
In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy




Abstract
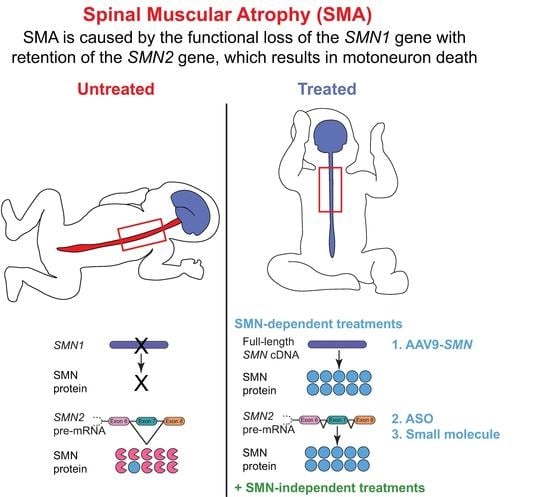
:
1. Genetics
2. Clinical Manifestations
3. Impact of Motor Impairment on Quality of Life
4. The SMN Protein
5. Temporal Requirements of SMN
6. Lower α-Motoneuron Pathologies
6.1. Motoneuron Somas
6.2. Motoneuron Terminals
7. Other Cell and Tissue Types Vulnerable to SMN Deficiency
7.1. Skeletal Muscle
7.2. Schwann Cells
7.3. Astrocytes
7.4. Heart
7.5. Additional Susceptible Cell and Tissue Types
8. The Quest for Effective SMA Therapies: FDA-Approved SMN-Dependent Therapeutics
8.1. Nusinersen: An Antisense Oligonucleotide (ASO)
8.2. Onasemnogene Abeparvovec-Xioi: A Self-Complimentary Adeno-Associated Virus (scAAV9)
8.3. Risdiplam: An Oral, Brain-Penetrant Small Molecule
8.4. Cost–Benefit Considerations for Approved SMN-Based Therapies
9. The Quest for Additional SMA Therapies: SMN-Independent Therapeutics
9.1. Neuroprotective Strategies
9.2. Muscle-Directed Strategies
9.3. Drugs Targeting Neuromuscular Function
9.4. Endogenous SMN-Independent Protective Modifiers
9.5. Physical Therapy Strategies
9.6. Biomedical Devices
10. Future Directions for SMA Therapies
Finding a Cure through Complementary Treatment: NMJs Are Crucial Targets
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.; Androphy, E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997, 61, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef] [Green Version]
- McAndrew, P.E.; Parsons, D.W.; Simard, L.R.; Rochette, C.; Ray, P.N.; Mendell, J.R.; Prior, T.W.; Burghes, A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997, 60, 1411–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, B.T. Spinal muscular atrophies. Pediatr. Clin. N. Am. 2015, 62, 743–766. [Google Scholar] [CrossRef]
- Faravelli, I.; Nizzardo, M.; Comi, G.P.; Corti, S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat. Rev. Neurol. 2015, 11, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Munsat, T.L. Workshop Report: International SMA Collaboration. Neuromuscul. Disord. 1991, 1, 81. [Google Scholar] [CrossRef]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef]
- Zerres, K.; Davies, K.E. 59th ENMC International Workshop: Spinal Muscular Atrophies: Recent progress and revised diagnostic criteria, 17–19 April 1998, Soestduinen, The Netherlands. Neuromuscul. Disord. 1999, 9, 272–278. [Google Scholar] [CrossRef]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef]
- Darras, B.T.; Finkel, R.S. Natural History of Spinal Muscular Atrophy. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 399–421. [Google Scholar]
- Dubowitz, V. Very severe spinal muscular atrophy (SMA type 0): An expanding clinical phenotype. Eur. J. Paediatr. Neurol. 1999, 3, 49–51. [Google Scholar] [CrossRef]
- MacLeod, M.J.; Taylor, J.E.; Lunt, P.W.; Mathew, C.G.; Robb, S.A. Prenatal onset spinal muscular atrophy. Eur. J. Paediatr. Neurol. 1999, 3, 65–72. [Google Scholar] [CrossRef]
- Rudnik-Schoneborn, S.; Vogelgesang, S.; Armbrust, S.; Graul-Neumann, L.; Fusch, C.; Zerres, K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve 2010, 42, 144–147. [Google Scholar] [CrossRef]
- Carrasco, D.; Magoulas, P.; Scull, J.C.; Jarrell, J.A.; Lalani, S.R.; Wangler, M.F. Digital necrosis in an infant with severe spinal muscular atrophy. Neurol. Genet. 2019, 5, e361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnik-Schoneborn, S.; Goebel, H.H.; Schlote, W.; Molaian, S.; Omran, H.; Ketelsen, U.; Korinthenberg, R.; Wenzel, D.; Lauffer, H.; Kreiss-Nachtsheim, M.; et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology 2003, 60, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schoneborn, S.; Heller, R.; Berg, C.; Betzler, C.; Grimm, T.; Eggermann, T.; Eggermann, K.; Wirth, R.; Wirth, B.; Zerres, K. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. 2008, 45, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Harding, B.N.; Kariya, S.; Monani, U.R.; Chung, W.K.; Benton, M.; Yum, S.W.; Tennekoon, G.; Finkel, R.S. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J. Neuropathol. Exp. Neurol. 2015, 74, 15–24. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schoneborn, S.; Forrest, E.; Lusakowska, A.; Borkowska, J.; Hausmanowa-Petrusewicz, I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J. Neurol. Sci. 1997, 146, 67–72. [Google Scholar] [CrossRef]
- Montes, J.; McDermott, M.P.; Mirek, E.; Mazzone, E.S.; Main, M.; Glanzman, A.M.; Duong, T.; Young, S.D.; Salazar, R.; Pasternak, A.; et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression. PLoS ONE 2018, 13, e0199657. [Google Scholar] [CrossRef]
- Piepers, S.; van den Berg, L.H.; Brugman, F.; Scheffer, H.; Ruiterkamp-Versteeg, M.; van Engelen, B.G.; Faber, C.G.; de Visser, M.; van der Pol, W.L.; Wokke, J.H. A natural history study of late onset spinal muscular atrophy types 3b and 4. J. Neurol. 2008, 255, 1400–1404. [Google Scholar] [CrossRef]
- Mongiovi, P.; Dilek, N.; Garland, C.; Hunter, M.; Kissel, J.T.; Luebbe, E.; McDermott, M.P.; Johnson, N.; Heatwole, C. Patient Reported Impact of Symptoms in Spinal Muscular Atrophy (PRISM-SMA). Neurology 2018, 91, e1206–e1214. [Google Scholar] [CrossRef]
- Wan, H.W.Y.; Carey, K.A.; D’Silva, A.; Vucic, S.; Kiernan, M.C.; Kasparian, N.A.; Farrar, M.A. Health, wellbeing and lived experiences of adults with SMA: A scoping systematic review. Orphanet J. Rare Dis. 2020, 15, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Bastida, J.; Pena-Longobardo, L.M.; Aranda-Reneo, I.; Tizzano, E.; Sefton, M.; Oliva-Moreno, J. Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J. Rare Dis. 2017, 12, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjorth, E.; Kreicbergs, U.; Sejersen, T.; Lovgren, M. Parents’ advice to healthcare professionals working with children who have spinal muscular atrophy. Eur. J. Paediatr. Neurol. 2018, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; McGraw, S.; Henne, J.; Jarecki, J.; Hobby, K.; Yeh, W.S. Understanding the experiences and needs of individuals with Spinal Muscular Atrophy and their parents: A qualitative study. BMC Neurol. 2015, 15, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, R.; Belter, L.; Wasnock, M.; Nazarelli, A.; Jarecki, J. Evaluating Benefit-risk Decision-making in Spinal Muscular Atrophy: A First-ever Study to Assess Risk Tolerance in the SMA Patient Community. Clin. Ther. 2019, 41, 943–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGraw, S.; Qian, Y.; Henne, J.; Jarecki, J.; Hobby, K.; Yeh, W.S. A qualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. 2017, 17, 68. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Lucibello, S.; Perulli, M.; Coratti, G.; de Sanctis, R.; Pera, M.C.; Pane, M.; Montes, J.; de Vivo, D.C.; Darras, B.T.; et al. Longitudinal natural history of type I spinal muscular atrophy: A critical review. Orphanet J. Rare Dis. 2020, 15, 84. [Google Scholar] [CrossRef]
- Wang, C.H.; Finkel, R.S.; Bertini, E.S.; Schroth, M.; Simonds, A.; Wong, B.; Aloysius, A.; Morrison, L.; Main, M.; Crawford, T.O.; et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child Neurol. 2007, 22, 1027–1049. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Nurputra, D.K.; Lai, P.S.; Harahap, N.I.; Morikawa, S.; Yamamoto, T.; Nishimura, N.; Kubo, Y.; Takeuchi, A.; Saito, T.; Takeshima, Y.; et al. Spinal muscular atrophy: From gene discovery to clinical trials. Ann. Hum. Genet. 2013, 77, 435–463. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Day, J.W.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.; Schultz, M.; et al. AVXS-101 phase 3 study in spinal muscular atrophy type 1. J. Neurol. Neurosurg. Psychiatry 2019, 90, e8. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 2020, 1, CD006282. [Google Scholar] [CrossRef] [PubMed]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [Green Version]
- Luhrmann, R.; Kastner, B.; Bach, M. Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim. Biophys. Acta 1990, 1087, 265–292. [Google Scholar] [CrossRef]
- Lerner, M.R.; Steitz, J.A. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1979, 76, 5495–5499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatnik, A.J.; McGovern, V.L.; Le, T.T.; Iyer, C.C.; Kaspar, B.K.; Burghes, A.H.M. Conditional deletion of SMN in cell culture identifies functional SMN alleles. Hum. Mol. Genet. 2021, 29, 3477–3492. [Google Scholar] [CrossRef]
- So, B.R.; Zhang, Z.; Dreyfuss, G. The Function of Survival Motor Neuron Complex and Its Role in Spinal Muscular Atrophy Pathogenesis. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 99–111. [Google Scholar]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Pellizzoni, L.; Kataoka, N.; Charroux, B.; Dreyfuss, G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 1998, 95, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Baccon, J.; Pellizzoni, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J. Biol. Chem. 2002, 277, 31957–31962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhler, D.; Raker, V.; Luhrmann, R.; Fischer, U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: Implications for spinal muscular atrophy. Hum. Mol. Genet. 1999, 8, 2351–2357. [Google Scholar] [CrossRef]
- Carissimi, C.; Baccon, J.; Straccia, M.; Chiarella, P.; Maiolica, A.; Sawyer, A.; Rappsilber, J.; Pellizzoni, L. Unrip is a component of SMN complexes active in snRNP assembly. FEBS Lett. 2005, 579, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Carissimi, C.; Saieva, L.; Baccon, J.; Chiarella, P.; Maiolica, A.; Sawyer, A.; Rappsilber, J.; Pellizzoni, L. Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J. Biol. Chem. 2006, 281, 8126–8134. [Google Scholar] [CrossRef] [Green Version]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J. Cell Biol. 1999, 147, 1181–1194. [Google Scholar] [CrossRef]
- Charroux, B.; Pellizzoni, L.; Perkinson, R.A.; Yong, J.; Shevchenko, A.; Mann, M.; Dreyfuss, G. Gemin4: A novel component of the SMN complex that is found in both gems and nucleoli. J. Cell Biol. 2000, 148, 1177–1186. [Google Scholar] [CrossRef]
- Fischer, U.; Liu, Q.; Dreyfuss, G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997, 90, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Gubitz, A.K.; Mourelatos, Z.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J. Biol. Chem. 2002, 277, 5631–5636. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Buhler, D.; Pillai, R.; Lottspeich, F.; Fischer, U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol 2001, 3, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L.; Baccon, J.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J. Biol. Chem. 2002, 277, 7540–7545. [Google Scholar] [CrossRef] [Green Version]
