Abstract
In the present paper, we describe the biological activity of the newly designed and synthesized series N-substituted 3,4-pyrroledicarboximides 2a–2p. The compounds 2a–2p were obtained in good yields by one-pot, three-component condensation of pyrrolo[3,4-c]pyrrole scaffold (1a–c) with secondary amines and an excess of formaldehyde solution in C2H5OH. The structural properties of the compounds were characterized by 1H NMR, 13C NMR FT-IR, MS, and elemental analysis. Moreover, single crystal X-ray diffraction has been recorded for compound 2h. The colorimetric inhibitor screening assay was used to obtain their potencies to inhibit COX-1 and COX-2 enzymes. According to the results, all of the tested compounds inhibited the activity of COX-1 and COX-2. Theoretical modeling was also applied to describe the binding properties of compounds towards COX-1 and COX-2 cyclooxygenase isoform. The data were supported by QSAR study.
1. Introduction
An important group of scaffolds for the synthesis of bioactive compounds such as anti-microbial [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15], antitumor [16,17,18,19,20,21,22,23], antihyperlipidemic, hypoglycemic [24,25], and carbonic anhydrase inhibitors [26,27,28,29,30,31] are cyclic imides. The imide core is also present in compounds whose activity is related to CNS activity [32,33,34,35,36]. In particular, much attention has been paid to their ability to inhibit cyclooxygenases, anti-inflammatory, and analgetic activity [37,38,39,40,41,42,43,44,45,46,47]. To date, several drugs with cyclic imide structure have been approved, including lurasidone, buspirone, ethosuximide, mesuximide, and the IMiDs class includes thalidomide and its analogues lenalidomide, pomalidomide, apremilast (Figure 1).
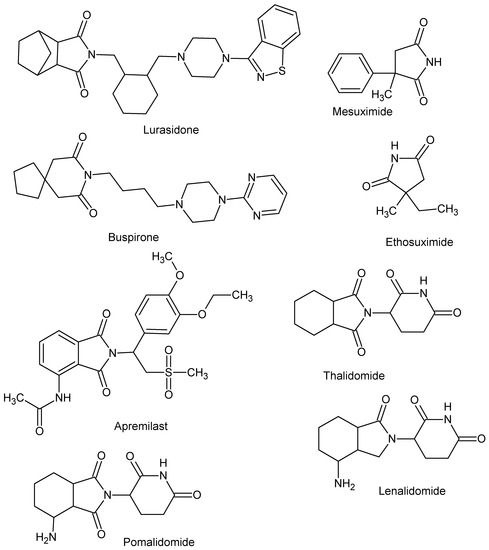
Figure 1.
Drugs with cyclic imide structure.
The structure of many significant natural products, for example cladoniamide A, lamprolobine, migrastatine, julocrotine, is based on the cyclic imide motif [48]. One of the natural precursors used in the synthesis of imide compounds is phyllanthimide isolated from leaves of Phyllanthus sellowianus (Euphorbiaceae) [49]. The properties of the imide derivatives are strongly dependent on the nature of substituent in the imide ring. For example, the size and electrophilic character of substituents influence their steric properties [50]. The presence of specific groups with nitrogen and oxygen atoms is responsible for pharmacological consequences [51,52]. Additionally, imide based compounds are often neutral and hydrophobic, which determines their ability to penetrate biological membranes [9,50,53].
We have recently reported on the synthesis and COX-1/COX-2 inhibition of N-substituted cyclic imides (Figure 2) [39].
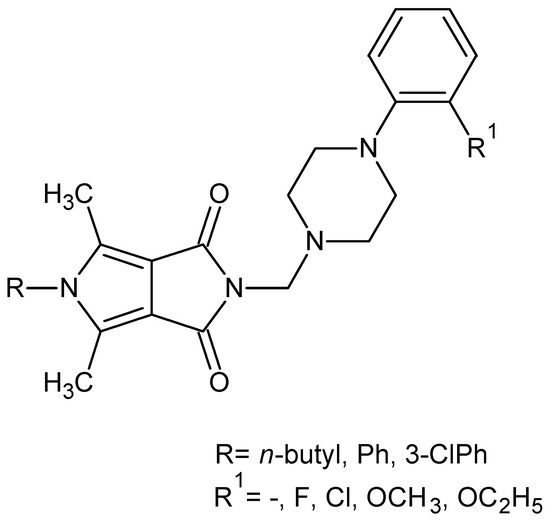
Figure 2.
Pyrrolo[3,4-c]pyrroles with activity as a COX-1 and COX-2 inhibitors.
The obtained structures (Figure 2) inhibited less COX-2 and more COX-1 than the reference drug—meloxicam. Therefore, their selectivity ratio (COX-2/COX-1) was more than twice as high as meloxicam [39].
Based on the fragmental data in this paper, we undertook further modifications of the 4-arylpiperazinyl fragment (Figure 2) in order to increase COX-2 inhibitions. Structural modifications were achieved by varying the 4-substituent on the piperazine moiety and the synthesis of analogues with 4-arylpiperidine or morpholine pharmacophore for the structure–activity relationship (SAR) studies. In all the new compounds, we retained the one-carbon central alkyl chain as an optimal spacer.
The new set of compounds was investigated in terms of their activity as potential COX-1 and COX-2 inhibitors by colorimetric inhibitor screening assay [54]. Moreover, the binding mode of designed derivatives was obtained based on molecular docking results and previously reported data.
2. Results and Discussion
2.1. Chemistry
The new derivatives 2a–2p were obtained as a result of a one-step synthesis, as shown in Scheme 1. The key intermediates, imides 1a–1c, were prepared from ethyl α,β-diacetylsuccinate in a five-step synthesis according to known methods [39,55]. The preparation of the final compounds 2a–2p involved condensation of the substrate 1a-1c and the appropriate derivatives of 1-substituted piperazine 2a–2j (arylpiperazines 2a–2e, heteroarylpiperazine 2f, cyclohexylpiperazines 2g–2j, hydroxyethylpiperazine 2j), piperazine 2k, morpholine 2l and 4-arylpiperidine 2m–2p (Scheme 1).
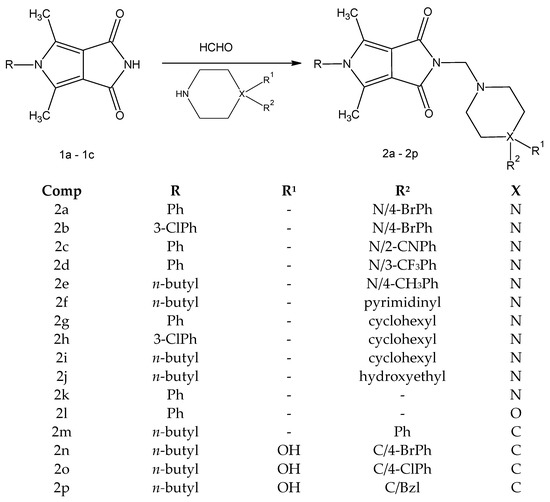
Scheme 1.
Synthetic route of the target compounds 2a–2p.
The reactions were carried out under mild conditions and gave, in several cases, the desired products in high yield (70–84%). The piperazine, piperidine, and morpholine intermediates were commercially available with at least 97% purity.
The crude products were purified by crystallization from ethanol. The identity of the new compounds was established by their FT-IR, 1H NMR, 13C NMR, elemental analyses, and MS. The 1H NMR spectra of 2a–2p revealed that the presence of the methylene group of the Mannich base showed at δ∼4.5 ppm (s, 2H). The obtained values of elemental analysis were in consonance with theoretical information. Additionally, the structure of 2h, taken as an example, was unambiguously established by X-ray crystallography (Figure 3).
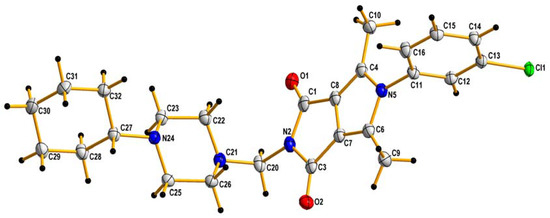
Figure 3.
X-ray structures of 2h. Displacement ellipsoids are drawn at the 50% probability level. H atoms are shown as small spheres of arbitrary radii.
The crystal data and structure refinement for 2h are summarized in Table 1.

Table 1.
Crystal and experimental data.
In general, the bond lengths and angles do not exhibit surprising features [56].
In the crystal lattice, a C-H…O type hydrogen bond is present which can additionally stabilize the crystal lattice (see Supplementary Materials). The angle in the N21-C20-N2 molecule is equal to 117.1(2)° and therefore the molecule is generally V-shaped. A similar particle shape was observed for newly designed pyrrolo[3,4-c]pyrrole derivatives, where the angle in the N12-C11-N2 molecule was equal to 116.4(5)° [39]. In addition, the presence of atom Cl in the benzene ring causes it to align itself perpendicularly to the flat core of the pyrrolo[3,4-c]pyrrole.
