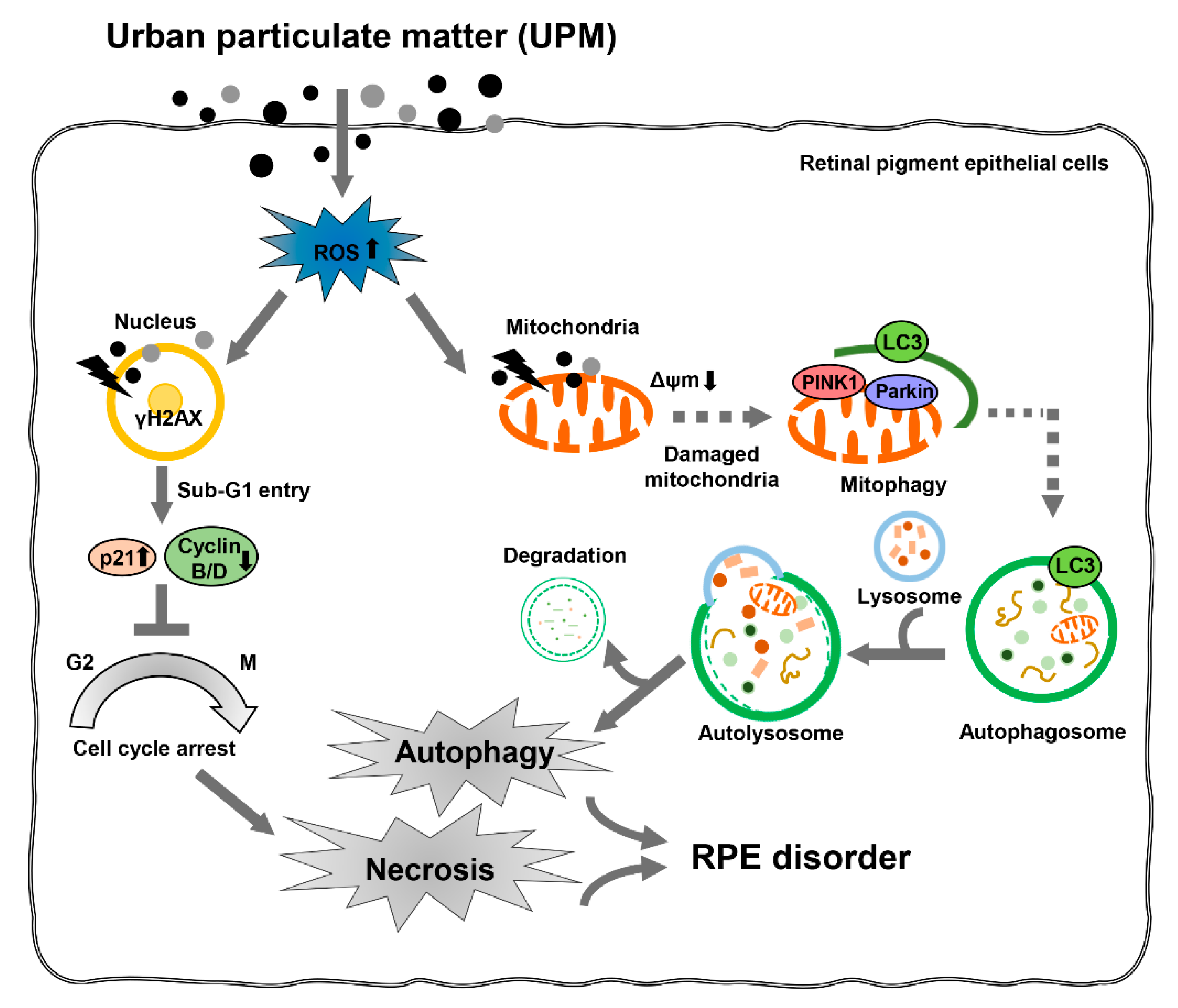
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Q.; Li, J.; Yang, J.; Chen, Y.; Li, Y.; Li, S.; Xie, C.; Chen, C.; Wang, L.; Wang, L.; et al. Seasonal characterization of aerosol composition and sources in a polluted city in Central China. Chemosphere 2020, 258, 127310. [Google Scholar] [CrossRef]
- Ngarambe, J.; Joen, S.J.; Han, C.H.; Yun, G.Y. Exploring the relationship between particulate matter, CO, SO2, NO2, O3 and urban heat island in Seoul, Korea. J. Hazard. Mater. 2021, 403, 123615. [Google Scholar] [CrossRef]
- Kelly, F.J. Urban air quality and health: Two steps forward, one step back. Eur. Respir. J. 2019, 53, 1900280. [Google Scholar] [CrossRef]
- Bakolis, I.; Hammoud, R.; Stewart, R.; Beevers, S.; Dajnak, D.; MacCrimmon, S.; Broadbent, M.; Pritchard, M.; Shiode, N.; Fecht, D.; et al. Mental health consequences of urban air pollution: Prospective population-based longitudinal survey. Soc. Psychiatry Psychiatr. Epidemiol. 2020, 1–13. [Google Scholar] [CrossRef]
- Crobeddu, B.; Aragao-Santiago, L.; Bui, L.C.; Boland, S.; Squiban, B.A. Oxidative potential of particulate matter 2.5 as predictive indicator of cellular stress. Environ. Pollut. 2017, 230, 125–133. [Google Scholar] [CrossRef]
- Atkinson, R.W.; Fuller, G.W.; Anderson, H.R.; Harrison, R.M.; Armstrong, B. Urban ambient particle metrics and health: A time-series analysis. Epidemiology 2010, 21, 501–511. [Google Scholar] [CrossRef]
- Ritz, B.; Hoffmann, B.; Peters, A. The effects of fine dust, ozone, and nitrogen dioxide on health. Dtsch. Arztebl. Int. 2019, 51–52, 881–886. [Google Scholar] [CrossRef]
- Mu, G.; Zhou, M.; Wang, B.; Cao, L.; Yang, S.; Qiu, W.; Nie, X.; Ye, Z.; Zhou, Y.; Chen, W. Personal PM2.5 exposure and lung function: Potential mediating role of systematic inflammation and oxidative damage in urban adults from the general population. Sci. Total Environ. 2020, 755, 142522. [Google Scholar] [CrossRef]
- Lu, Y.; Lin, S.; Fatmi, Z.; Malashock, D.; Hussain, M.M.; Siddique, A.; Carpenter, D.O.; Lin, Z.; Khwaja, H.A. Assessing the association between fine particulate matter (PM2.5) constituents and cardiovascular diseases in a mega-city of Pakistan. Environ. Pollut. 2019, 252, 1412–1422. [Google Scholar] [CrossRef]
- Shahbaz, M.A.; Martikainen, M.V.; Rönkkö, T.J.; Komppula, M.; Jalava, P.I.; Roponen, M. Urban air PM modifies differently immune defense responses against bacterial and viral infections in vitro. Environ. Res. 2020, 192, 110244. [Google Scholar] [CrossRef]
- Chen, X.; Guo, J.; Huang, Y.; Liu, S.; Huang, Y.; Zhang, Z.; Zhang, F.; Lu, Z.; Li, F.; Zheng, J.C.; et al. Urban airborne PM2.5-activated microglia mediate neurotoxicity through glutaminase-containing extracellular vesicles in olfactory bulb. Environ. Pollut. 2020, 264, 114716. [Google Scholar] [CrossRef]