- Pellizzoni, L.; Yong, J.; Dreyfuss, G. Essential role for the SMN complex in the specificity of snRNP assembly. Science 2002, 298, 1775–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. Spinal muscular atrophy: The role of SMN in axonal mRNA regulation. Brain Res. 2012, 1462, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; Vercelli, A. Neuromuscular Junctions as Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef] [Green Version]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Goncalves Ido, C.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Rathod, R.; Havlicek, S.; Frank, N.; Blum, R.; Sendtner, M. Laminin induced local axonal translation of beta-actin mRNA is impaired in SMN-deficient motoneurons. Histochem. Cell Biol. 2012, 138, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.M.; Shen, H.; Barry, D.M.; Garcia, V.B.; Rose, F.F., Jr.; Lorson, C.L.; Garcia, M.L. The spinal muscular atrophy mouse model, SMADelta7, displays altered axonal transport without global neurofilament alterations. Acta Neuropathol. 2011, 122, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Sasaki, Y.; Wen, Z.; Bassell, G.J.; Zheng, J.Q. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat. Neurosci. 2006, 9, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, B.; Krober, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Hosseini Barkooie, S.M.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013, 22, 1328–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowerman, M.; Anderson, C.L.; Beauvais, A.; Boyl, P.P.; Witke, W.; Kothary, R. SMN, profilin IIa and plastin 3: A link between the deregulation of actin dynamics and SMA pathogenesis. Mol. Cell. Neurosci. 2009, 42, 66–74. [Google Scholar] [CrossRef]
- Giesemann, T.; Rathke-Hartlieb, S.; Rothkegel, M.; Bartsch, J.W.; Buchmeier, S.; Jockusch, B.M.; Jockusch, H. A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J. Biol. Chem. 1999, 274, 37908–37914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bergeijk, J.; Rydel-Konecke, K.; Grothe, C.; Claus, P. The spinal muscular atrophy gene product regulates neurite outgrowth: Importance of the C terminus. FASEB J. 2007, 21, 1492–1502. [Google Scholar] [CrossRef]
- Fan, L.; Simard, L.R. Survival motor neuron (SMN) protein: Role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 2002, 11, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef]
- Gogliotti, R.G.; Quinlan, K.A.; Barlow, C.B.; Heier, C.R.; Heckman, C.J.; Didonato, C.J. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 2012, 32, 3818–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, T.L.; Kong, L.; Wang, X.; Osborne, M.A.; Crowder, M.E.; Van Meerbeke, J.P.; Xu, X.; Davis, C.; Wooley, J.; Goldhamer, D.J.; et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 2012, 32, 8703–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, V.L.; Iyer, C.C.; Arnold, W.D.; Gombash, S.E.; Zaworski, P.G.; Blatnik, A.J., 3rd; Foust, K.D.; Burghes, A.H. SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNDelta7 mouse model of SMA. Hum. Mol. Genet. 2015, 24, 5524–5541. [Google Scholar] [CrossRef] [Green Version]
- Paez-Colasante, X.; Seaberg, B.; Martinez, T.L.; Kong, L.; Sumner, C.J.; Rimer, M. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS ONE 2013, 8, e75866. [Google Scholar] [CrossRef] [Green Version]
- Park, G.H.; Maeno-Hikichi, Y.; Awano, T.; Landmesser, L.T.; Monani, U.R. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J. Neurosci. 2010, 30, 12005–12019. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Coovert, D.D.; Burghes, A.H. Animal models of spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 2451–2457. [Google Scholar] [CrossRef] [Green Version]
- Gabanella, F.; Butchbach, M.E.; Saieva, L.; Carissimi, C.; Burghes, A.H.; Pellizzoni, L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2007, 2, e921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabanella, F.; Carissimi, C.; Usiello, A.; Pellizzoni, L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 2005, 14, 3629–3642. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.M.; Kariya, S.; Patruni, S.; Osborne, M.A.; Liu, D.; Henderson, C.E.; Li, D.K.; Pellizzoni, L.; Rojas, J.; Valenzuela, D.M.; et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Investig. 2011, 121, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Burlet, P.; Huber, C.; Bertrandy, S.; Ludosky, M.A.; Zwaenepoel, I.; Clermont, O.; Roume, J.; Delezoide, A.L.; Cartaud, J.; Munnich, A.; et al. The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum. Mol. Genet. 1998, 7, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Schrank, B.; Kralewski, M.; Rossoll, W.; Sendtner, M. Reduced survival motor neuron (Smn) gene dose in mice leads to motor neuron degeneration: An animal model for spinal muscular atrophy type III. Hum. Mol. Genet. 2000, 9, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.K.; Gibbs, R.M.; Feng, Z.; Ko, C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012, 21, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Sendtner, M. Developmental regulation of SMN expression: Pathophysiological implications and perspectives for therapy development in spinal muscular atrophy. Gene Ther. 2017, 24, 506–513. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; DiDonato, C.J.; McGovern, V.L.; Arnold, W.D. Mammalian Models of Spinal Muscular Atrophy. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 241–260. [Google Scholar]
- Gavrilina, T.O.; McGovern, V.L.; Workman, E.; Crawford, T.O.; Gogliotti, R.G.; DiDonato, C.J.; Monani, U.R.; Morris, G.E.; Burghes, A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008, 17, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Fu, A.K.; Ip, N.Y. Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 2012, 35, 441–453. [Google Scholar] [CrossRef]
- Duque, S.I.; Arnold, W.D.; Odermatt, P.; Li, X.; Porensky, P.N.; Schmelzer, L.; Meyer, K.; Kolb, S.J.; Schumperli, D.; Kaspar, B.K.; et al. A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 2015, 77, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Murray, L.M.; Beauvais, A.; Pinheiro, B.; Kothary, R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 2012, 22, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Gogliotti, R.G.; Cardona, H.; Singh, J.; Bail, S.; Emery, C.; Kuntz, N.; Jorgensen, M.; Durens, M.; Xia, B.; Barlow, C.; et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013, 22, 4084–4101. [Google Scholar] [CrossRef] [Green Version]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; DiDonato, C.J. A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Michaud, M.; Arnoux, T.; Bielli, S.; Durand, E.; Rotrou, Y.; Jablonka, S.; Robert, F.; Giraudon-Paoli, M.; Riessland, M.; Mattei, M.G.; et al. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 2010, 38, 125–135. [Google Scholar] [CrossRef]
- Monani, U.R.; Sendtner, M.; Coovert, D.D.; Parsons, D.W.; Andreassi, C.; Le, T.T.; Jablonka, S.; Schrank, B.; Rossoll, W.; Prior, T.W.; et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.M.; Beauvais, A.; Gibeault, S.; Courtney, N.L.; Kothary, R. Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun. 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [Green Version]
- Nichterwitz, S.; Nijssen, J.; Storvall, H.; Schweingruber, C.; Comley, L.H.; Allodi, I.; Lee, M.V.; Deng, Q.; Sandberg, R.; Hedlund, E. LCM-seq reveals unique transcriptional adaptation mechanisms of resistant neurons and identifies protective pathways in spinal muscular atrophy. Genome Res. 2020, 30, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Broner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef]
- Fayzullina, S.; Martin, L.J. Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of Spinal Muscular Atrophy (SMA). PLoS ONE 2014, 9, e93329. [Google Scholar] [CrossRef] [PubMed]
- Huo, Q.; Kayikci, M.; Odermatt, P.; Meyer, K.; Michels, O.; Saxena, S.; Ule, J.; Schumperli, D. Splicing changes in SMA mouse motoneurons and SMN-depleted neuroblastoma cells: Evidence for involvement of splicing regulatory proteins. RNA Biol. 2014, 11, 1430–1446. [Google Scholar] [CrossRef] [Green Version]
- Baumer, D.; Lee, S.; Nicholson, G.; Davies, J.L.; Parkinson, N.J.; Murray, L.M.; Gillingwater, T.H.; Ansorge, O.; Davies, K.E.; Talbot, K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009, 5, e1000773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O’Donovan, M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, K.A.; Reedich, E.J.; Arnold, W.D.; Puritz, A.C.; Cavarsan, C.F.; Heckman, C.J.; DiDonato, C.J. Hyperexcitability precedes motoneuron loss in the Smn(2B/-) mouse model of spinal muscular atrophy. J. Neurophysiol. 2019, 122, 1297–1311. [Google Scholar] [CrossRef]
- Ling, K.K.; Lin, M.Y.; Zingg, B.; Feng, Z.; Ko, C.P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE 2010, 5, e15457. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.; Pearson, K.G. The role of proprioceptive feedback in the regulation and adaptation of locomotor activity. Adv. Exp. Med. Biol. 2002, 508, 343–355. [Google Scholar] [CrossRef]
- Windhorst, U. Muscle proprioceptive feedback and spinal networks. Brain Res. Bull. 2007, 73, 155–202. [Google Scholar] [CrossRef] [PubMed]
- Davidoff, R.A. Skeletal muscle tone and the misunderstood stretch reflex. Neurology 1992, 42, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Dietz, V.; Sinkjaer, T. Spastic movement disorder: Impaired reflex function and altered muscle mechanics. Lancet Neurol. 2007, 6, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Bhatia, K.; Kannan, A.; Gangwani, L. Molecular Mechanisms of Neurodegeneration in Spinal Muscular Atrophy. J. Exp. Neurosci. 2016, 10, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 6986–7004. [Google Scholar] [CrossRef] [Green Version]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of p53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576. [Google Scholar] [CrossRef] [Green Version]
- Courtney, N.L.; Mole, A.J.; Thomson, A.K.; Murray, L.M. Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death Dis. 2019, 10, 515. [Google Scholar] [CrossRef]
- Reedich, E.J.; Kalski, M.; Armijo, N.; Cox, G.A.; DiDonato, C.J. Spinal motor neuron loss occurs through a p53-and-p21-independent mechanism in the Smn2B/− mouse model of spinal muscular atrophy. Exp. Neurol. 2021, 337, 113587. [Google Scholar] [CrossRef]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Park, G.H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marce, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef]
- Neve, A.; Trub, J.; Saxena, S.; Schumperli, D. Central and peripheral defects in motor units of the diaphragm of spinal muscular atrophy mice. Mol. Cell. Neurosci. 2016, 70, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Torres-Benito, L.; Neher, M.F.; Cano, R.; Ruiz, R.; Tabares, L. SMN requirement for synaptic vesicle, active zone and microtubule postnatal organization in motor nerve terminals. PLoS ONE 2011, 6, e26164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, R.; Lopez-Manzaneda, M.; Arumugam, S.; Tabares, L. Synaptotagmin-2, and -1, linked to neurotransmission impairment and vulnerability in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 4703–4716. [Google Scholar] [CrossRef] [PubMed]
- Voigt, T.; Meyer, K.; Baum, O.; Schumperli, D. Ultrastructural changes in diaphragm neuromuscular junctions in a severe mouse model for Spinal Muscular Atrophy and their prevention by bifunctional U7 snRNA correcting SMN2 splicing. Neuromuscul. Disord. 2010, 20, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor neuron mitochondrial dysfunction in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.C.; Denton, K.R.; Wang, Z.B.; Zhang, X.; Li, X.J. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis. Model. Mech. 2016, 9, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.G.; Deguise, M.O.; Murray, L.M.; Yazdani, A.; De Repentigny, Y.; Boudreau-Lariviere, C.; Kothary, R. Myogenic program dysregulation is contributory to disease pathogenesis in spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 4249–4259. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R.; Casanas, J.J.; Torres-Benito, L.; Cano, R.; Tabares, L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010, 30, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, R.; Tabares, L. Neurotransmitter release in motor nerve terminals of a mouse model of mild spinal muscular atrophy. J. Anat. 2014, 224, 74–84. [Google Scholar] [CrossRef]