2.2. Biological Tests
2.2.1. Cyclooxygenase Inhibition
The researched compounds inhibited both the COX-1 and the COX-2 enzymes (Table 2). Compounds 2b and 2c had similar COX selectivity ratios and showed stronger activity than meloxicam, which was used as a reference, in the same concentration. The compounds 2a–2c, 2j, and 2m blocked the COX-1 isoform more strongly than the reference compound. In the case of the COX-2 enzyme, all tested structures displayed higher activity than the meloxicam.
Table 2.
COX inhibition calculated for COX-1 and COX-2 enzymes after incubation for 2 min with the tested compounds at a concentration of 100 µM and COX selectivity ratio; SRB assay—IC50 values calculated for all tested compounds; data are presented as mean (SD).
2.2.2. Viability of Cell Cultures
All tested compounds 2a–2p showed no cytotoxic activity in the tested concentration range (Table 2). The viability of NHDF cells decreased with increasing concentration, but only for compound 2g, the calculated IC50 value was lower than the highest concentration used. Based on the IC50 values, it can be assumed that the tested compounds may show a pro-proliferative effect on human dermal fibroblasts (NHDF cells).
2.3. Computational Studies
2.3.1. The Analysis of Lipinski and Veber Rules
In the next step, using the SwissAdme website [57] physicochemical properties of the final compounds 2a–2p were determined based on Lipinski and Veber rules (Table 3).
Table 3.
Drug-likeness parameters, according to Lipinski and Veber rules.
The Lipinski and Veber rules are used to evaluate drug-likeness or to determine if compounds possess chemical and physical properties to be an orally active drug in humans. Compounds that do not meet at least two of the criteria of the Lipinski rules may cause absorption or permeability problems. The criteria of Lipinski rules are molecular weight (MW) ≤ 500 Da, lipophilicity values (log P) ≤ 5, number of hydrogen bond donors (NHD) ≤ 5, number of hydrogen bond acceptors (NHA) ≤ 10. Veber rules are rotatable bonds (NBR) ≤ 10 and polar surface area (PSA) ≤ 140 Å2 [58,59].
The 14 tested compounds comply with Lipinski’s rule of five and contained less than five hydrogen bond donors, less than 10 hydrogen bond acceptors, molecular weights below 500 Da, and logP values < 5. Moreover, in line with the Veber rule, these molecules had the number of rotating bonds (NBR) less than 10 and polar surface area (PSA) values lower than 140 Å2.
2.3.2. Structure–Activity Relationship (SAR) of N-Substituted 3,4-Pyrroledicarboximides 2a–2p
Despite the limited number of compounds tested, on the bases of the results presented in Table 2, we formulated some SAR.
For SAR discussion, the compounds were divided into two series—derivatives of piperazine (2a–2k and 2l) and piperidine 2m–2p. We included compound 2l in the piperazine group because we treated it as an analogue of a 2k compound in which an oxygen atom replaced the nitrogen atom in the piperazine ring.
The SAR of N-substituted 3,4-pyrroledicarboximides (Table 2) shows the following:
- All compounds 2a–2p inhibited COX-2 a stronger than meloxicam, which was used as a reference.
- Within the piperazine derivatives the best COX selectivity ratio IC50 (COX-2)/IC50 (COX-1) had compounds 2b and 2c. Compound 2b does not meet the Lipinski and Veber rules. However, it should be emphasized that these rules define only general properties that describe potential orally administered drugs. Therefore, the predictive ability of these rules may be limited in some cases, and one should be careful during interpretation such type of results.
- Replacement of aryl substituent in a series of piperazine derivatives with heteroaryl or cycloalkyl substituent (2g–2i) leads to a significant reduction in COX-1 inhibition while maintaining COX-2 inhibition higher than that of the reference drug. The 2h compound has the lowest COX-1 inhibition value.
- Introduction of hydrophilic hydroxyethyl substituent (2j) leads to a decrease of inhibitory activity in relation to both enzymes.
- Replacement of the 4-arylpiperazine moiety with 4-arylpiperidine leads to a loss of COX-2 inhibition.
- Introduction of group OH in a series of arylpiperidine derivatives significantly reduces the COX-1 inhibition (compare 2m to 2n–2p).
2.3.3. Molecular Docking Studies
Molecular docking simulations were performed in order to predict the binding mode of all designed compounds to the main binding sites of both enzymes. The obtained data were presented in Figure 4 and Figure 5 in the manuscript and Supplementary Materials (S2a–S2p).
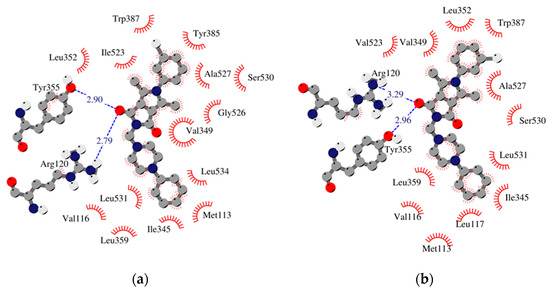
Figure 4.
Schematic 2D diagrams of protein–ligand interactions 2h. Hydrophobic contacts are represented by an arc with spokes radiating towards the ligand atoms they contact. Free enthalpy of binding: (a) COX-1_h2 −10.53 [kcal/mol], (b) COX-2_h2 −11.92 [kcal/mol].
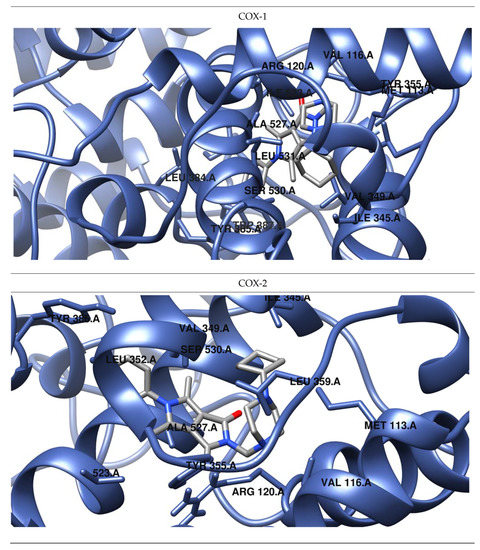
Figure 5.
Predicted orientations of the lowest energy conformations of 2h towards COX-1 and COX-2. The docked ligand was zoomed graphically for each complex.
The results of our study show that most of the tested inhibitors (2a–2c, 2f–2i, 2m–2n, 2o–2p) are more effective against COX-2, which is consistent with the results of enzymatic measurements. In the mentioned cases, the binding manner to the active center of COX-2 and the potency of binding are similar. Most of them can bind to the additional binding pocket of COX-2 including Leu352, Ser353, Tyr355, Phe518, and Val523 amino acid residues which arise due to the conformation of Tyr355 induced by the presence of Val523 instead of Ile523.
Compounds 2g–2i containing a cyclohexylpiperazine substituent take the same orientation in the binding site of an enzyme (Figure 4 and Figure 5). All derivatives show significant inhibitory activity towards COX-2, among which 2h demonstrates to be the most potent compound (inhibition constant 1.84 nM). Binding free energy of the most stable complex (2h - COX-2) was established as −11.9 kcal/mol (Table 4). All compounds can form two hydrogen bonds, between the oxygen of pyrrolo[3,4-c]pyrrole moiety and Tyr355 and Arg120 residues. As determined, cyclohexylpiperazine rings form hydrophobic interactions with Val116, Leu117, Ile345, Leu531, and Leu359 amino acids. Benzene and chlorobenzene rings of 2g and 2h are exposed towards hydrophobic and polar amino acid residues (Leu352, Trp387, Ala527, and Ser530), whereas the aliphatic substituent of 2i is located near Val522, Leu352, Ser352, Met522, and Ala527 of COX-2.
Table 4.
Free energy of binding and binding mode of all considered compounds to COX-1.