- Kitakaze, T.; Yoshioka, Y.; Furuyashiki, T.; Ashida, H. Enzymatically synthesized glycogen protects inflammation induced by urban particulate matter in normal human epidermal keratinocytes. J. Clin. Biochem. Nutr. 2020, 67, 29–35. [Google Scholar] [CrossRef]
- Gutiérrez, A.M.; Giuliani, D.; Porta, A.A.; Andrinolo, D. Relationship between ocular surface alterations and concentrations of aerial particulate matter. J. Ophthalmic Vis. Res. 2019, 14, 419–427. [Google Scholar] [CrossRef]
- Fu, Q.; Mo, Z.; Lyu, D.; Zhang, L.; Qin, Z.; Tang, Q.; Yin, H.; Xu, P.; Wu, L.; Lou, X.; et al. Air pollution and outpatient visits for conjunctivitis: A case-crossover study in Hangzhou, China. Environ. Pollut. 2017, 231, 1344–1350. [Google Scholar] [CrossRef]
- Mo, Z.; Fu, Q.; Lyu, D.; Zhang, L.; Qin, Z.; Tang, Q.; Yin, H.; Xu, P.; Wu, L.; Wang, X.; et al. Impacts of air pollution on dry eye disease among residents in Hangzhou, China: A case-crossover study. Environ. Pollut. 2019, 246, 183–189. [Google Scholar] [CrossRef]
- Kang, W.S.; Choi, H.; Jang, G.; Lee, K.H.; Kim, E.; Kim, K.J.; Jeong, G.Y.; Kim, J.S.; Na, C.S.; Kim, S. Long-term exposure to urban particulate matter on the ocular surface and the incidence of deleterious changes in the cornea, conjunctiva and retina in rats. Int. J. Mol. Sci. 2020, 21, 4976. [Google Scholar] [CrossRef]
- Lee, T.G.; Hyun, S.W.; Jo, K.; Park, B.; Lee, I.S.; Song, S.J.; Kim, C.S. Achyranthis radix extract improves urban particulate matter-induced dry eye disease. Int. J. Environ. Res. Public Health 2019, 16, 3229. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Calderón-Garcidueñas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.H.; Hsu, P.Y.; Lin, C.J.; Lin, C.L.; Juo, S.H.; Liang, C.L. Traffic-related air pollutants increase the risk for age-related macular degeneration. J. Investig. Med. 2019, 67, 1076–1081. [Google Scholar] [CrossRef]
- Louwies, T.; Panis, L.I.; Kicinski, M.; De Boever, P.; Nawrot, T.S. Retinal microvascular responses to short-term changes in particulate air pollution in healthy adults. Environ. Health Perspect. 2013, 121, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.C.; Huang, C.C.; Chin, W.S.; Chen, B.Y.; Chan, C.C.; Guo, Y.L. Association between air pollution exposure and diabetic retinopathy among diabetics. Environ. Res. 2020, 181, 108960. [Google Scholar] [CrossRef]
- Provost, E.B.; Panis, L.I.; Saenen, N.D.; Kicinski, M.; Louwies, T.; Vrijens, K.; De Boever, P.; Nawrot, T.S. Recent versus chronic fine particulate air pollution exposure as determinant of the retinal microvasculature in school children. Environ. Res. 2017, 159, 103–110. [Google Scholar] [CrossRef]