- Jablonka, S.; Beck, M.; Lechner, B.D.; Mayer, C.; Sendtner, M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007, 179, 139–149. [Google Scholar] [CrossRef] [Green Version]
- McGovern, V.L.; Gavrilina, T.O.; Beattie, C.E.; Burghes, A.H. Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet. 2008, 17, 2900–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.L.; Chen, T.H.; Hsu, Y.Y.; Cheng, Y.H.; Juang, B.T.; Jong, Y.J. Selective Neuromuscular Denervation in Taiwanese Severe SMA Mouse Can Be Reversed by Morpholino Antisense Oligonucleotides. PLoS ONE 2016, 11, e0154723. [Google Scholar] [CrossRef]
- Thomson, S.R.; Nahon, J.E.; Mutsaers, C.A.; Thomson, D.; Hamilton, G.; Parson, S.H.; Gillingwater, T.H. Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLoS ONE 2012, 7, e52605. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Croizat, B.; Lagrange, M.C.; Warter, J.M.; Poindron, P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet 1995, 345, 694–695. [Google Scholar] [CrossRef]
- Walker, M.P.; Rajendra, T.K.; Saieva, L.; Fuentes, J.L.; Pellizzoni, L.; Matera, A.G. SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet. 2008, 17, 3399–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berciano, M.T.; Castillo-Iglesias, M.S.; Val-Bernal, J.F.; Lafarga, V.; Rodriguez-Rey, J.C.; Lafarga, M.; Tapia, O. Mislocalization of SMN from the I-band and M-band in human skeletal myofibers in spinal muscular atrophy associates with primary structural alterations of the sarcomere. Cell Tissue Res. 2020, 381, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.P.; Monani, U.R. Muscle-specific SMN reduction reveals motor neuron-independent disease in spinal muscular atrophy models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef] [Green Version]
- Iyer, C.C.; McGovern, V.L.; Murray, J.D.; Gombash, S.E.; Zaworski, P.G.; Foust, K.D.; Janssen, P.M.; Burghes, A.H. Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNDelta7 mouse model of SMA. Hum. Mol. Genet. 2015, 24, 6160–6173. [Google Scholar] [CrossRef] [Green Version]
- Guettier-Sigrist, S.; Coupin, G.; Braun, S.; Rogovitz, D.; Courdier, I.; Warter, J.M.; Poindron, P. On the possible role of muscle in the pathogenesis of spinal muscular atrophy. Fundam. Clin. Pharmacol. 2001, 15, 31–40. [Google Scholar] [CrossRef]
- Shafey, D.; Cote, P.D.; Kothary, R. Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 2005, 311, 49–61. [Google Scholar] [CrossRef]
- Arnold, A.S.; Gueye, M.; Guettier-Sigrist, S.; Courdier-Fruh, I.; Coupin, G.; Poindron, P.; Gies, J.P. Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab. Investig. 2004, 84, 1271–1278. [Google Scholar] [CrossRef]
- Bricceno, K.V.; Martinez, T.; Leikina, E.; Duguez, S.; Partridge, T.A.; Chernomordik, L.V.; Fischbeck, K.H.; Sumner, C.J.; Burnett, B.G. Survival motor neuron protein deficiency impairs myotube formation by altering myogenic gene expression and focal adhesion dynamics. Hum. Mol. Genet. 2014, 23, 4745–4757. [Google Scholar] [CrossRef] [Green Version]
- Hayhurst, M.; Wagner, A.K.; Cerletti, M.; Wagers, A.J.; Rubin, L.L. A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Dev. Biol. 2012, 368, 323–334. [Google Scholar] [CrossRef]
- Osborne, M.; Gomez, D.; Feng, Z.; McEwen, C.; Beltran, J.; Cirillo, K.; El-Khodor, B.; Lin, M.Y.; Li, Y.; Knowlton, W.M.; et al. Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum. Mol. Genet. 2012, 21, 4431–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.I.; Mikesh, M.; Smith, I.; Rimer, M.; Thompson, W. Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev. Biol. 2011, 356, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.; Aghamaleky Sarvestany, A.; Roche, S.L.; Symes, R.C.; Gillingwater, T.H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2014, 23, 2235–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, G.; Powis, R.A.; Jones, R.A.; Groen, E.J.; Shorrock, H.K.; Lane, F.M.; Zheng, Y.; Sherman, D.L.; Brophy, P.J.; Gillingwater, T.H. Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 2853–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindt, H.; Feng, Z.; Mazzasette, C.; Glascock, J.J.; Valdivia, D.; Pyles, N.; Crawford, T.O.; Swoboda, K.J.; Patitucci, T.N.; Ebert, A.D.; et al. Astrocytes influence the severity of spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 4094–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.A.; Stucky, C.L.; Ebert, A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Araujo, A.; Araujo, M.; Swoboda, K.J. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J. Pediatr. 2009, 155, 292–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, J.; Martinez-Hernandez, R.; Also-Rallo, E.; Alias, L.; Barcelo, M.J.; Amenedo, M.; Medina, C.; Senosiain, R.; Calaf, J.; Baiget, M.; et al. Ultrasound evaluation of fetal movements in pregnancies at risk for severe spinal muscular atrophy. Neuromuscul. Disord. 2011, 21, 97–101. [Google Scholar] [CrossRef]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Biondi, O.; Lopes, P.; Desseille, C.; Branchu, J.; Chali, F.; Ben Salah, A.; Pariset, C.; Chanoine, C.; Charbonnier, F. Physical exercise reduces cardiac defects in type 2 spinal muscular atrophy-like mice. J. Physiol. 2012, 590, 5907–5925. [Google Scholar] [CrossRef]
- Bevan, A.K.; Hutchinson, K.R.; Foust, K.D.; Braun, L.; McGovern, V.L.; Schmelzer, L.; Ward, J.G.; Petruska, J.C.; Lucchesi, P.A.; Burghes, A.H.; et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 2010, 19, 3895–3905. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.R.; DiDonato, C.J. ECG in neonate mice with spinal muscular atrophy allows assessment of drug efficacy. Front. Biosci. (Elite Ed.) 2015, 7, 107–116. [Google Scholar]
- Heier, C.R.; Satta, R.; Lutz, C.; DiDonato, C.J. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 2010, 19, 3906–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Maxwell, G.K.; Szunyogova, E.; Shorrock, H.K.; Gillingwater, T.H.; Parson, S.H. Developmental and degenerative cardiac defects in the Taiwanese mouse model of severe spinal muscular atrophy. J. Anat. 2018, 232, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreml, J.; Riessland, M.; Paterno, M.; Garbes, L.; Rossbach, K.; Ackermann, B.; Kramer, J.; Somers, E.; Parson, S.H.; Heller, R.; et al. Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 2013, 21, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Habibi, J.; Ma, L.; Glascock, J.J.; Sowers, J.R.; Lorson, C.L. Partial restoration of cardio-vascular defects in a rescued severe model of spinal muscular atrophy. J. Mol. Cell. Cardiol. 2012, 52, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shababi, M.; Habibi, J.; Yang, H.T.; Vale, S.M.; Sewell, W.A.; Lorson, C.L. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 2010, 19, 4059–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Wan, B.; Feng, P.; Sun, J.; Rigo, F.; Bennett, C.F.; Akerman, M.; Krainer, A.R.; Hua, Y. Downregulation of Survivin contributes to cell-cycle arrest during postnatal cardiac development in a severe spinal muscular atrophy mouse model. Hum. Mol. Genet. 2018, 27, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Chen, C.L.; Ting, C.H.; Lin-Chao, S.; Hwu, W.L.; Dodge, J.C.; Passini, M.A.; Cheng, S.H. Systemic administration of a recombinant AAV1 vector encoding IGF-1 improves disease manifestations in SMA mice. Mol. Ther. 2014, 22, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Kumada, S.; Uchiyama, A.; Saito, K.; Osawa, M.; Yagishita, A.; Kurata, K.; Hayashi, M. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings. Brain Dev. 2004, 26, 53–56. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Shababi, M.; Lorson, C.L.; Rudnik-Schoneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell. Mol. Life Sci. 2016, 73, 1003–1020. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.-P.; Wang, G.-S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as Just a Motor Neuron Disease. Pediatr. Neurol. 2020, 109, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [Green Version]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Chen, T.H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.S.; Jarecki, J.; MacKenzie, A.; Chen, K.S. Spinal Muscular Atrophy Therapeutics Development. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.-P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 263–281. [Google Scholar]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef]
- Singh, N.N.; Lee, B.M.; DiDonato, C.J.; Singh, R.N. Mechanistic principles of antisense targets for the treatment of spinal muscular atrophy. Future Med. Chem. 2015, 7, 1793–1808. [Google Scholar] [CrossRef] [Green Version]
- Verma, A. Recent Advances in Antisense Oligonucleotide Therapy in Genetic Neuromuscular Diseases. Ann. Indian Acad. Neurol. 2018, 21, 3–8. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O’Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef] [Green Version]
- D’Ydewalle, C.; Sumner, C.J. Spinal Muscular Atrophy Therapeutics: Where do we Stand? Neurotherapeutics 2015, 12, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Ramos, D.M.; d’Ydewalle, C.; Gabbeta, V.; Dakka, A.; Klein, S.K.; Norris, D.A.; Matson, J.; Taylor, S.J.; Zaworski, P.G.; Prior, T.W.; et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J. Clin. Investig. 2019, 129, 4817–4831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuluaga Sanchez, S.; Purser, M.; Mader, G.; Gould, I.G.; Knight, C.; Johnson, N.B.; Patel, M.; Jensen, J.; Odom, T. Improved quality of life and life-years in patients with infantile-onset SMA following treatment with nusinersen. Value Health 2019, 22, S337. [Google Scholar] [CrossRef]
- Hagenacker, T.; Wurster, C.D.; Gunther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef]
- Jochmann, E.; Steinbach, R.; Jochmann, T.; Chung, H.Y.; Rodiger, A.; Neumann, R.; Mayer, T.E.; Kirchhof, K.; Loudovici-Krug, D.; Smolenski, U.C.; et al. Experiences from treating seven adult 5q spinal muscular atrophy patients with Nusinersen. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420907803. [Google Scholar] [CrossRef] [Green Version]
- Arnold, W.; McGovern, V.L.; Sanchez, B.; Li, J.; Corlett, K.M.; Kolb, S.J.; Rutkove, S.B.; Burghes, A.H. The neuromuscular impact of symptomatic SMN restoration in a mouse model of spinal muscular atrophy. Neurobiol. Dis. 2016, 87, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Crawford, T.; Sumner, C.; Finkel, R.; De Vivo, D.; Oskoui, M.; Tizzano, E.; Zhao, G.; Petrillo, M.; Stebbins, C.; Farwell, W. Phosphorylated neurofilament heavy chain (pNF-H) levels in infants and children with SMA: Evaluation of pNF-H as a potential biomarker of SMA disease activity. Neuromuscul. Disord. 2018, 28, S110–S111. [Google Scholar] [CrossRef]
- Darras, B.; Finkel, R.; Mercuri, E.; Sumner, C.; Oskoui, M.; Tizzano, E.; Ryan, M.; Zhao, G.; Petrillo, M.; Stebbins, C.; et al. Association of phosphorylated neurofilament heavy chain (pNF-H) with nusinersen treatment of SMA: Analyses from the ENDEAR and CHERISH studies. Neuromuscul. Disord. 2018, 28, S31. [Google Scholar] [CrossRef]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porensky, P.N.; Burghes, A.H. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum. Gene Ther. 2013, 24, 489–498. [Google Scholar] [CrossRef] [Green Version]
- SPINRAZA [Package Insert]; Biogen Inc.: Cambridge, MA, USA, 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf (accessed on 3 February 2021).