The presence of a hydroxy pyridine ring (2n–2p) strongly affects the way of binding. In this case, the strongest inhibitor of COX-2 is compound 2n (free energy of binding −10.3 kcal/mol). Two hydrogen bonds are created between the hydroxyl pyridine ring of 2n and Asp120 and Tyr355 amino acid residues and hydrophobic interactions with Val89, Val116, Arg120, Leu359, and Tyr355. According to a molecular docking study compound, 2p is a more potent inhibitor of COX-2 in terms of free energy of binding (−10 kcal/mol). There is also the possibility of H-bonding interactions which involved oxygen atom of pyrrolo[3,4-c]pyrrole moiety and Tyr355 of COX-2. In the vicinity of the aliphatic chain of 2p hydrophobic and polar amino acids are present (Leu352, Leu384, Tyr385, Trp387, Met522, Val532, and Gly526). Compound 2o exhibits a unique binding configuration in the binding site of COX-2. As presented in Figure 6 it can form two hydrogen bonds with Val116 and Ser530, and the hydroxyl pyridine group forms hydrophobic interactions Leu93, Val116, Arg120, Leu359, and Ala527. Additionally, an aliphatic chain of 2o forms hydrophobic interactions with Leu352, Leu384, Trp387, Phe518, Met522, Gly526.
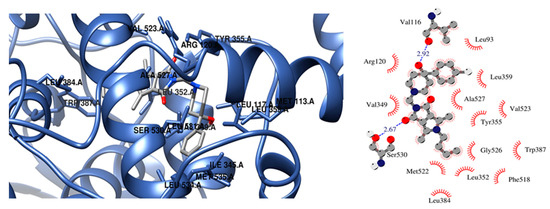
Figure 6.
Configuration in the binding site of COX-2 for compounds 2o.
Compounds 2a–2c, 2f, 2m are selective inhibitors of COX-2 (binding energy ranges from −10.4 to −11.3 kcal/mol). In the case of 2a, one hydrogen bond with Ser530 is formed. Compound binding to the active site of COX-2 is exposed towards hydrophobic amino acids residues (Leu93, Val116, Val349, Leu352, Met522, Val523, Gly526, Ala527), which are mainly involved in van der Waals type interactions. The obtained results show that compound 2f is almost located in the same binding place as meloxicam and is a more effective inhibitor of COX-2 (inhibition constant 5.57 nM). In this case, it occupies an additional binding cavity of COX-2 and is located in close proximity of Leu352, Tyr355, Phe518, and Val523.
The data indicate that compounds 2d, 2j, 2l, 2e, and 2k are not selective inhibitors of cyclooxygenases. Although the binding manner to both enzymes differs significantly, the free energy of binding ranges from −8.2 to −9.7 kcal/mol (Table 4 and Table 5).
Table 5.
Free energy of binding and binding mode of all considered compounds to COX-2.
Compound 2d forms hydrogen bonding three hydrogen bonds with Lys360, Asp352, Leu531 and Ser530, Leu531, Met535 of COX-1 and COX-2, respectively. The aliphatic substituent of 2d was observed to be encompassed by a number of polar and hydrophobic amino acids of COX-1, namely, Leu352, Trp387, Phe518, Met 522, Ile523, Gly526, and Ser530. In the case of COX-2 in close proximity of chain are additionally located Leu384, Tyr385, and Val523 instead of Ile523.
The replacement of benzene rings with aliphatic chains decreased the affinity of compound 2j to cyclooxygenases. In both cases docking study showed hydrophobic interactions with Ile345, Val349, Leu352, Trp387, Met522, Gly526, Ala527, Ser530, Leu534 (see Figure S2j in Supplementary Materials). The orientation of 2l in the binding sites of COX-1 and COX-2 is practically the same. In both cases one hydrogen bonds between compound and oxygen atom od -OH group of Ser530. Benzene moiety occupied the binding pocket of enzymes formed by Leu384, Trp387, Phe518, Met522, and Gly526. Moreover, the morpholine ring occupied the hydrophobic pocket formed by Met113, Val349, Leu531, Leu534, Leu535 in the case of COX-1 and Met113, Leu117, Ile345, Leu531 in the case of COX-2.
2.3.4. QSAR Analysis of Biological Properties
The biological activity of all designed compounds was estimated using 3D/4D QSAR model with restricted docking protocol and was expressed as the probability to be active. Analgetic activity, anti-inflammatory activity in vivo oedema paw carrageenin, anti-inflammatory activity combined action, antioxidant activity, LOX inhibitory activity were evaluated. The data are presented in Supplementary Materials. Compounds 2o, 2p, and 2e show anti-oxidant activity (over 60%). The very high probability of LOX inhibition exhibit compounds 2d, 2h, 2f, 2c, 2b, 2m, 2j, 2p, and 2e. Most of the synthesized derivatives have anti-inflammatory activity, additionally compounds 2b–2f, 2n, and 2m can be used as analgetic agents.
3. Materials and Methods
3.1. Chemistry
3.1.1. Instrumentation and Chemicals
All chemicals, reagents, and solvents used in the current study were purchased from commercial suppliers (Chemat, Gdańsk, Poland; Archem, Łany, Poland; Alchem Wrocław, Poland) and used without further purification. Dry solvents were obtained according to the standard procedure. Progress of the reaction was monitored by the thin-layer chromatography (TLC) technique on silica-gel-60-F254-coated TLC plates (Fluka Chemie GmbH, Switzerland) and visualized by UV light at 254 nm. The melting points of received products were determined by an open capillary method on Electrothermal Mel-Temp 1101D apparatus (Cole-Parmer, Vernon Hills, IL, USA) and were uncorrected. The 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Bruker 300 MHz NMR spectrometer (Bruker Analytische Messtechnik GmbH, Rheinstetten, Germany) in CDCl3 using tetramethylsilane (TMS) as an internal reference. Spectral data includes chemical shifts in ppm, multiplicities, constant couplings in Hz, number of protons, protons’ positions. Multiplicities are abbreviated as follow: s (singlet), d (doublet), t (triplet), and m (multiplet). Spectra were recorded and read using TopSpin 3.6.2 Bruker Daltonik, GmbH, Bremen, Germany). The infrared (IR) spectra were determined on a Nicolet iS50 FT-IR Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Samples were applied as solids, and frequencies are reported in cm−1. Spectra were read using OMNIC Spectra 2.0 Thermo Fisher Scientific, Waltham, MA, USA). Mass spectra were recorded using a Bruker Daltonics Compact ESI-mass spectrometer (Bruker Daltonik, GmbH, Bremen, Germany). The instrument was operated in positive ion mode. Analyzed compounds were dissolved in a mixture of chloroform and methanol. Elemental analyses for carbon, nitrogen, and hydrogen were carried out on a Carlo Erba NA-1500 analyzer (Thermo Fisher Scientific, Waltham, MA, USA), and obtained results were within ± 0.4% of the theoretical values calculated for corresponding formulas.
3.1.2. General Procedure for Preparation of N-Substituted 3,4-Pyrroledicarboximides 2a–2p
A solution of 2 mmol of pyrroledicarboximide 1a–1c [39,55], 0.4 mL of 37% formaldehyde (w/v), and 2 mmol of appropriate amine in ethanol (25 mL) was refluxed for 3–5 h. The course of the reaction was controlled by TLC. The reaction was cooled, and the precipitate was filtered off. The crude product was purified by crystallization in ethanol.
4,6-dimethyl-5-phenyl-2{[4-(p-bromophenyl)-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2a).
- 2a: from 1a and 1-(p-bromophenyl)piperazine. Yield 72%, m.p. 213–215 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.18, 2.20 (2x s, 6H, 4,6–CH3), 2.75–2.85 (m, 4H, 2xCH2-piperazine), 3.15–3.25 (m, 4H, 2xCH2-piperazine), 4.58 (s, 2H, CH2), 6.73 (d, 2H, ArH, J = 9 Hz), 7.20–7.23 (m, 2H, ArH), 7.30–7.33 (m, 2H, ArH), 7.52–7.54 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.05, 135.88, 131.84, 129.94, 129.64, 127.80, 117.75, 116.31, 58.31, 50.28, 48.99, 11.89
- FT-IR (selected lines, ϒmax, cm−1): 1688 (C=O), 1747 (C=O)
- ESI-MS (m/z): calcd. for C25H25BrN4O2 [M+H]+: 494.4034; found: 494.3202
4,6-dimethyl-5-(o-chlorophenyl)-2-{[4-(p-bromophenyl)-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2b).
- 2b: from 1c and 1-(p-bromophenyl)piperazine. Yield 78%, m.p. 230–232 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.19, 2.22 (2x s, 6H, 4,6–CH3), 2.80–2.90 (m, 4H, 2xCH2-piperazine), 3.15–3.30 (m, 4H,2xCH2-piperazine), 4.58 (s, 2H, CH2), 6.73–6.76 (m, 2H, ArH), 7.11–7.15 (m, 1H, ArH), 7.25–7.32 (m, 3H, ArH), 7.50–7.53 (m, 2H ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.82, 150.23, 137.00, 135.66, 131.84, 130.95, 130.04, 128.17, 126.17, 117.74, 116.62, 58.36, 50.28, 48.97, 11.87
- FT-IR (selected lines, ϒmax, cm−1): 1694 (C=O), 1752 (C=O)
- ESI-MS (m/z): calcd. for C25H24BrClN4O2 [M+H]+: 528.8485; found: 529.0797
4,6-dimethyl-5-phenyl-2{[4-(o-cyanophenyl)-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2c).