- Kim, S.; Park, H.; Park, H.; Joung, B.; Kim, E. The acute respiratory exposure by intratracheal instillation of Sprague-Dawley rats with diesel particulate matter induces retinal thickening. Cutan. Ocul. Toxicol. 2016, 5, 275–280. [Google Scholar] [CrossRef]
- Tien, P.T.; Lin, H.J.; Tsai, Y.Y.; Lim, Y.P.; Chen, C.S.; Chang, C.Y.; Lin, C.J.; Chen, J.J.; Wu, S.M.; Huang, Y.J.; et al. Perfluorooctanoic acid in indoor particulate matter triggers oxidative stress and inflammation in corneal and retinal cells. Sci. Rep. 2020, 10, 15702. [Google Scholar] [CrossRef]
- Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Park, C.; Hong, S.H.; Kim, G.Y.; Song, K.S.; Hyun, J.W.; et al. Diesel particulate matter2.5 promotes epithelial-mesenchymal transition of human retinal pigment epithelial cells via generation of reactive oxygen species. Environ. Pollut. 2020, 262, 114301. [Google Scholar] [CrossRef]
- Mori, I.; Sun, Z.; Ukachi, M.; Nagano, K.; McLeod, C.W.; Cox, A.G.; Nishikawa, M. Development and certification of the new NIES CRM 28: Urban aerosols for the determination of multielements. Anal. Bioanal. Chem. 2008, 391, 1997–2003. [Google Scholar] [CrossRef]
- Choi, Y.H. Trans-cinnamaldehyde protects C2C12 myoblasts from DNA damage, mitochondrial dysfunction and apoptosis caused by oxidative stress through inhibiting ROS production. Genes Genom. 2020. [Google Scholar] [CrossRef]
- Zhang, N.; Li, F.; Gao, J.; Zhang, S.; Wang, Q. Osteopontin accelerates the development and metastasis of bladder cancer via activating JAK1/STAT1 pathway. Genes Genom. 2020, 42, 467–475. [Google Scholar] [CrossRef]
- Hwangbo, H.; Kim, S.Y.; Lee, H.; Park, S.H.; Hong, S.H.; Park, C.; Kim, G.Y.; Leem, S.H.; Hyun, J.W.; Cheong, J.; et al. Auranofin enhances sulforaphane-mediated apoptosis in hepatocellular carcinoma Hep3B cells through inactivation of the PI3K/Akt signaling pathway. Biomol. Ther. 2020, 28, 443–455. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Hong, Y.; Lee, H.; Tran, Q.; Kim, C.; Kwon, S.H.; Park, J.; Park, J.; Kim, S.H. Myristoylated TMEM39AS41, a cell-permeable peptide, causes lung cancer cell death. Toxicol. Res. 2020, 36, 123–130. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.J.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Hong, S.H.; Jeong, J.W.; Han, M.H.; Choi, S.H.; et al. Induction of G2/M cell cycle arrest and apoptosis by genistein in human bladder cancer T24 cells through inhibition of the ROS-dependent PI3k/Akt signal transduction pathway. Antioxidants 2019, 8, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willermain, F.; Scifo, L.; Weber, C.; Caspers, L.; Perret, J.; Delporte, C. Potential interplay between hyperosmolarity and inflammation on retinal pigmented epithelium in pathogenesis of diabetic retinopathy. Int. J. Mol. Sci. 2018, 19, 1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin. Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Hanus, J.; Kalinowska-Herok, M.; Widlak, P. The major apoptotic endonuclease DFF40/CAD is a deoxyribose-specific and double-strand-specific enzyme. Apoptosis 2008, 13, 377–382. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.M.; Huang, D.Y.; Sekar, P.; Hsu, S.H.; Lin, W.W. Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J. Biomed. Sci. 2019, 26, 40. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between autophagy and the ubiquitin-proteasome system and its role in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, E.; Lotery, A.J.; Tumbarello, D.A.; Ratnayaka, J.A. Impaired cargo clearance in the retinal pigment epithelium (RPE) underlies irreversible blinding diseases. Cells 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanus, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liang, Y.; Murphy, S.F.; Huang, A.; Shen, H.; Kelly, D.F.; Sobrado, P.; Sheng, Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015, 11, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.H.; Wu, Y.F.; Wang, P.L.; Wu, Y.P.; Li, Z.Y.; Zhao, Y.; Zhou, J.S.; Zhu, C.; Cao, C.; Mao, Y.Y.; et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy 2016, 12, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Zhang, F.; Rui, W.; Long, F.; Wang, L.; Feng, Z.; Chen, D.; Ding, W. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol. Vitr. 2013, 27, 1762–1770. [Google Scholar] [CrossRef]
- Fu, Q.; Lyu, D.; Zhang, L.; Qin, Z.; Tang, Q.; Yin, H.; Lou, X.; Chen, Z.; Yao, K. Airborne particulate matter (PM2.5) triggers autophagy in human corneal epithelial cell line. Environ. Pollut. 2017, 227, 314–322. [Google Scholar] [CrossRef]
- Lyu, D.; Chen, Z.; Almansoob, S.; Chen, H.; Ye, Y.; Song, F.; Zhang, L.; Qin, Z.; Tang, Q.; Yin, H.; et al. Transcriptomic profiling of human corneal epithelial cells exposed to airborne fine particulate matter (PM2.5). Ocul. Surf. 2020, 18, 554–564. [Google Scholar] [CrossRef]
- Pearce, A.K.; Humphrey, T.C. Integrating stress-response and cell-cycle checkpoint pathways. Trends Cell Biol. 2001, 11, 426–433. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair 2004, 3, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, P.; Tyers, M. How cells coordinate growth and division. Curr. Biol. 2004, 14, 1014–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtieri, M.; Ovrevik, J.; Mollerup, S.; Asare, N.; Longhin, E.; Dahlman, H.J.; Camatini, M.; Holme, J.A. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA, mitochondria, AhR binding and spindle organization. Mutat. Res. 2011, 713, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Longhin, E.; Holme, J.A.; Gutzkow, K.B.; Arlt, V.M.; Kucab, J.E.; Camatini, M.; Gualtieri, M. Cell cycle alterations induced by urban PM2.5 in bronchial epithelial cells: Characterization of the process and possible mechanisms involved. Part. Fibre. Toxicol. 2013, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Shi, Y.; Asweto, C.O.; Feng, L.; Yang, X.; Zhang, Y.; Hu, H.; Duan, J.; Sun, Z. Fine particle matters induce DNA damage and G2/M cell cycle arrest in human bronchial epithelial BEAS-2B cells. Environ. Sci. Pollut. Res. Int. 2017, 24, 25071–25081. [Google Scholar] [CrossRef]
- Zhang, J.; Ghio, A.J.; Gao, M.; Wei, K.; Rosen, G.D.; Upadhyay, D. Ambient particulate matter induces alveolar epithelial cell cycle arrest: Role of G1 cyclins. FEBS Lett. 2007, 581, 5315–5320. [Google Scholar] [CrossRef] [Green Version]
- Al Bitar, S.; Gali-Muhtasib, H. The role of the cyclin dependent kinase inhibitor p21cip1/waf1 in targeting cancer: Molecular mechanisms and novel therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [Green Version]
- Lezaja, A.; Altmeyer, M. Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle 2018, 17, 24–32. [Google Scholar] [CrossRef]
- Fragkos, M.; Jurvansuu, J.; Beard, P. H2AX is required for cell cycle arrest via the p53/p21 pathway. Mol. Cell Biol. 2009, 29, 2828–2840. [Google Scholar] [CrossRef] [Green Version]
- Leni, Z.; Künzi, L.; Geiser, M. Air pollution causing oxidative stress. Curr. Opin. Toxicol. 2020, 20–21, 1–8. [Google Scholar] [CrossRef]
- Lodovici, M.; Bigagli, E. Oxidative stress and air pollution exposure. J. Toxicol. 2011, 2011, 487074. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Cho, H.K.; Shin, H.J.; Park, K.H.; Lim, H.B. Comparison of mutagenic activities of various ultra-fine particles. Toxicol. Res. 2018, 34, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Garçon, G. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Li, W.; Niu, B.; Li, J.; Sun, J.; Qin, M.; Zhou, Z. Mitochondrial dysfunction in endothelial cells induced by airborne fine particulate matter (<2.5 μm). J. Appl. Toxicol. 2019, 39, 1424–1432. [Google Scholar] [CrossRef]
- Bhargava, A.; Tamrakar, S.; Aglawe, A.; Lad, H.; Srivastava, R.K.; Mishra, D.K.; Tiwari, R.; Chaudhury, K.; Goryacheva, I.Y.; Mishra, P.K. Ultrafine particulate matter impairs mitochondrial redox homeostasis and activates phosphatidylinositol 3-kinase mediated DNA damage responses in lymphocytes. Environ. Pollut. 2018, 234, 406–419. [Google Scholar] [CrossRef]