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Schmelzer, L.; Braun, L.; McGovern, V.; Likhite, S.; Michels, O.; Govoni, A.; Fitzgerald, J.; Morales, P.; et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: A dose-response study in mice and nonhuman primates. Mol. Ther. 2015, 23, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Crawford, T.O. Two breakthrough gene-targeted treatments for spinal muscular atrophy: Challenges remain. J. Clin. Investig. 2018, 128, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- ZOLGENSMA [Package Insert]; AveXis, Inc.: Bannockburn, IL, USA, 2019. Available online: https://www.fda.gov/media/126109/download (accessed on 3 February 2021).
- Dyer, O. Health ministers condemn Novartis lottery for Zolgensma, the world’s most expensive drug. BMJ 2020, 368, m580. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; Servais, L.; Day, J.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 16-month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with type 1 spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef]
- Mercuri, E.; Barisic, N.; Boespflug-Tanguy, O.; Deconinck, N.; Kostera-Pruszczyk, A.; Masson, R.; Mazzone, E.; Nascimento, A.; Saito, K.; Vlodavets, D.; et al. SUNFISH Part 2: Efficacy and Safety of Risdiplam (RG7916) in Patients with Type 2 or Non-Ambulant Type 3 Spinal Muscular Atrophy (SMA) (1260). Neurology 2020, 94, 1260. [Google Scholar]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Genentech Inc. Evrysdi™ (Risdiplam): US Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213535s000lbl.pdf (accessed on 3 February 2021).
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Pagliarulo, N. Zolgensma Set a New Drug Pricing Bar. Insurers Show Some Signs of Pushback. BioPharma Dive. 2019. Available online: https://www.biopharmadive.com/news/zolgensma-set-a-new-drug-pricing-bar-insurers-show-some-signs-of-pushback/558101/ (accessed on 3 February 2021).
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Dixon, S.; Naik, R.; Noone, J.M.; Buchenberger, J.D.; Whitmire, S.M.; Mills, R.; Arnold, W. Early experiences of nusinersen for the treatment of spinal muscular atrophy: Results from a large survey of patients and caregivers. Muscle Nerve 2020, 1–9. [Google Scholar] [CrossRef]
- Ellis, A.G.; Mickle, K.; Herron-Smith, S.; Kumar, V.M.; Cianciolo, L.; Seidner, M.; Thokala, P.; Stevenson, M.; Rind, D.; Pearson, S.D. Spinraza® and Zolgensma® for Spinal Muscular Atrophy: Effectiveness and Value; Institute for Clinical and Economic Review: Boston, MA, USA, 2019. [Google Scholar]
- Starner, C.I.; Gleason, P.P. Spinal Muscular Atrophy Therapies: ICER Grounds the Price to Value Conversation in Facts. J. Manag. Care Spec. Pharm. 2019, 25, 1306–1308. [Google Scholar] [CrossRef]
- Darras, B.T.; De Vivo, D.C. Precious SMA natural history data: A benchmark to measure future treatment successes. Neurology 2018, 91, 337–339. [Google Scholar] [CrossRef]
- Glascock, J.J.; Osman, E.Y.; Wetz, M.J.; Krogman, M.M.; Shababi, M.; Lorson, C.L. Decreasing disease severity in symptomatic, Smn(-/-);SMN2(+/+), spinal muscular atrophy mice following scAAV9-SMN delivery. Hum. Gene Ther. 2012, 23, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Muller-Felber, W.; Vill, K.; Schwartz, O.; Glaser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Roschinger, W.; Becker, M.; Durner, J.; et al. Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening—Opportunity or Burden? J. Neuromuscul. Dis. 2020, 7, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, L.; Solari, A.; Vita, G.; Bertini, E.; Minetti, C.; Mongini, T.; Mazzoni, E.; Angelini, C.; Morandi, L. Role of gabapentin in spinal muscular atrophy: Results of a multicenter, randomized Italian study. J. Child. Neurol. 2003, 18, 537–541. [Google Scholar] [CrossRef]
- Miller, R.G.; Moore, D.H.; Dronsky, V.; Bradley, W.; Barohn, R.; Bryan, W.; Prior, T.W.; Gelinas, D.F.; Iannaccone, S.; Kissel, J.; et al. A placebo-controlled trial of gabapentin in spinal muscular atrophy. J. Neurol. Sci. 2001, 191, 127–131. [Google Scholar] [CrossRef]
- Dimitriadi, M.; Kye, M.J.; Kalloo, G.; Yersak, J.M.; Sahin, M.; Hart, A.C. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci. 2013, 33, 6557–6562. [Google Scholar] [CrossRef] [Green Version]
- Haddad, H.; Cifuentes-Diaz, C.; Miroglio, A.; Roblot, N.; Joshi, V.; Melki, J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 2003, 28, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Abbara, C.; Estournet, B.; Lacomblez, L.; Lelievre, B.; Ouslimani, A.; Lehmann, B.; Viollet, L.; Barois, A.; Diquet, B. Riluzole pharmacokinetics in young patients with spinal muscular atrophy. Br. J. Clin. Pharmacol. 2011, 71, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Pang, K.; Chen, Y.; Wang, S.; Li, H.; Xu, Y.; Han, F.; Yao, H.; Liu, H.; Lopes-Rodrigues, V.; et al. TrkB agonistic antibodies superior to BDNF: Utility in treating motoneuron degeneration. Neurobiol. Dis. 2019, 132, 104590. [Google Scholar] [CrossRef] [PubMed]