- 2c: from 1a and 1-(o-cyanophenyl)piperazine. Yield 70%, m.p. 203–205 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.19, 2.25 (2x s, 6H, 4,6–CH3), 2.85–2.95 (m, 4H, 2xCH2-piperazine), 3.15–3.30 (m, 4H, 2xCH2-piperazine), 4.59 (s, 2H, CH2), 6.97–7.10 (m, 2H, ArH), 7.24–7.28 (m, 2H, ArH), 7.44–7.60 (m, 5H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.03, 155.90, 136.00, 134.73, 133.76, 130.07, 129.89, 129.54, 127.87, 121.79, 118.87, 118.38, 116.42, 106.17, 58.26, 51.78, 50.55, 11.90
- FT-IR (selected lines, ϒmax, cm−1): 1694 (C=O), 1752 (C=O)
- ESI-MS (m/z): calcd. for C26H25N5O2 [M+H]+: 440.5168; found: 440.2015
4,6-dimethyl-5-phenyl-2{[4-(m-trifluoromethylphenyl)-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2d).
- 2d: from 1a and 1-(m-trifluoromethylphenyl)piperazine. Yield 82%, m.p. 232–235 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.19 (s, 6H, 4,6–CH3), 2.83–2.86 (m, 4H, 2xCH2-piperazine), 3.23–3.26 (m, 4H, 2xCH2-piperazine), 4.58 (s, 2H, CH2), 7.02–7.08 (m, 3H, ArH), 7.21–7.24 (m, 2H ArH), 7.28–7.35 (m, 1H, ArH), 7.54–7.57 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.07, 151.42, 135.92, 131.57, 131.16, 130.08, 129.92, 129.60, 129.49, 127.80, 126.12, 118.75, 116.37, 115.64, 115.59, 112.18, 112.13, 58.41, 50.36, 48.77, 11.86
- FT-IR (selected lines, ϒmax, cm−1): 1686 (C=O), 1745 (C=O)
- ESI-MS (m/z): calcd. for C26H25F3N4O2 [M+H]+ 483.5053; found: 483.1990
4,6-dimethyl-5-n-butyl-2-{[4-(p-methylphenyl)-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3-(2H,5H)-dione (2e).
- 2e: from 1b and 1-(p-methylphenyl)piperazine. Yield 80%, m.p. 115–117 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.99 (t, 3H, CH3, J = 7.2 Hz), 1.37–1.44 (m, 2H, CH2), 1.62–1.68 (m, 2H, CH2), 2.26 (s, 3H, CH3), 2.41 (s, 6H, 4,6–CH3), 2.70–2.85 (m, 4H, 2xCH2-piperazine), 3.10–3.20 (m, 4H, 2xCH2-piperazine), 3.77 (t, 2H, CH2, J = 7.8 Hz), 4.54 (s, 2H, CH2), 6.80 (d, 2H, ArH, J = 9 Hz), 7.04 (d, 2H, ArH, J = 9 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 166.11, 129.58, 128.72, 116.60, 116.13, 58.23, 50.40, 49.82, 43.88, 32.45, 20.40, 20.02, 13.70, 11.35
- FT-IR (selected lines, ϒmax, cm−1): 1694 (C=O), 1730 (C=O)
- ESI-MS (m/z): calcd. for C24H32N4O2 [M+H]+: 409.5443; found: 409.2565
4,6-dimethyl-5-n-butyl-2-{[4-(2-pyrimidinyl)piperazin-1-yl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2f).
- 2f: from 1b and 1-(2-pyrimidinyl)piperazine. Yield 71%, m.p. 203–205 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3, J = 7.5Hz), 1.24–1.44 (m, 2H, CH2), 1.57–1.67 (m, 2H, CH2), 2.38 (s, 6H, 4,6–CH3), 2.65–2.71 (m, 4H, 2xCH2-piperazine), 3.75 (t, 2H, CH2, J = 7.5Hz), 3.80–3.82 (m, 4H, 2xCH2-piperazine), 4.52 (s, 2H, CH2), 6.43 (t, 1H, 5-H pyrimidine, J = 4.5 Hz), 8.25 (d, 2H, 4,6-H pyrimidine, J = 4.8 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.93, 161.56, 157.63, 128.72, 116.06, 109.65, 58.42, 50.46, 43.86, 43.50, 32.43, 19.99, 13.68, 11.34
- FT-IR (selected lines, ϒmax, cm−1): 1689 (C=O), 1738 (C=O)
- ESI-MS (m/z): calcd. for C21H28N6O2 [M+H]+ 397.4938; found: 397.2313
4,6-dimethyl-5-phenyl-2-[(4-cyclohexyl-1-piperazinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2g).
- 2g: from 1a and 1-cyclohexylpiperazine. Yield 75%, m.p. 140–142 °C.
- 1H NMR (300 MHz, CDCl3) δ: 1.21–1.26 (m, 6H, 3xCH2), 1.62–1.88 (m, 4H, 2xCH2), 2.16 (s, 6H, 4,6–CH3), 2.50–2.65 (m. 4H, 2xCH2-piperazine), 2.70–2.74 (m, 4H, 2xCH2-piperazine), 4.52 (s, 2H, CH2), 7.19–7.26 (m, 2H, ArH), 7.54–7.56 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.95, 137.09, 135.59, 130.91, 129.62, 128.19, 126.24, 116.83, 63.50, 58.27, 50.93, 48.89, 28.86, 26.23, 25.84, 11.78
- FT-IR (selected lines, ϒmax, cm−1): 1691 (C=O), 1746 (C=O)
- ESI-MS (m/z): calcd. for C25H32N4O2 [M+H]+ 421.5549; found: 421.2578
4,6-dimethyl-5-(o-chlorophenyl)-2-[(4-cyclohexyl-1-piperazinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2h).
- 2h: from 1c and 1-cyclohexylpiperazine. Yield 72%, m.p. 199–201 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.99–1.24 (m, 4H, CH2), 1.59–1.63 (m, 2H, CH2), 1.76–1.87 (m, 4H, CH2), 2.18 (s, 6H, 4,6–CH3), 2.50–2.60 (m. 4H, 2xCH2-piperazine), 2.65–2.71 (m, 4H, 2xCH2-piperazine), 4.53 (s, 2H, CH2), 7.14–7.15 (m, 1H, ArH), 7.25–7.27 (m, 1H, ArH), 7.51–7.55 (m, 2H ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.96, 137.11, 135.59, 130.90, 129.95, 129.58, 128.19, 126.24, 116.85, 63.43, 58.33, 50.97, 48.92, 28.96, 26.31, 25.88, 11.77
- FT-IR (selected lines, ϒmax, cm−1): 1695 (C=O), 1756 (C=O)
- ESI-MS (m/z): calcd. for C25H31ClN4O2 [M+H]+: 455.9900; found: 455.2193
4,6-dimethyl-5-n-butyl-2-[(4-cyclohexyl-1-piperazinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2i).
- 2i: from 1b and 1-cyclohexylpiperazine. Yield 70%, m.p. 103–105 °C.
- 1H NMR (300 MHz, CDCl3): δ: 0.98 (t, 3H, CH3, J = 7.2 Hz), 1.20–1.25 (m, 4H, 2xCH2), 1.35–1.42 (m, 2H, CH2), 1.60–1.63 (m, 4H, 2xCH2), 1.76–1.95 (m, 4H, 2xCH2), 2.37 (s, 6H, 4,6–CH3), 2.40–2.72 (m. 8H, 4xCH2-piperazine), 3.75 (t, 2H, CH2, J = 7.2 Hz), 4.47 (s, 2H, CH2)
- 13C NMR (75 MHz, CDCl3) δ: 166.17, 129.12, 116.18, 43.85, 32.43, 28.85, 25.76, 20.02, 13.69, 11.41, 11.32
- FT-IR (selected lines, ϒmax, cm−1): 1685 (C=O), 1743 (C=O)
- ESI-MS (m/z): calcd. for C23H36N4O2 [M+H]+: 401.5653; found: 401.2901
4,6-dimethyl-5-n-butyl-2-{[4-hydroxyethyl-1-piperazinyl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2j).