- Cui, Y.H.; Hu, Z.X.; Gao, Z.X.; Song, X.L.; Feng, Q.Y.; Yang, G.; Li, Z.J.; Pan, H.W. Airborne particulate matter impairs corneal epithelial cells migration via disturbing FAK/RhoA signaling pathway and cytoskeleton organization. Nanotoxicology 2018, 12, 312–324. [Google Scholar] [CrossRef]
- Somayajulu, M.; Ekanayaka, S.; McClellan, S.A.; Bessert, D.; Pitchaikannu, A.; Zhang, K.; Hazlett, L.D. Airborne particulates affect corneal homeostasis and immunity. Invest. Ophthalmol. Vis. Sci. 2020, 61, 23. [Google Scholar] [CrossRef]
- Sachdeva, K.; Do, D.C.; Zhang, Y.; Hu, X.; Chen, J.; Gao, P. Environmental exposures and asthma development: Autophagy, mitophagy, and cellular senescence. Front. Immunol. 2019, 10, 2787. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Qiu, Y.N.; Wang, G.H.; Zhou, F.; Hao, J.J.; Tian, L.; Guan, L.F.; Geng, X.K.; Ding, Y.C.; Wu, H.W.; Zhang, K.Z. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol. Environ. Saf. 2019, 167, 178–187. [Google Scholar] [CrossRef]
- Wang, H.T.; Lin, J.H.; Yang, C.H.; Haung, C.H.; Weng, C.W.; Maan-Yuh Lin, A.; Lo, Y.L.; Chen, W.S.; Tang, M.S. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kim, D.H.; Kim, J.-H.; Park, S.-K.; Jeong, J.-W.; Kim, M.-Y.; Hong, S.-H.; Song, K.S.; Kim, G.-Y.; Hyun, J.W.; et al. Urban Aerosol Particulate Matter Promotes Necrosis and Autophagy via Reactive Oxygen Species-Mediated Cellular Disorders that Are Accompanied by Cell Cycle Arrest in Retinal Pigment Epithelial Cells. Antioxidants 2021, 10, 149. https://doi.org/10.3390/antiox10020149
Lee H, Kim DH, Kim J-H, Park S-K, Jeong J-W, Kim M-Y, Hong S-H, Song KS, Kim G-Y, Hyun JW, et al. Urban Aerosol Particulate Matter Promotes Necrosis and Autophagy via Reactive Oxygen Species-Mediated Cellular Disorders that Are Accompanied by Cell Cycle Arrest in Retinal Pigment Epithelial Cells. Antioxidants. 2021; 10(2):149. https://doi.org/10.3390/antiox10020149
Chicago/Turabian StyleLee, Hyesook, Da Hye Kim, Jeong-Hwan Kim, Seh-Kwang Park, Ji-Won Jeong, Mi-Young Kim, Seok-Ho Hong, Kyoung Seob Song, Gi-Young Kim, Jin Won Hyun, and et al. 2021. "Urban Aerosol Particulate Matter Promotes Necrosis and Autophagy via Reactive Oxygen Species-Mediated Cellular Disorders that Are Accompanied by Cell Cycle Arrest in Retinal Pigment Epithelial Cells" Antioxidants 10, no. 2: 149. https://doi.org/10.3390/antiox10020149
APA StyleLee, H., Kim, D. H., Kim, J.-H., Park, S.-K., Jeong, J.-W., Kim, M.-Y., Hong, S.-H., Song, K. S., Kim, G.-Y., Hyun, J. W., & Choi, Y. H. (2021). Urban Aerosol Particulate Matter Promotes Necrosis and Autophagy via Reactive Oxygen Species-Mediated Cellular Disorders that Are Accompanied by Cell Cycle Arrest in Retinal Pigment Epithelial Cells. Antioxidants, 10(2), 149. https://doi.org/10.3390/antiox10020149