- Haase, G.; Kennel, P.; Pettmann, B.; Vigne, E.; Akli, S.; Revah, F.; Schmalbruch, H.; Kahn, A. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat. Med. 1997, 3, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Ikeda, K.; Klinkosz, B.; Cedarbaum, J.M.; Wong, V.; Lindsay, R.M. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 1994, 265, 1107–1110. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Houenou, L.J.; Johnson, J.E.; Lin, L.F.; Li, L.; Lo, A.C.; Newsome, A.L.; Prevette, D.M.; Wang, S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature 1995, 373, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Sendtner, M.; Holtmann, B.; Kolbeck, R.; Thoenen, H.; Barde, Y.A. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 1992, 360, 757–759. [Google Scholar] [CrossRef]
- Sendtner, M.; Schmalbruch, H.; Stockli, K.A.; Carroll, P.; Kreutzberg, G.W.; Thoenen, H. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 1992, 358, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Sendtner, M.; Stockli, K.A.; Thoenen, H. Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J. Cell Biol. 1992, 118, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Murdocca, M.; Malgieri, A.; Luchetti, A.; Saieva, L.; Dobrowolny, G.; de Leonibus, E.; Filareto, A.; Quitadamo, M.C.; Novelli, G.; Musaro, A.; et al. IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol. Med. 2012, 18, 1076–1085. [Google Scholar] [CrossRef]
- Bosch-Marce, M.; Wee, C.D.; Martinez, T.L.; Lipkes, C.E.; Choe, D.W.; Kong, L.; Van Meerbeke, J.P.; Musaro, A.; Sumner, C.J. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum. Mol. Genet. 2011, 20, 1844–1853. [Google Scholar] [CrossRef]
- Shababi, M.; Glascock, J.; Lorson, C.L. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 2011, 22, 135–144. [Google Scholar] [CrossRef]
- Tsai, L.K.; Chen, Y.C.; Cheng, W.C.; Ting, C.H.; Dodge, J.C.; Hwu, W.L.; Cheng, S.H.; Passini, M.A. IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol. Dis. 2012, 45, 272–279. [Google Scholar] [CrossRef]
- Dombert, B.; Balk, S.; Luningschror, P.; Moradi, M.; Sivadasan, R.; Saal-Bauernschubert, L.; Jablonka, S. BDNF/trkB Induction of Calcium Transients through Cav2.2 Calcium Channels in Motoneurons Corresponds to F-actin Assembly and Growth Cone Formation on beta2-Chain Laminin (221). Front. Mol. Neurosci. 2017, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; Balk, S.; Franco-Espin, J.; Ojeda, J.; Hennlein, L.; Drexl, H.; Dombert, B.; Clausen, J.D.; Torres-Benito, L.; Saal-Bauernschubert, L.; et al. R-Roscovitine Improves Motoneuron Function in Mouse Models for Spinal Muscular Atrophy. iScience 2020, 23, 100826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.T.; Chen, C.S.; Jong, Y.J.; Chang, F.R.; Lo, Y.C. Loganin possesses neuroprotective properties, restores SMN protein and activates protein synthesis positive regulator Akt/mTOR in experimental models of spinal muscular atrophy. Pharmacol. Res. 2016, 111, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Yokomaku, D.; Richards, M.; Hori, H.; Adachi, N.; Kunugi, H. Functional interactions between steroid hormones and neurotrophin BDNF. World J. Biol. Chem. 2010, 1, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pinto, A.M.; Wan, L.; Wang, W.; Berg, M.G.; Oliva, I.; Singh, L.N.; Dengler, C.; Wei, Z.; Dreyfuss, G. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 19348–19353. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell. Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Bordet, T.; Berna, P.; Abitbol, J.L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Bertini, E.; Comi, G.; Kirschner, J.; Lusakowska, A.; Mercuri, E.; Scoto, M.; van der Pol, W.L.; Vuillerot, C.; Burdeska, A.; et al. Long-term follow-up of patients with type 2 and non-ambulant type 3 spinal muscular atrophy (SMA) treated with olesoxime in the OLEOS trial. Neuromuscul. Disord. 2020, 30, 959–969. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [Green Version]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Zhu, X.; Hadhazy, M.; Wehling, M.; Tidball, J.G.; McNally, E.M. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 2000, 474, 71–75. [Google Scholar] [CrossRef]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef] [Green Version]
- Rose, F.F., Jr.; Mattis, V.B.; Rindt, H.; Lorson, C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009, 18, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindt, H.; Buckley, D.M.; Vale, S.M.; Krogman, M.; Rose, F.F., Jr.; Garcia, M.L.; Lorson, C.L. Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 2012, 22, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Wee, C.D.; Warsing, L.C.; Choe, D.W.; Ng, A.S.; Lutz, C.; Wagner, K.R. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 2009, 18, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin inhibition in combination with antisense oligonucleotide therapy improves outcomes in spinal muscular atrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef]