- 2j: from 1b and 1-(2-hydroxyethyl)piperazine. Yield 80%, m.p. 129–131 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.99 (t, 3H, CH3, J = 7.2 Hz), 1.34–1.41 (m, 2H, CH2), 1.60–1.65 (m, 2H, CH2), 2.41 (s, 6H, 4,6–CH3), 2.60–2.65 (m, 6H, 2xCH2-piperazine+ CH2), 2.70–2.77 (m, 4H, 2xCH2-piperazine), 3.65 (t, 2H, CH2, J = 5.4Hz), 3.74 (t, 2H, CH2, J = 7.8 Hz), 4.48 (s, 2H, CH2)
- 13C NMR (75 MHz, CDCl3) δ: 165.99, 128.89, 116.05, 59.90, 57.91, 56.96, 53.07, 49.27, 43.91, 32.46, 20.05, 13.69, 11.38
- FT-IR (selected lines, ϒmax, cm−1): 1685 (C=O), 1737 (C=O), 3199 (OH)
- ESI-MS (m/z): calcd. for C19H33N5O3 [M+H]+ 363.4743; found: 363.2362
4,6-dimethyl-5-phenyl-2-[(piperazinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2k).
- 2k: from 1a and piperazine. Yield 84%, m.p. 288–290 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.19 (s, 6H, 4,6–CH3), 2.60–2.75 (m. 4H, 2xCH2-piperazine), 3.60–3.75 (m, 4H, 2xCH2-piperazine), 4.50 (s, 2H, CH2), 7.21–7.26 (m, 2H, ArH), 7.54–7.57 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.02, 135.98, 129.89, 129.55, 127.85, 116.41, 58.27, 50.46, 11.84
- FT-IR (selected lines, ϒmax, cm−1): 1686 (C=O), 1747 (C=O)
- ESI-MS (m/z): calcd. for C19H22N4O2 [M+H]+ 339.4113; found: 339.4114
4,6-dimethyl-5-phenyl-2-[(morpholinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2l).
- 2l: from 1a and morpholine. Yield 73%, m.p. 133–135 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.18 (s, 6H, 4,6–CH3), 2.60–2.75 (m, 4H, 2xCH2-piperazine), 3.60–3.75 (m, 4H, 2xCH2-piperazine), 4.50 (s, 2H, CH2), 7.21–7.26 (m, 2H, ArH), 7.54–7.57 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.99, 136.00, 129.93, 129.61, 127.81, 116.35, 67.01, 58.71, 50.89, 11.88
- FT-IR (selected lines, ϒmax, cm−1): 1687 (C=O), 1746 (C=O)
- ESI-MS (m/z): calcd. for C19H21N3O3 [M+H]+: 340.3961; found: 340.1622
4,6-dimethyl-5-n-butyl-2-[(4-phenyl-1-piperidinyl)methyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2m).
- 2m: from 1b and 4-phenyl-1-piperidine. Yield 80%, m.p. 232–235 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.99 (t, 3H, CH3, J = 7.5 Hz), 1.35–1.40 (m, 2H, CH2), 1.45–1.58 (m, 2H, CH2), 1.60–1.73 (m, 4H, CH2), 1.80–1.90 (m, 2H, CH2), 2.42 (s, 6H, 4,6–CH3), 3.10–3.21 (m, 4H, 2XCH2), 3.78 (t, 2H, CH2, J = 7.8 Hz), 4.51 (s, 2H, CH2), 7.10–7.39 (m, 5H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.17, 128.53, 116.24, 48.81, 43.83, 32.43, 25.75, 20.00, 13.68, 11.30
- FT-IR (selected lines, ϒmax, cm−1): 1686 (C=O), 1747 (C=O)
- ESI-MS (m/z): calcd. for C24H40N4O2 [M+H]+;394.8590 found: 394.2452
4,6-dimethyl-5-n-butyl-2-{[4-(p-bromophenyl)-4-hydroxypiperidin-1-yl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2n).
- 2n: from 1b and 4-(p-bromopheny)l-4-hydroxypiperidine. Yield 74%, m.p. 169–171 °C.
- 1H NMR (300 MHz, CDCl3) δ: 1.00 (t, 3H, CH3, J = 7.2 Hz), 1.38–1.48 (m, 2H, CH2), 1.62–1.75 (m, 4H, CH2), 2.00–2.20 (m, 2H, CH2), 2.42 (s, 3H, 4–CH3), 2.43 (s, 3H, 6–CH3), 2.65–2.75 (m. 2H, CH2), 2.85–3.10 (m, 2H, CH2), 3.79 (t, 2H, CH2, J = 7.8 Hz), 4.54 (s, 2H, CH2), 7.20–7.32 (m, 2H, ArH), 7.42–7.45 (m, 2H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.92, 146.57, 133.25, 128.38, 126.10, 46.88, 43.91, 32.48, 20.04, 13.69, 11.36
- FT-IR (selected lines, ϒmax, cm−1): 1680 (C=O), 1733 (C=O), 3524 (OH)
- ESI-MS (m/z): calcd. for C24H30BrN3O3 [M+H]+;489.4251 found: 489.1990
4,6-dimethyl-5-n-butyl-2-{[4-(p-chlorophenyl)-4-hydroxypiperidin-1-yl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2o).
- 2o: from 1b and 4-(p-chlorophenyl)-4-hydroxypiperidine. Yield 78%, m.p. 170–172 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.98 (t, 3H, CH3, J = 7.2 Hz), 1.30–1.45 (m, 2H, CH2), 1.62–1.71 (m, 4H, 2xCH2), 2.00–2.20 (m, 2H, CH2), 2.38 (s, 6H, 4,6–CH3), 2.60–2.75 (m, 2H, CH2), 2.85–2.95 (m, 2H, CH2), 3.76 (t, 2H, CH2, J = 7.8 Hz), 4.49 (s, 2H, CH2), 7.33–7.36 (m, 2H, ArH), 7.42–7.46 (m, 2H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 166.08, 146.87, 132.75, 128.75, 128.36, 126.11, 116.14, 98.74, 70.67, 58.51, 46.87, 43.90, 38.30, 32.47, 20.04, 13.70, 11.37
- FT-IR (selected lines, ϒmax, cm−1): 1680 (C=O), 1733 (C=O), 3517 (OH)
- ESI-MS (m/z): calcd. for C24H30ClN3O3 [M+H]+: 444.9741; found: 444.2015
4,6-dimethyl-5-n-butyl-2-{[4-benzyl-4-hydroxypiperidin-1-yl]methyl}pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (2p).
- 2p: from 1b and 4-benzyl-4-hydroxypiperidine, Yield 83%, m.p. 141–143 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.98 (t, 3H, CH3, J = 7.2 Hz), 1.35–1.40 (m, 2H, CH2), 1.45–1.58 (m, 2H CH2), 1.60–1.73 (m, 4H, 2xCH2), 2.38 (s, 6H, 4,6–CH3), 2.50–2.60 (m, 2H, 2XCH2), 2.71 (s, 2H, CH2), 2.71–2.78 (m, 2H, CH2), 3.75 (t, 2H, CH2, J = 7.8 Hz), 4.45 (s, 2H, CH2), 7.15–7.18 (m, 2H, ArH), 7.22–7.29 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.92, 136.37, 130.54, 128.75, 128.27, 126.63, 116.12, 68.72, 58.36, 48.88, 46.82, 43.88, 36.49, 32.46, 20.03, 13.69, 11.34
- FT-IR (selected lines, ϒmax, cm−1): 1682 (C=O), 1742 (C=O), 3504 (OH)
- ESI-MS (m/z): calcd. for C25H33N3O3 [M+H]+ 424.5556; found: 424.2565.
3.2. Crystallography
3.2.1. X-ray Structure Determinations of 2h
X-ray Experimental Details
Crystals of 2h suitable for single-crystal X-ray diffraction analysis were obtained by dissolution in ethanol/2-propanol followed by slow evaporation of the solvent at room temperature. Crystallographic measurements for 1 was collected with Κ-geometry diffractometers: Xcalibur Gemini with graphite monochromatized Cu-Kα radiation (λ = 1.5418 Å) at 100(2) K, using an Oxford Cryosystems cooler. Data collection, cell refinement, data reduction, and analysis were carried out with CrysAlisPro [60]. Analytical absorption correction was applied to data with the use of CrysAlisPro. The crystal structures were solved using SHELXS [61] and refined on F2 by a full-matrix least squares technique with SHELXL-2015 [62] with anisotropic thermal parameters for all the ordered non-H atoms. In the final refinement cycles, H atoms were repositioned in their calculated positions and treated as riding atoms, with C-H = 0.95–0.98 Å, and with Uiso(H) = 1.2 Ueq(C) for CH, CH2 and 1.5 Ueq(C) for CH3. All figures were made using DIAMOND program [63]. CCDC 2036201 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by e-mailing data_request@ccdc.cam.ac.uk, or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44(0)1223-336033.