- Dagbay, K.B.; Treece, E.; Streich, F.C., Jr.; Jackson, J.W.; Faucette, R.R.; Nikiforov, A.; Lin, S.C.; Boston, C.J.; Nicholls, S.B.; Capili, A.D.; et al. Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015. J. Biol. Chem. 2020, 295, 5404–5418. [Google Scholar] [CrossRef] [Green Version]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.J.; Hartman, J.J.; Hinken, A.C.; Muci, A.R.; Kawas, R.; Driscoll, L.; Godinez, G.; Lee, K.H.; Marquez, D.; Browne, W.F., IV; et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat. Med. 2012, 18, 452–455. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Edell, S.; Ferrufino, C.; Thomsen, K.; Hwee, D.T.; Morgan, B.P.; Malik, F.I.; Chin, E.R. The Fast Skeletal Muscle Troponin Activator Reldesemtiv in Combination with Nusinersen Improves Muscle Function in a Mouse Model of Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Edell, S.; Ferrufino, C.; Thomsen, K.; Hwee, D.T.; Morgan, B.P.; Malik, F.I.; Chin, E.R. The Fast Skeletal Muscle Troponin Activator Reldesemtiv in Combination with SMN-C1 Improves Muscle Function in a Mouse Model of Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Van der Pol, W.L.; Wadman, R.I.; Van den Berg, L.H.; Vrancken, A.F.J. Dysfunction of the neuromuscular junction in patients with spinal muscular atrophy type 2 and 3. Neuromuscul. Disord. 2012, 22, 871–872. [Google Scholar] [CrossRef]
- Vrbova, G.; Slawinska, U. Critical period of neuromuscular development: Importance for a new treatment of SMA. Neuromuscul. Disord. 2018, 28, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Tarr, T.B.; Malick, W.; Liang, M.; Valdomir, G.; Frasso, M.; Lacomis, D.; Reddel, S.W.; Garcia-Ocano, A.; Wipf, P.; Meriney, S.D. Evaluation of a novel calcium channel agonist for therapeutic potential in Lambert-Eaton myasthenic syndrome. J. Neurosci. 2013, 33, 10559–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarr, T.B.; Lacomis, D.; Reddel, S.W.; Liang, M.; Valdomir, G.; Frasso, M.; Wipf, P.; Meriney, S.D. Complete reversal of Lambert-Eaton myasthenic syndrome synaptic impairment by the combined use of a K+ channel blocker and a Ca2+ channel agonist. J. Physiol. 2014, 592, 3687–3696. [Google Scholar] [CrossRef] [PubMed]
- Ojala, K.S.; Liang, M.; Wipf, P.; Meriney, S.D. A Novel, Peripherally-Targeted Treatment for Ameliorating the Neuromuscular Weakness Caused by Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Anaheim, CA, USA, 28 June–1 July 2019. [Google Scholar]
- Ojala, K.S.; Kaufhold, C.J.; Davey, M.R.; Liang, M.; Wipf, P.; Meriney, S.D. GV-58, a selective calcium channel gating modifier, can increase muscle strength and neurotransmission in untreated and ASO-treated SMA model mice. In Proceedings of the Cure SMA Conference, Orlando, FL, USA, 11–14 June 2020. [Google Scholar]
- Bernal, S.; Alias, L.; Barcelo, M.J.; Also-Rallo, E.; Martinez-Hernandez, R.; Gamez, J.; Guillen-Navarro, E.; Rosell, J.; Hernando, I.; Rodriguez-Alvarez, F.J.; et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 2010, 47, 640–642. [Google Scholar] [CrossRef] [Green Version]
- Bernal, S.; Also-Rallo, E.; Martinez-Hernandez, R.; Alias, L.; Rodriguez-Alvarez, F.J.; Millan, J.M.; Hernandez-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef]
- Burghes, A.H.; Ingraham, S.E.; Kote-Jarai, Z.; Rosenfeld, S.; Herta, N.; Nadkarni, N.; DiDonato, C.J.; Carpten, J.; Hurko, O.; Florence, J.; et al. Linkage mapping of the spinal muscular atrophy gene. Hum. Genet. 1994, 93, 305–312. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alias, L.; March, F.; Vencesla, A.; Rodriguez-Alvarez, F.J.; Aller, E.; Fernandez, R.M.; Borrego, S.; Millan, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Cobben, J.M.; van der Steege, G.; Grootscholten, P.; de Visser, M.; Scheffer, H.; Buys, C.H. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 1995, 57, 805–808. [Google Scholar] [PubMed]
- DiDonato, C.J.; Morgan, K.; Carpten, J.D.; Fuerst, P.; Ingraham, S.E.; Prescott, G.; McPherson, J.D.; Wirth, B.; Zerres, K.; Hurko, O.; et al. Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am. J. Hum. Genet. 1994, 55, 1218–1229. [Google Scholar]
- Hahnen, E.; Forkert, R.; Marke, C.; Rudnik-Schoneborn, S.; Schonling, J.; Zerres, K.; Wirth, B. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet. 1995, 4, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejowska, M.; Borkowska, J.; Zimowski, J.; Kostera-Pruszczyk, A.; Milewski, M.; Jurek, M.; Sielska, D.; Kostyk, E.; Nyka, W.; Zaremba, J.; et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur. J. Hum. Genet. 2008, 16, 930–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedrzejowska, M.; Milewski, M.; Zimowski, J.; Borkowska, J.; Kostera-Pruszczyk, A.; Sielska, D.; Jurek, M.; Hausmanowa-Petrusewicz, I. Phenotype modifiers of spinal muscular atrophy: The number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease. Acta Biochim. Pol. 2009, 56, 103–108. [Google Scholar] [CrossRef]
- Oprea, G.E.; Krober, S.; McWhorter, M.L.; Rossoll, W.; Muller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Pane, M.; Lapenta, L.; Abiusi, E.; de Sanctis, R.; Luigetti, M.; Palermo, C.; Ranalli, D.; Fiori, S.; Tiziano, F.D.; Mercuri, E. Longitudinal assessments in discordant twins with SMA. Neuromuscul. Disord. 2017, 27, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Prior, T.W.; Swoboda, K.J.; Scott, H.D.; Hejmanowski, A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A 2004, 130A, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frebourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadman, R.I.; Jansen, M.D.; Curial, C.A.D.; Groen, E.J.N.; Stam, M.; Wijngaarde, C.A.; Medic, J.; Sodaar, P.; van Eijk, K.R.; Huibers, M.M.H.; et al. Analysis of FUS, PFN2, TDP-43, and PLS3 as potential disease severity modifiers in spinal muscular atrophy. Neurol. Genet. 2020, 6, e386. [Google Scholar] [CrossRef] [Green Version]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Lohr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef] [Green Version]
- Torres-Benito, L.; Schneider, S.; Rombo, R.; Ling, K.K.; Grysko, V.; Upadhyay, A.; Kononenko, N.L.; Rigo, F.; Bennett, C.F.; Wirth, B. NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice. Am. J. Hum. Genet. 2019, 105, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrafiah, A.; Karyka, E.; Coldicott, I.; Iremonger, K.; Lewis, K.E.; Ning, K.; Azzouz, M. Plastin 3 Promotes Motor Neuron Axonal Growth and Extends Survival in a Mouse Model of Spinal Muscular Atrophy. Mol. Ther. Methods Clin. Dev. 2018, 9, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Delanote, V.; Vandekerckhove, J.; Gettemans, J. Plastins: Versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 2005, 26, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engqvist-Goldstein, A.E.; Drubin, D.G. Actin assembly and endocytosis: From yeast to mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 287–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Kaifer, K.A.; Villalon, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.T.; Wolman, M.; Granato, M.; Beattie, C.E. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J. Neurosci. 2012, 32, 5074–5084. [Google Scholar] [CrossRef] [Green Version]
- McGovern, V.L.; Massoni-Laporte, A.; Wang, X.; Le, T.T.; Le, H.T.; Beattie, C.E.; Rich, M.M.; Burghes, A.H. Plastin 3 Expression Does Not Modify Spinal Muscular Atrophy Severity in the 7 SMA Mouse. PLoS ONE 2015, 10, e0132364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.Y.; Mikhail, A.; Ljubicic, V. Mechanisms of exercise-induced survival motor neuron expression in the skeletal muscle of spinal muscular atrophy-like mice. J. Physiol. 2019, 597, 4757–4778. [Google Scholar] [CrossRef]
- Grondard, C.; Biondi, O.; Armand, A.S.; Lecolle, S.; Della Gaspera, B.; Pariset, C.; Li, H.; Gallien, C.L.; Vidal, P.P.; Chanoine, C.; et al. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2005, 25, 7615–7622. [Google Scholar] [CrossRef] [Green Version]
- Biondi, O.; Grondard, C.; Lecolle, S.; Deforges, S.; Pariset, C.; Lopes, P.; Cifuentes-Diaz, C.; Li, H.; della Gaspera, B.; Chanoine, C.; et al. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2008, 28, 953–962. [Google Scholar] [CrossRef]
- Charbonnier, F. Exercise-induced neuroprotection in SMA model mice: A means for determining new therapeutic strategies. Mol. Neurobiol. 2007, 35, 217–223. [Google Scholar] [CrossRef]
- Bora, G.; Subasi-Yildiz, S.; Yesbek-Kaymaz, A.; Bulut, N.; Alemdaroglu, I.; Tunca-Yilmaz, O.; Topaloglu, H.; Karaduman, A.A.; Erdem-Yurter, H. Effects of Arm Cycling Exercise in Spinal Muscular Atrophy Type II Patients: A Pilot Study. J. Child Neurol. 2018, 33, 209–215. [Google Scholar] [CrossRef]
- Houdebine, L.; D’Amico, D.; Bastin, J.; Chali, F.; Desseille, C.; Rumeau, V.; Soukkari, J.; Oudot, C.; Rouquet, T.; Bariohay, B.; et al. Low-Intensity Running and High-Intensity Swimming Exercises Differentially Improve Energy Metabolism in Mice With Mild Spinal Muscular Atrophy. Front. Physiol. 2019, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Chali, F.; Desseille, C.; Houdebine, L.; Benoit, E.; Rouquet, T.; Bariohay, B.; Lopes, P.; Branchu, J.; Della Gaspera, B.; Pariset, C.; et al. Long-term exercise-specific neuroprotection in spinal muscular atrophy-like mice. J. Physiol. 2016, 594, 1931–1952. [Google Scholar] [CrossRef] [Green Version]
- Lewelt, A.; Krosschell, K.J.; Stoddard, G.J.; Weng, C.; Xue, M.; Marcus, R.L.; Gappmaier, E.; Viollet, L.; Johnson, B.A.; White, A.T.; et al. Resistance strength training exercise in children with spinal muscular atrophy. Muscle Nerve 2015, 52, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, K.L.; Hansen, R.S.; Preisler, N.; Thogersen, F.; Berthelsen, M.P.; Vissing, J. Training improves oxidative capacity, but not function, in spinal muscular atrophy type III. Muscle Nerve 2015, 52, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Bartels, B.; Montes, J.; van der Pol, W.L.; de Groot, J.F. Physical exercise training for type 3 spinal muscular atrophy. Cochrane Database Syst. Rev. 2019, 3, CD012120. [Google Scholar] [CrossRef] [Green Version]