3.3. Pharmacology
3.3.1. Determination of COX-1/COX-2-Inhibition
Material and Methods
Cell Line
The NHDF cell line (from LONZA) was used to determine the cytotoxicity effect of newly synthesized compounds. The cells were incubated in DMEM without phenol red supplemented with 10% fetal bovine serum (FBS), 2 mM L glutamine, 1.25 µg/mL amphotericin B, and 100 µg/mL gentamicin. The medium prepared in this way was stored at 4–8 °C. The NHDF cells were cultivated at 37 °C in 5% CO2 and 95% humidity. The cells were passaged, or the medium was changed twice a week.
3.3.2. Tested Compounds
The 15 newly synthesized compounds were dissolved in DMSO to the final concentration of 10 mM (stock solutions) and stored at −20 °C for up to 6 months. The working solution was dissolved in a culture medium to a final concentration of 10, 50, and 100 µM (the DMSO concentration did not reach 1%).
3.3.3. SRB Assay
The SRB assay was performed to evaluate the cytotoxicity effect of newly synthesized compounds on NHDF cells. The cells were seeded at a density of 10,000 cells/well on the 96-well plates and left at 37 °C in the 5% CO2 incubator for 24 h to allow cells to adhere to the well surface. After removing the supernatant, tested compounds were applied for 48 h. The 10% w/v cold TCA solution was added to the culture plate for 1 h at 4–8 °C to fix the cells. The plates were washed four times with running water and air-dried at RT. The 0.4% SRB dye solution in 1% acetic acid was applied for 30 min at RT. The plates were then rinsed with 1% (v/v) acetic acid five times and air-dried again at RT. The 10 mM Trizma base was supplied to dissolve protein-bound dye for 30 min, and the absorbance was measured at 565 nm using Varioskan LUX microplate reader (Thermo Scientific).
3.3.4. Cyclooxygenase Inhibition Assay
The COX peroxidase activity of all tested compounds at a concentration of 100 µM was assessed using a ready-to-use kit (Cayman, cat. no. 701050). The peroxidase absorbance was measured after 2 min incubation at RT using Varioskan LUX (λ = 590 nm). Based on the obtained results, the IC50 values (concentrations at which 50% inhibition of enzyme activity was observed) were calculated separately for COX-1 and COX-2 enzymes. There were also calculated the ratios of IC50 values (IC50(COX-2)/IC50(COX-1)), which allowed assessing the selectivity of cyclooxygenase inhibition [64].
3.3.5. Statistical Analysis
All results were presented as IC50 values ± SD. All biological assays were performed in three independent replications. Significance was assessed with parametric tests—ANOVA and Tukey’s post-hoc. The significance level was assumed as p < 0.05.
3.4. Molecular Modeling
The structures of designed compounds were optimized at the CAM-B3LYP/6-31++G** level of theory with the polarizable continuum model (PCM) including solvent effects [65,66,67] using the Gaussian 09 program [68]. Molecular docking was performed using AutoDock4.2 package, and a standard protocol was followed to predict the binding mode and the free energy of binding [69]. The following equation expresses the free energy of binding, which characterizes the affinity of protein-ligand complexes:
ΔG binding = [ΔG intermolecular + ΔG internal + ΔG tors] − ΔG unbound
The crystallographic structures of COX-1 (PDB ID: 4O1Z) and COX-2 (PDB ID: 4M11) co-crystallized with meloxicam originated from Protein Data Bank [70]. The polar hydrogen atoms and solvent parameters were added to the chain A of cyclooxygenases and Gasteiger charges for each of the atoms were declared. The binding place was defined using the grid of 80 × 80 × 80 points with 0.375 Å spacing. The grid center was established in the active site according to the crystalized inhibitor location. The Lamarckian genetic algorithm (LGA) implemented with local search and 200 runs for each complex was used during the docking procedure. All calculations included a population of 150 individuals with 27,000 generations and 250,000 energy evaluations. The validation protocol was performed by docking of meloxicam into the crystal structures of cyclooxygenases and the comparison of its position with a crystal structure.
Binding modes of designed compounds were visualized using Chimera and LIGPLOT v.4.5.3 programs [71,72]. The simulations of biological properties were performed using the 3D/4DQSAR BiS/MC and CoCon algorithms from ChemoSophia Company [73,74].
4. Conclusions
The newly designed N-substituted 3,4-pyrroledicarboximides derivatives were tested in terms of their inhibitory activity against COX-1 and COX-2 enzymes. Additionally, their cytotoxicity was determined. The analyzed compounds exhibit an inhibitory effect on both enzymes. Taking into account the results of the colorimetric study, we can assume that all compounds inhibit COX-2 more strongly than meloxicam. The results also indicate that 2b, 2c derivatives proved to be the most effective towards the COX-2 enzyme. The 2h compound, which is the weakest of all studied structures, inhibits the COX-1 enzyme, with COX-2 inhibition value higher than the reference drug, also deserves attention.
The molecular docking studies indicate that all designed compounds can bind to the hydrophobic pocket created by polar and hydrophobic amino acid residues. The hydrophobic and van der Waals forces are the origin of stabilization of protein–ligand complexes. Therefore, compounds of this type can be very promising cyclooxygenase inhibitors; however, they still require further research.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/22/3/1410/s1.
Author Contributions
Conceptualization, A.R.; methodology, A.R., Ż.C., B.W., K.G. and A.K.; formal analysis, A.R.; investigation, A.R., Ż.C., K.G., B.W., A.K.; writing—original draft preparation, A.R., Ż.C., A.K.; writing—review and editing, A.R., Ż.C., K.G., B.W., A.K.; visualization, A.R., Ż.C., A.K.; supervision, A.R.; project administration A.R.; funding acquisition K.G. All authors have read and agreed to the published version of the manuscript.
Funding
The publication was prepared under the project financed from the funds granted by the Ministry of Science and Higher Education in the “Regional Initiative of Excellence” program for the years 2019–2022, project number 016/RID/2018/19, the amount of funding 11 998 121.30 PLN.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Theoretical modeling was performed in Wroclaw Center of Networking and Supercomputing (WCSS). Molecular graphics were done using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco, support No. NIH P41-GM103311.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| COX | cyclooxygenase |
| CNS | central nervous system |
| IMIDs | Immunomodulatory imide drugs |
| Ph | phenyl |
| Bzl | benzyl |
| RT | room temperature |
| SRB | sulforhodamine B |
| QSAR | Quantitative structure–activity relationship |
| LOX | lipoxygenase |
| MS | mass spectra |
| DMSO | dimethyl sulfoxide |
| NHDF | normal human dermal fibroblasts |
References
- Lamie, P.F.; Philoppes, J.N.; El-Gendy, A.O.; Rarova, L.; Gruz, J. Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents. Molecules 2015, 20, 16620–16642. [Google Scholar] [CrossRef] [PubMed]
- Pareshkumar, P.; Gohel, V.; Purohit, D.; Patolia, V.N. Synthesis and Biological Evaluation of Some New Chalcone and Isoxazole Derivatives. Int. J. Sci. Technoledge 2014, 2, 138–141. [Google Scholar]
- Dhivare, R.S.; Rajput, S.S. Microwave Assisted Solvent Free Synthesis and Antifungal Evaluation Of3, 5-Bis-(4-Hydroxy-3-Methoxybenzylidene)-N-phenylpiperidine-2, 6-Dione derived from N-Phenyl Glutarimides. Int. J. ChemTech Res. 2016, 9, 325–331. [Google Scholar]
- Al-Azzawi, A.M. Synthesis, Characterization and Evaluation of Antibacterial Activity of Several New 3,4-Dimethyl Maleimides and 1,8-Naphthalimides Containing 1,3,4-Oxadiazole Ring. Al-Nahrain J. Sci. 2011, 14, 15–29. [Google Scholar] [CrossRef]
- Khalil, A.E.G.M.; Berghot, M.A.; Gouda, M.A. Synthesis and Study of Some New N-Substituted Imide Derivatives as Potential Antibacterial Agents. Chem. Pap. 2010, 64, 637–644. [Google Scholar] [CrossRef]
- Azzawi, A.; Al-Obiadi, K.K. Synthesis and anti-microbial screening of new bisschiff bases and their acetyl oxadiazole azetidinone derivatives derived from pyromelliticdiimide. Int. J. Res. Pharm. Chem. 2016, 6, 1–8. [Google Scholar]
- Dhivare, R.; Rajput, S. Synthesis and anti-microbial activity of five membered cyclic imide derivatives of mono, di and tri substituted aromatic amines and naphtyl amine. World J. Pharm. Res. 2015, 4, 1650–1658. [Google Scholar]
- Ahmed, H.E.A.; Abdel-Salam, H.A.; Shaker, M.A. Synthesis, Characterization, Molecular Modeling, and Potential Antimicrobial and Anticancer Activities of Novel 2-Aminoisoindoline-1,3-Dione Derivatives. Bioorg. Chem. 2016, 66, 1–11. [Google Scholar] [CrossRef]
- Bansode, T.N.; Shelke, J.V.; Dongre, V.G. Synthesis and Antimicrobial Activity of Some New N-Acyl Substituted Phenothiazines. Eur. J. Med. Chem. 2009, 44, 5094–5098. [Google Scholar] [CrossRef]
- Navakoski De Oliveira, K.; Chiaradia, L.D.; Alves Martins, P.G.; Mascarello, A.; Sechini Cordeiro, M.N.; Carvalho Guido, R.V.; Andricopulo, A.D.; Yunes, R.A.; Nunes, R.J.; Vernal, J.; et al. Sulfonyl-Hydrazones of Cyclic Imides Derivatives as Potent Inhibitors of the Mycobacterium Tuberculosis Protein Tyrosine Phosphatase B (PtpB). MedChemComm 2011, 2, 500–504. [Google Scholar] [CrossRef]
- Kim, B.S.; Moon, S.S.; Hwang, B.K. Isolation, Antifungal Activity, and Structure Elucidation of the Glutarimide Antibiotic, Streptimidone, Produced by Micromonospora Coerulea. J. Agric. Food Chem. 1999, 47, 3372–3380. [Google Scholar] [CrossRef] [PubMed]
- Lahore, S.; Aiwale, S.T.; Sardi, P.; Dallavalle, S. Synthesis of Natural Maleimides Farinomaleins C-E and Evaluation of Their Antifungal Activity. Tetrahedron Lett. 2014, 55, 4196–4198. [Google Scholar] [CrossRef]
- Akgün, H.; Karamelekoglu, I.; Berk, B.; Kurnaz, I.; Saribiyik, G.; Öktem, S.; Kocagöz, T. Synthesis and Antimycobacterial Activity of Some Phthalimide Derivatives. Bioorg. Med. Chem. 2012, 20, 4149–4154. [Google Scholar] [CrossRef]
- Gayoso, C.W.; Lima, E.D.O.; De Souza, E.L.; Cechinel Filho, V.; Trajano, V.N.; Pereira, F.D.O.; Lima, I.O. Antimicrobial Effectiveness of Maleimides on Fungal Strains Isolated from Onychomycosis. Braz. Arch. Biol. Technol. 2006, 49, 661–664. [Google Scholar] [CrossRef]
- Sortino, M.; Cechinel Filho, V.; Corrêa, R.; Zacchino, S. N-Phenyl and N-Phenylalkyl-Maleimides Acting against Candida spp.: Time-to-Kill, Stability, Interaction with Maleamic Acids. Bioorg. Med. Chem. 2008, 16, 560–568. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M. Novel and Versatile Methodology for Synthesis of Cyclic Imides and Evaluation of Their Cytotoxic, DNA Binding, Apoptotic Inducing Activities and Molecular Modeling Study. Eur. J. Med. Chem. 2007, 42, 614–626. [Google Scholar] [CrossRef]
- Collin, X.; Robert, J.M.; Wielgosz, G.; Le Baut, G.; Bobin-Dubigeon, C.; Grimaud, N.; Petit, J.Y. New Anti-Inflammatory N-Pyridinyl(Alkyl)Phthalimides Acting as Tumor Necrosis Factor-α Production Inhibitors. Eur. J. Med. Chem. 2001, 36, 639–649. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Alanazi, A.M.; Abdel-Aziz, N.I.; Al-Suwaidan, I.A.; El-Sayed, M.A.A.; El-Sherbeny, M.A.; Abdel-Aziz, A.A.M. Synthesis, Molecular Modeling Study, Preliminary Antibacterial, and Antitumor Evaluation of N-Substituted Naphthalimides and Their Structural Analogues. Med. Chem. Res. 2013, 22, 2360–2375. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, N.; Roy, P.; Sondhi, S.M.; Sharma, A. Synthesis of Acridine Cyclic Imide Hybrid Molecules and Their Evaluation for Anticancer Activity. Med. Chem. Res. 2015, 24, 3272–3282. [Google Scholar] [CrossRef]
- Tumiatti, V.; Milelli, A.; Minarini, A.; Micco, M.; Gasperi Campani, A.; Roncuzzi, L.; Baiocchi, D.; Marinello, J.; Capranico, G.; Zini, M.; et al. Design, Synthesis, and Biological Evaluation of Substituted Naphthalene Imides and Diimides as Anticancer Agent ∞. J. Med. Chem. 2009, 52, 7873–7877. [Google Scholar] [CrossRef]
- Jindal, D.P.; Bedi, V.; Jit, B.; Karkra, N.; Guleria, S.; Bansal, R.; Palusczak, A.; Hartmann, R.W. Synthesis and Study of Some New N-Substituted Imide Derivatives as Potential Anticancer Agents. Il Farmaco 2005, 60, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Radice, T.; Antonino, M.; Beccalli, E.M.; Tinelli, S.; Zunino, F.; Corsini, A.; Pratesi, G.; Ragg, E.M.; Gelmi, M.L.; et al. Synthesis, Structural, and Biological Evaluation of Bis-Heteroarylmaleimides and Bis-Heterofused Imides. Bioorg. Med. Chem. 2011, 19, 5291–5299. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.M.; Rani, R.; Roy, P.; Agrawal, S.K.; Saxena, A.K. Microwave-Assisted Synthesis of N-Substituted Cyclic Imides and Their Evaluation for Anticancer and Anti-Inflammatory Activities. Bioorg. Med. Chem. Lett. 2009, 19, 1534–1538. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; Attia, S.M.; Al-Obaid, A.M.; Al-Omar, M.A.; El-Subbagh, H.I. Synthesis and Biological Evaluation of Some Novel Cyclic-Imides as Hypoglycaemic, Anti-Hyperlipidemic Agents. Eur. J. Med. Chem. 2011, 46, 4324–4329. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kongot, M.; Singh, V.; Maurya, N.; Patel, R.; Kumar Singhal, N.; Avecilla, F.; Kumar, A. Coumarin Tethered Cyclic Imides as Efficacious Glucose Uptake Agents and Investigation of Hit Candidate to Probe Its Binding Mechanism with Human Serum Albumin. Bioorg. Chem. 2019, 92, 103212. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Abdel-Aziz, A.A.M.; Sakr, H.M.; El-Azab, A.S.; Bua, S.; Supuran, C.T. Synthesis and Human/Bacterial Carbonic Anhydrase Inhibition with a Series of Sulfonamides Incorporating Phthalimido Moieties. Bioorg. Med. Chem. 2017, 25, 2524–2529. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.M.; Ayyad, R.R.; Ceruso, M.; Supuran, C.T. Inhibition of Carbonic Anhydrase Isoforms I, II, IV, VII and XII with Carboxylates and Sulfonamides Incorporating Phthalimide/Phthalic Anhydride Scaffolds. Bioorg. Med. Chem. 2016, 24, 20–25. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; Ceruso, M.; Supuran, C.T. Carbonic Anhydrase Inhibitory Activity of Sulfonamides and Carboxylic Acids Incorporating Cyclic Imide Scaffolds. Bioorg. Med. Chem. Lett. 2014, 24, 5185–5189. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; Abu El-Enin, M.A.; Almehizia, A.A.; Supuran, C.T.; Nocentini, A. Synthesis of Novel Isoindoline-1,3-Dione-Based Oximes and Benzenesulfonamide Hydrazones as Selective Inhibitors of the Tumor-Associated Carbonic Anhydrase IX. Bioorg. Chem. 2018, 80, 706–713. [Google Scholar] [CrossRef]
- Sethi, K.K.; Verma, S.M.; Tanç, M.; Purper, G.; Calafato, G.; Carta, F.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Synthesis and Inhibition of the Human Carbonic Anhydrase Isoforms I, II, IX and XII with Benzene Sulfonamides Incorporating 4- and 3-Nitrophthalimide Moieties. Bioorg. Med. Chem. 2014, 22, 1586–1595. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; Ghiaty, A.H.; Gratteri, P.; Supuran, C.T.; Nocentini, A. 4-Substituted Benzenesulfonamides Featuring Cyclic Imides Moieties Exhibit Potent and Isoform-Selective Carbonic Anhydrase II/IX Inhibition. Bioorg. Chem. 2019, 83, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Malinka, W.; Sieklucka-Dziuba, M.; Rajtar, G.; Rejdak, R.; Rejdak, K.; Kleinrok, Z. Synthesis of some N-substituted 3,4-pyrroledicarboximides as potential CNS depressive agents. Pharmazie 2000, 55, 9–16. [Google Scholar] [PubMed]