- Vita, G.L.; Stancanelli, C.; La Foresta, S.; Faraone, C.; Sframeli, M.; Ferrero, A.; Fattore, C.; Galbo, R.; Ferraro, M.; Ricci, G.; et al. Psychosocial impact of sport activity in neuromuscular disorders. Neurol. Sci. 2020, 41, 2561–2567. [Google Scholar] [CrossRef]
- Gandolla, M.; Antonietti, A.; Longatelli, V.; Pedrocchi, A. The Effectiveness of Wearable Upper Limb Assistive Devices in Degenerative Neuromuscular Diseases: A Systematic Review and Meta-Analysis. Front. Bioeng. Biotechnol. 2019, 7, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagengruber, A.; Vogel, J. Functional Tasks Performed by People with Severe Muscular Atrophy Using an sEMG Controlled Robotic Manipulator. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2018, 2018, 1713–1718. [Google Scholar] [CrossRef]
- Haumont, T.; Rahman, T.; Sample, W.; King, M.M.; Church, C.; Henley, J.; Jayakumar, S. Wilmington robotic exoskeleton: A novel device to maintain arm improvement in muscular disease. J. Pediatr. Orthop. 2011, 31, e44–e49. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Sample, W.; Seliktar, R.; Alexander, M.; Scavina, M. A body-powered functional upper limb orthosis. J. Rehabil. Res. Dev. 2000, 37, 675–680. [Google Scholar]
- Sanz-Merodio, D.; Puyuelo, G.; Ganguly, A.; Garces, E.; Goñi, A.; Garcia, E. EXOtrainer Project Clinical Evaluation of Gait Training with Exoskeleton in Children with Spinal Muscular Atrophy. In Advances in Robotics Research: From Lab to Market: ECHORD++: Robotic Science Supporting Innovation; Grau, A., Morel, Y., Puig-Pey, A., Cecchi, F., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 211–227. [Google Scholar] [CrossRef]
- Sakaki, T.; Aoki, K.; Ushijima, K.; Sakuragi, M. Robotic stretcher for spinal muscular atrophy patient: Preliminary tests of user controllability. In Proceedings of the 10th International Conference on Ubiquitous Robots and Ambient Intelligence (URAI), Jeju, Korea, 30 October–2 November 2013; pp. 156–160. [Google Scholar]
- Pera, M.C.; Luigetti, M.; Pane, M.; Coratti, G.; Forcina, N.; Fanelli, L.; Mazzone, E.S.; Antonaci, L.; Lapenta, L.; Palermo, C.; et al. 6MWT can identify type 3 SMA patients with neuromuscular junction dysfunction. Neuromuscul. Disord. 2017, 27, 879–882. [Google Scholar] [CrossRef]
- Wadman, R.I.; Vrancken, A.F.; van den Berg, L.H.; van der Pol, W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.M.; Montes, J.; Finkel, R.S. Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological Exam-Part 2: Experience from a nusinersen clinical study. Muscle Nerve 2018, 57, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.M. Nusinersen: Antisense oligonucleotide to increase SMN protein production in spinal muscular atrophy. Drugs Today (Barc) 2017, 53, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.D.; Severyn, S.; Linsenmayer, M.; Kelly, K.; Bartlett, A.; Tellez, M.; Heintzman, S.; Sterling, G.; Kolb, S.J.; Elsheikh, B. NMJ Transmission Failure in Adults with Spinal Muscular Atrophy. In Proceedings of the Cure SMA Conference, Orlando, FL, USA, 11–14 June 2020. [Google Scholar]
- Je, H.S.; Yang, F.; Ji, Y.; Potluri, S.; Fu, X.Q.; Luo, Z.G.; Nagappan, G.; Chan, J.P.; Hempstead, B.; Son, Y.J.; et al. ProBDNF and mature BDNF as punishment and reward signals for synapse elimination at mouse neuromuscular junctions. J. Neurosci. 2013, 33, 9957–9962. [Google Scholar] [CrossRef] [Green Version]
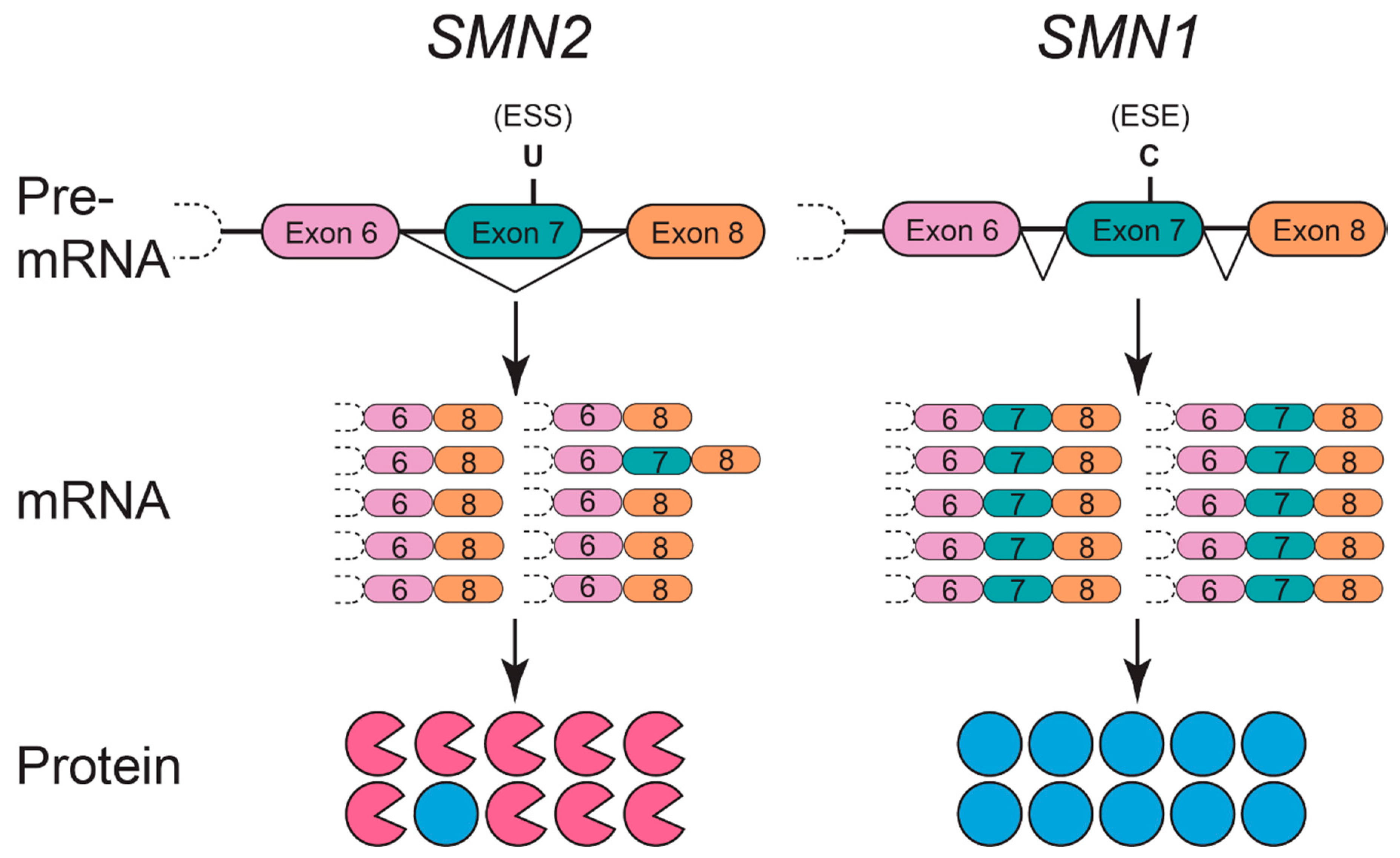
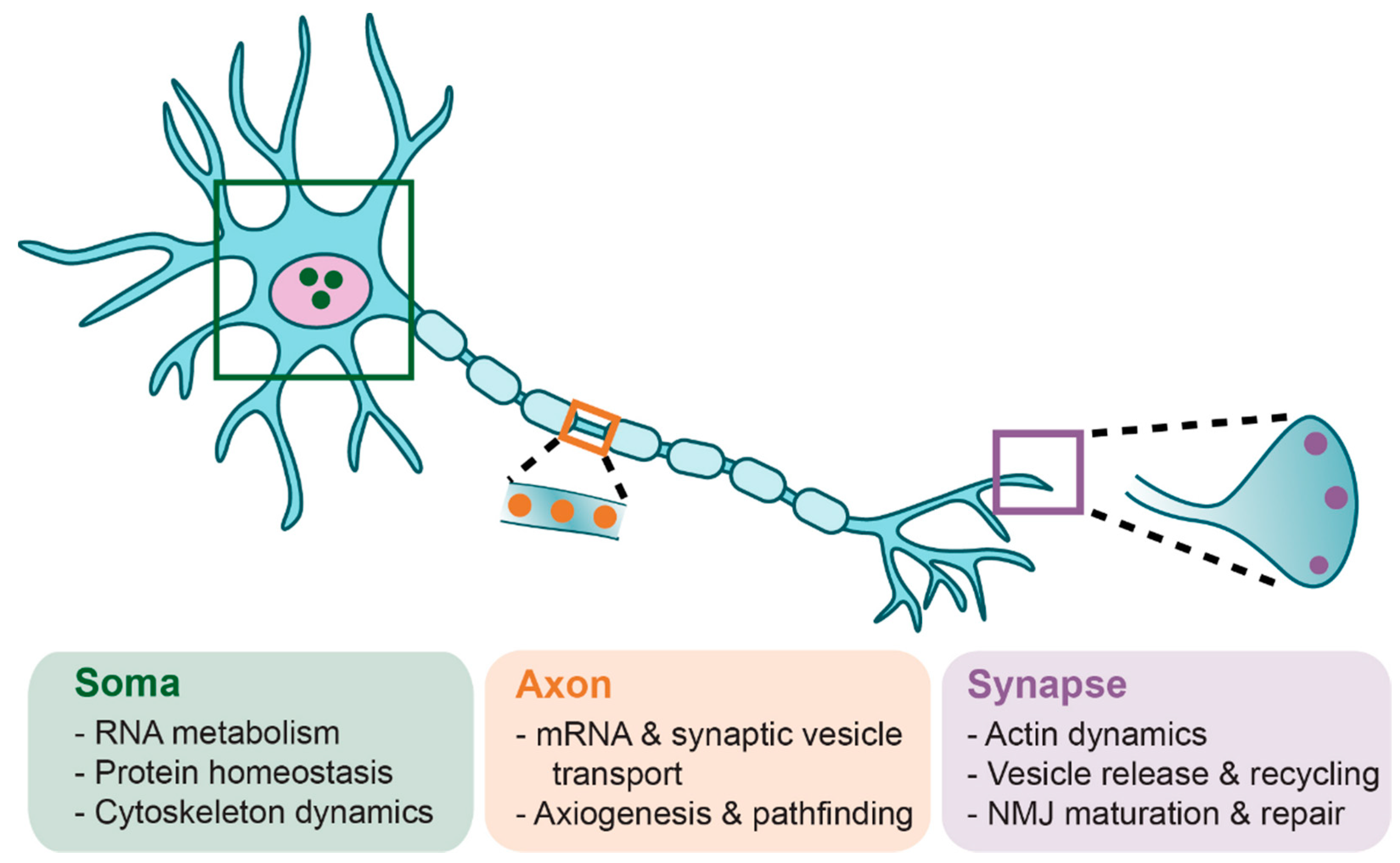
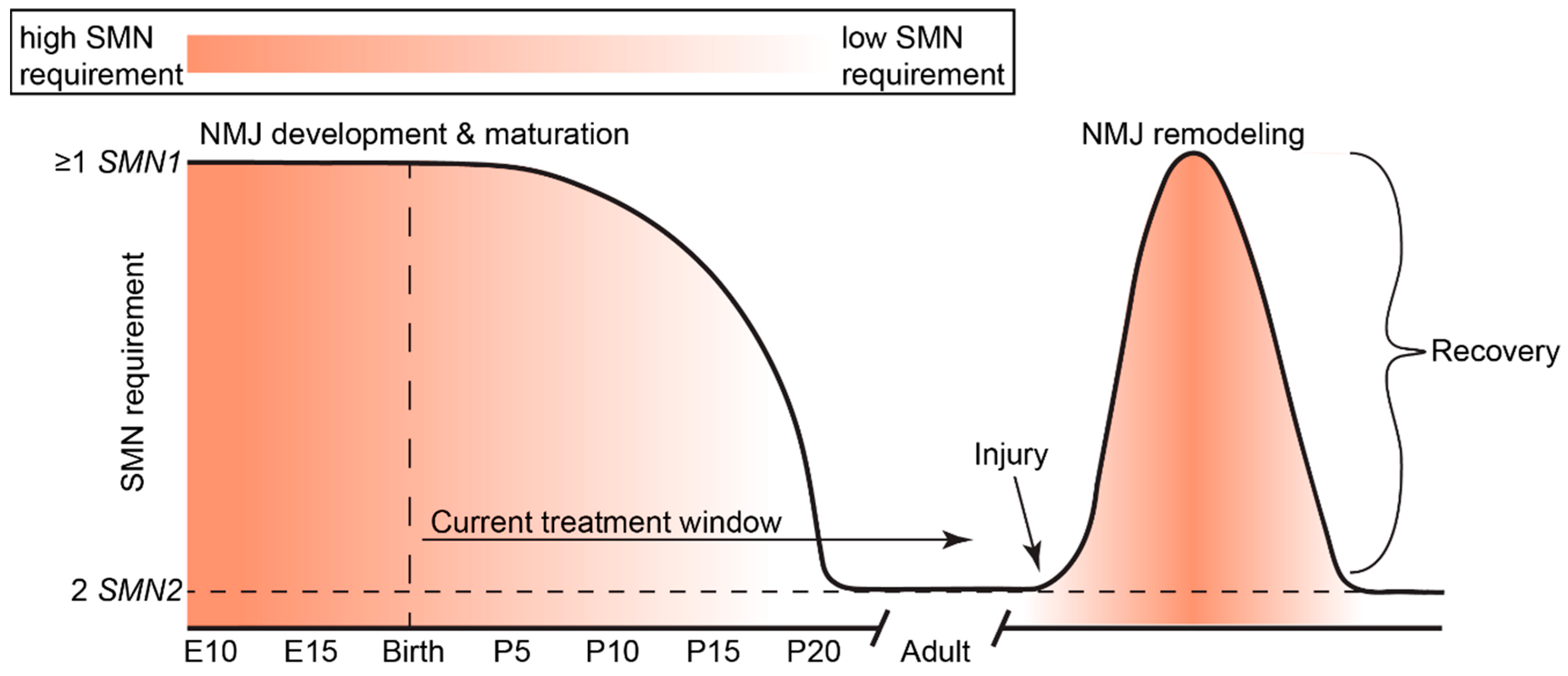
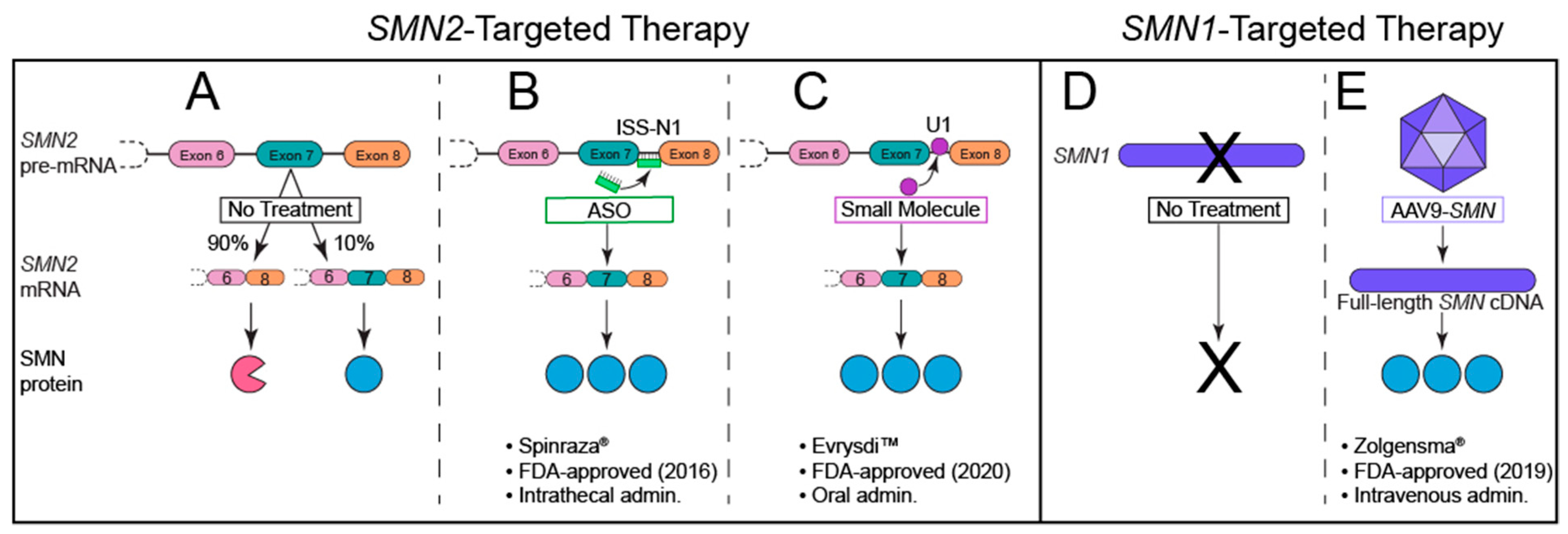

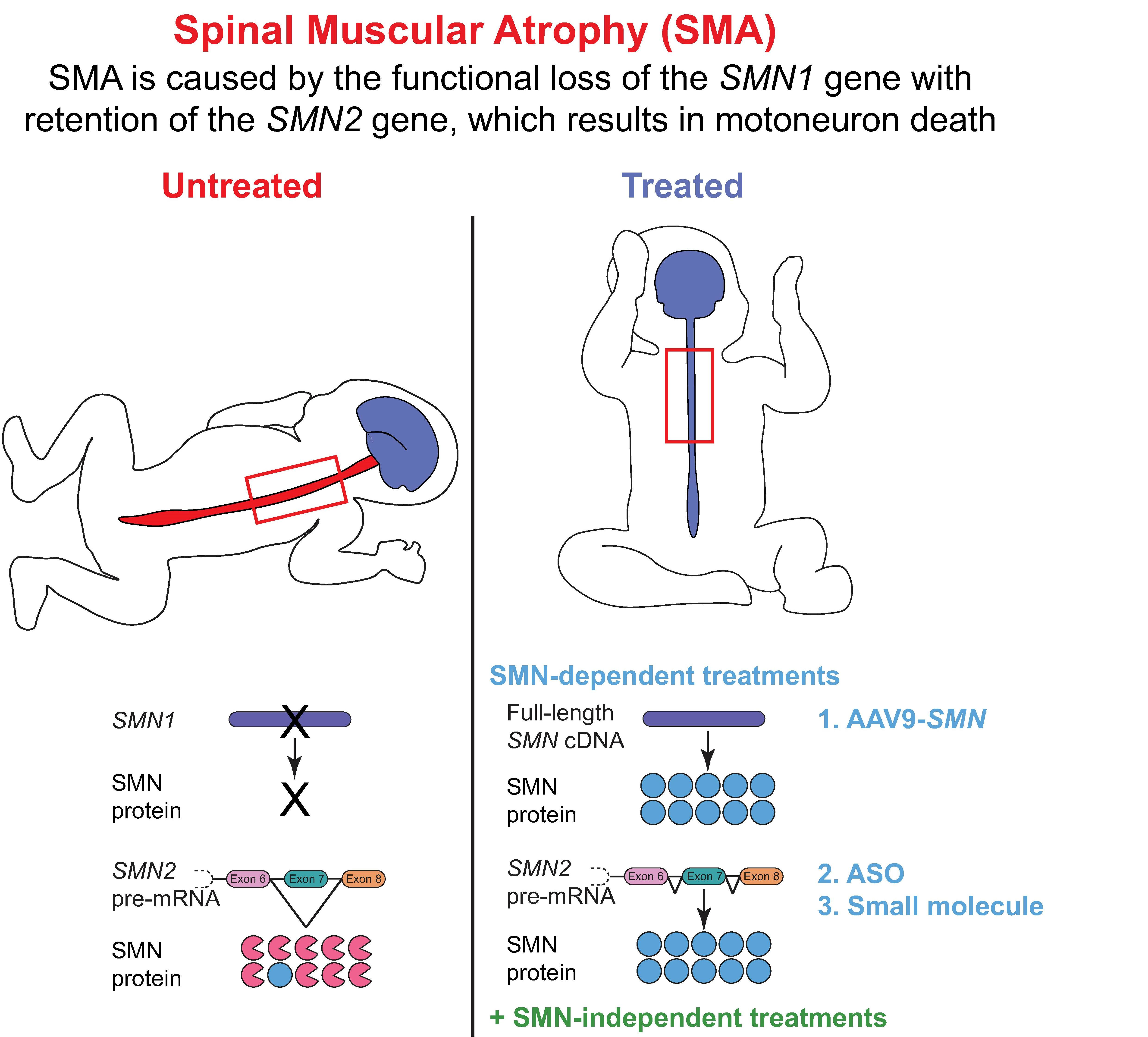{kind=link}
{kind=link}
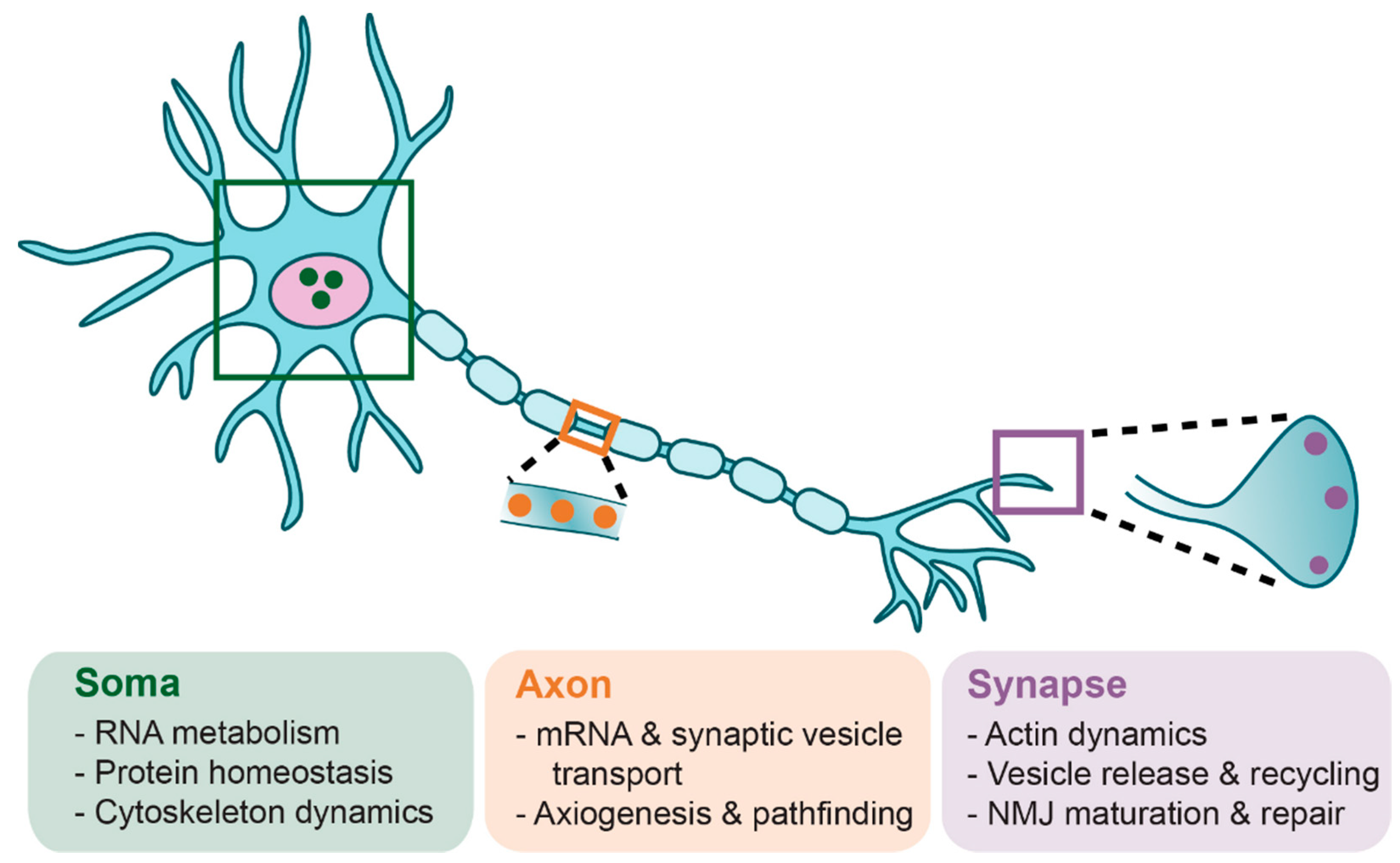{kind=link}
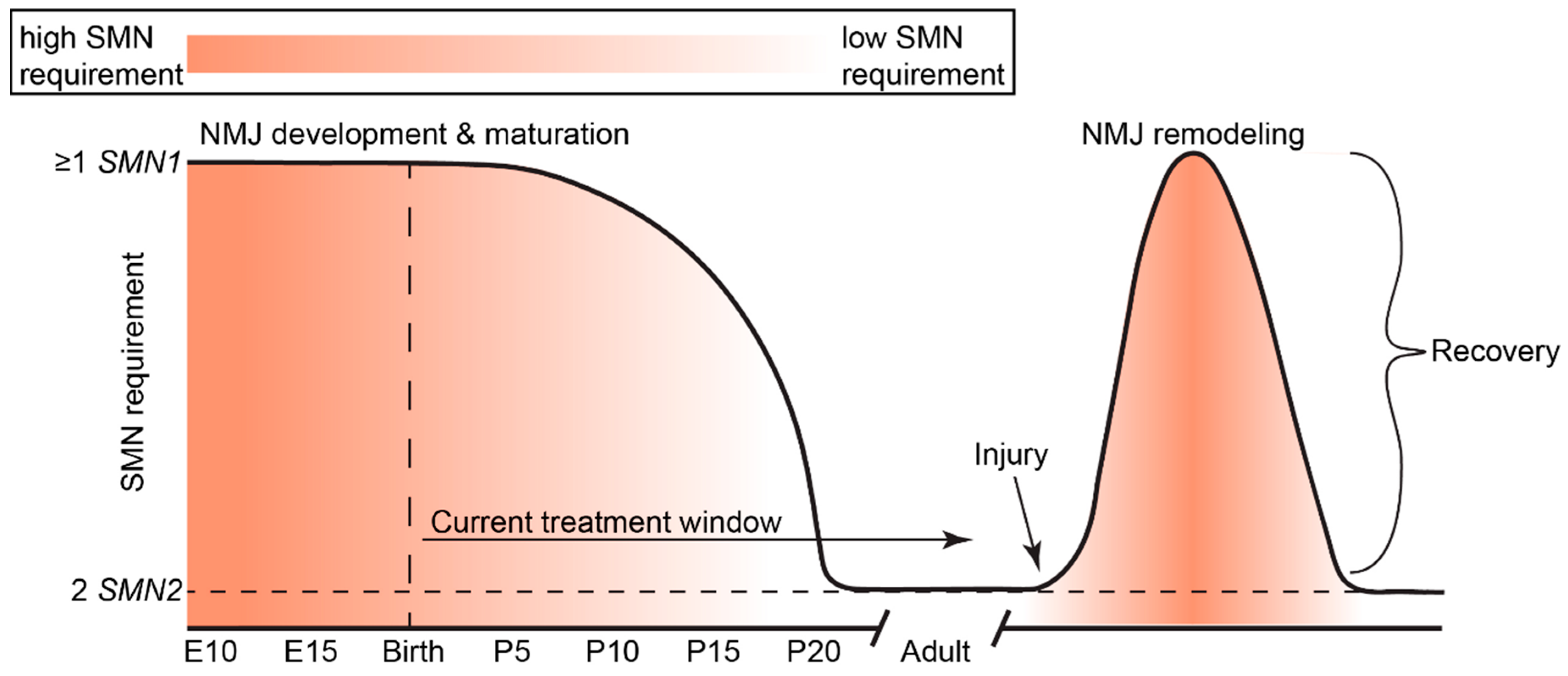{kind=link}
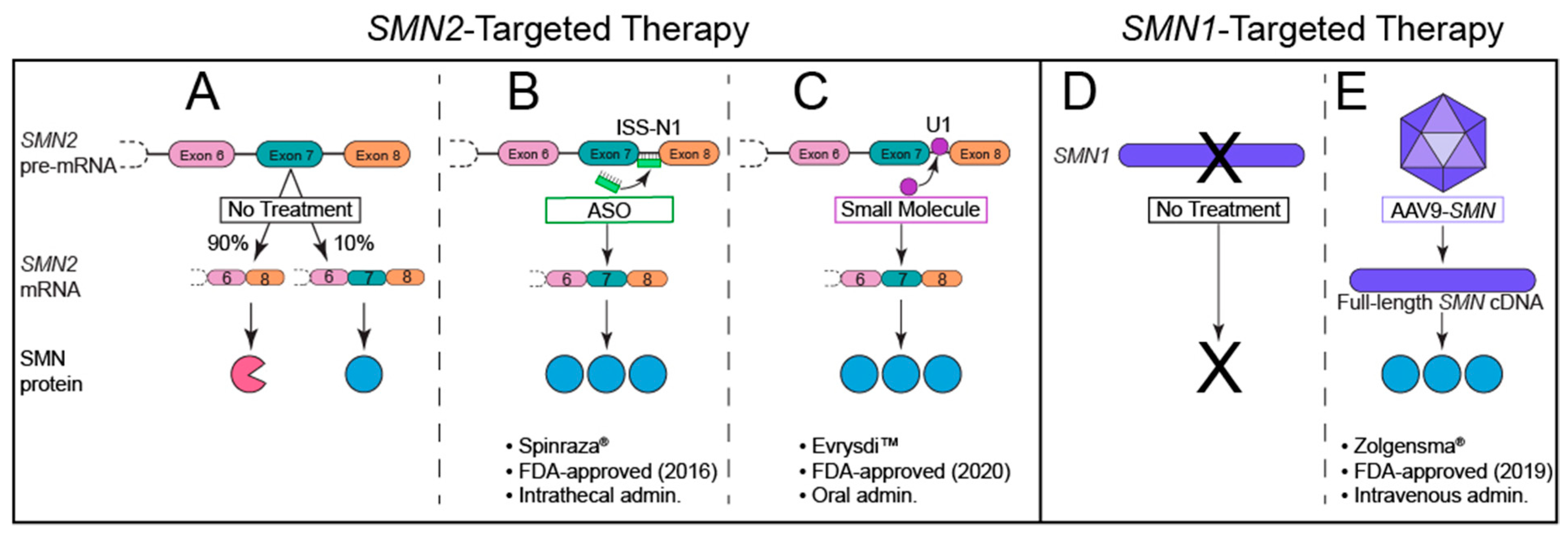{kind=link}
| SMA Type | Age of Symptom Onset | Defining Motor Function | Common Characteristics | Life Expectancy | SMN2 Copy Number |
|---|---|---|---|---|---|
| Type 0 | Prenatal | Respiratory support |
| ˂6 months | 1 |
| Type I | 0–6 months | Never sits unsupported |
| ˂2 years | 2 |
| Type II | 6–18 months | Sits; never stands independently |
| >2 years | 2–3 |
| Type III | ˃18 months | Walks |
| Adult | 3–4 |
| Type IV | ˃21 years | All motor function |
| Adult | ≥4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojala, K.S.; Reedich, E.J.; DiDonato, C.J.; Meriney, S.D. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sci. 2021, 11, 194. https://doi.org/10.3390/brainsci11020194
Ojala KS, Reedich EJ, DiDonato CJ, Meriney SD. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sciences. 2021; 11(2):194. https://doi.org/10.3390/brainsci11020194
Chicago/Turabian StyleOjala, Kristine S., Emily J. Reedich, Christine J. DiDonato, and Stephen D. Meriney. 2021. "In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy" Brain Sciences 11, no. 2: 194. https://doi.org/10.3390/brainsci11020194
APA StyleOjala, K. S., Reedich, E. J., DiDonato, C. J., & Meriney, S. D. (2021). In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sciences, 11(2), 194. https://doi.org/10.3390/brainsci11020194