- Norman, M.H.; Minick, D.J.; Rigdon, G.C. Effect of Linking Bridge Modifications on the Antipsychotic Profile of Some Phthalimide and Isoindolinone Derivatives. J. Med. Chem. 1996, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, Y.; Yang, F.; Wu, C.; Wang, Z.; Ye, B.; Jiang, X.; Zhao, Q.; Li, J.; Liu, Y.; et al. Synthesis and Biological Evaluation of a Series of Multi-Target N-Substituted Cyclic Imide Derivatives with Potential Antipsychotic Effect. Eur. J. Med. Chem. 2018, 145, 74–85. [Google Scholar] [CrossRef]
- Kossakowski, J.; Jarocka, M. Synthesis of New N-Substituted Cyclic Imides with an Expected Anxiolytic Activity. XVII. Derivatives of 1-ethoxybicyclo[2.2.2]-oct-5-one-2,3-dicarboximide. Il Farmaco 2001, 56, 785–789. [Google Scholar] [CrossRef]
- Kamiński, K.; Obniska, J.; Wiklik, B.; Atamanyuk, D. Synthesis and Anticonvulsant Properties of New Acetamide Derivatives of Phthalimide, and Its Saturated Cyclohexane and Norbornene Analogs. Eur. J. Med. Chem. 2011, 46, 4634–4641. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; AlSaif, N.A.; Alanazi, M.M.; El-Gendy, M.A.; Obaidullah, A.J.; Alkahtani, H.M.; Almehizia, A.A.; Al-Suwaidan, I.A. Synthesis, Anti-Inflammatory, Cytotoxic, and COX-1/2 Inhibitory Activities of Cyclic Imides Bearing 3-Benzenesulfonamide, Oxime, and β-Phenylalanine Scaffolds: A Molecular Docking Study. J. Enzym. Inhib. Med. Chem. 2020, 35, 610–621. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; Eltahir, K.E.H.; Asiri, Y.A. Synthesis, Anti-Inflammatory Activity and COX-1/COX-2 Inhibition of Novel Substituted Cyclic Imides. Part 1: Molecular Docking Study. Eur. J. Med. Chem. 2011, 46, 1648–1655. [Google Scholar] [CrossRef]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 Inhibition Activities and Molecular Docking Study of Newly Designed and Synthesized Pyrrolo[3,4-c]Pyrrole Mannich Bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef]
- Alanazi, A.M.; El-Azab, A.S.; Al-Suwaidan, I.A.; Eltahir, K.E.H.; Asiri, Y.A.; Abdel-Aziz, N.I.; Abdel-Aziz, A.A.M. Structure-Based Design of Phthalimide Derivatives as Potential Cyclooxygenase-2 (COX-2) Inhibitors: Anti-Inflammatory and Analgesic Activities. Eur. J. Med. Chem. 2015, 92, 115–123. [Google Scholar] [CrossRef]
- Pophale, R.A.; Deodhar, M. Synthesis and Evaluation of Novel Phthalimide Derivatives as Analgesic and Antiinflammatory Agents. Pharma Chem. 2010, 2, 185–193. [Google Scholar]
- Abu-Hashem, A.A.; Gouda, M.A. Synthesis, Anti-Inflammatory and Analgesic Evaluation of Certain New 3a,4,9,9a-Tetrahydro-4,9-Benzenobenz(f)Isoindole-1,3-Diones. Arch. Pharm. 2011, 344, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Al-Suwaidan, I.A.; Alanazi, A.M.; El-Azab, A.S.; Al-Obaid, A.M.; Eltahir, K.E.H.; Maarouf, A.R.; Abu El-Enin, M.A.; Abdel-Aziz, A.A.M. Molecular Design, Synthesis and Biological Evaluation of Cyclic Imides Bearing Benzenesulfonamide Fragment as Potential COX-2 Inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2013, 23, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- De Campos, F.; Corrêa, R.; De Souza, M.M.; Yunes, R.A.; Nunes, R.J.; Cechinel-Filho, V. Studies on New Cyclic Imides Obtained from Aminophenazone with Analgesic Properties/Potent Effects of a 3,4-Dichloromaleimide Derivative. ArzneimittelForschung/Drug Res. 2002, 52, 455–461. [Google Scholar] [CrossRef]
- Sano, H.; Noguchi, T.; Tanatani, A.; Hashimoto, Y.; Miyachi, H. Design and Synthesis of Subtype-Selective Cyclooxygenase (COX) Inhibitors Derived from Thalidomide. Bioorg. Med. Chem. 2005, 13, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.; Borchhardt, D.; Castilho, M. Classical and Fragment-Based Hologram Structure-Activity Relationships for a Series of Analgesic Cyclic Imides. Lett. Drug Des. Discov. 2008, 5, 57–64. [Google Scholar] [CrossRef]
- Said, S.A.; Amr, A.E.G.E.; Sabry, N.M.; Abdalla, M.M. Analgesic, Anticonvulsant and Anti-Inflammatory Activities of Some Synthesized Benzodiazipine, Triazolopyrimidine and Bis-Imide Derivatives. Eur. J. Med. Chem. 2009, 44, 4787–4792. [Google Scholar] [CrossRef]
- Lopchuk, J.M. Imide Natural Products. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 255–334. [Google Scholar] [CrossRef]
- Tempesta, M.S.; Corley, D.G.; Beutler, J.A.; Metral, C.J.; Yunes, R.A.; Giacomozzi, C.A.; Calixto, J.B. Phyllanthimide, a New Alkaloid from Phyllanthus Sellowianus. J. Nat. Prod. 1988, 51, 617–618. [Google Scholar] [CrossRef]
- Prado, S.R.T.; Cechinel-Filho, V.; Campos-Buzzi, F.; Corrêa, R.; Cadena, S.M.C.S.; De Oliveira, M.B.M. Biological Evaluation of Some Selected Cyclic Imides: Mitochondrial Effects and in Vitro Cytotoxicity. Z. Naturforsch. C 2004, 59, 663–672. [Google Scholar] [CrossRef][Green Version]
- Sultana, K.; Khan, H.; Shahid, K. Synthesis, Characterization and In Vitro Antibacterial Evaluation of Sn, Sb, and Zn Coordination Complexes of 2-(2-Methoxyphenyl)-1H-Isoindole-1, 3(2h)-Dione. Int. J. Pharm. Sci. Rev. Res. 2014, 28, 1–5. [Google Scholar]
- Marulasiddaiah, R.; Kalkhambkar, R.G.; Kulkarni, M.V. Synthesis and Biological Evaluation of Cyclic Imides with Coumarins and Azacoumarins. Open J. Med. Chem. 2012, 2, 89–97. [Google Scholar] [CrossRef][Green Version]
- Hargreaves, M.K.; Pritchard, J.G.; Dave, H.R. Cyclic Carboxylic Monoimides. Chem. Rev. 1970, 70, 439–469. [Google Scholar] [CrossRef]
- Szczȩśniak-Siega, B.; Gȩbczak, K.; Gȩbarowski, T.; Maniewska, J. Synthesis, COX-1/2 Inhibition and Antioxidant Activities of New Oxicam Analogues Designed as Potential Chemopreventive Agents. Acta Biochim. Pol. 2018, 65, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Malinka, W.; Bodalski, T. Synthesis of some 1-substituted-2,5-dimethylpyrrole-3,4-dicarboxyimides from α,β-diacetylsuccinate. Pol. J. Chem. 1994, 68, 297–307. [Google Scholar] [CrossRef]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A. Crystal Structure of 4,6-Dimethyl-5-(3-Chlorophenyl)-2-{[4-(3,4-Dichlorophenyl)-1-Piperazinyl]Methyl}-Pyrrolo[3,4-c]Pyrrole-1,3(2H,5H)-Dione. X-ray Struct. Anal. Online 2018, 34, 55–56. [Google Scholar] [CrossRef]
- SwissADME. Available online: https://www.swissadme.ch/ (accessed on 18 December 2020).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- CrysAlis PRO. Rigaku; Oxford Diffraction Ltd.: Yarnton, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Diamond—Crystal and Molecular Structure Visualization, Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Germany. Available online: https://www.crystalimpact.com/diamond/ (accessed on 18 December 2020).
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 Inhibition Activities and Molecular Docking Study of Isothiazolopyridine Derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site via a Two-Water-Mediated h-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Potemkin, V.; Grishina, M. Principles for 3D/4D QSAR Classification of Drugs. Drug Discov. Today 2008, 13, 952–959. [Google Scholar] [CrossRef]
- Potemkin, V.A.; Grishina, M.A.; Bartashevich, E.V. Modeling of Drug Molecule Orientation within a Receptor Cavity in the BiS Algorithm Framework. J. Struct. Chem. 2007, 48, 155–160. [Google Scholar] [CrossRef][Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).