
Field-Template, QSAR, Ensemble Molecular Docking, and 3D-RISM Solvation Studies Expose Potential of FDA-Approved Marine Drugs as SARS-CoVID-2 Main Protease Inhibitors

 , , and
, , and
Abstract
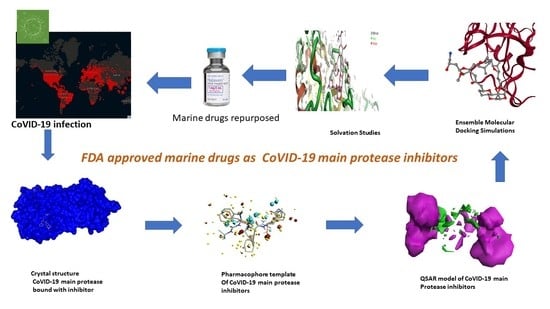
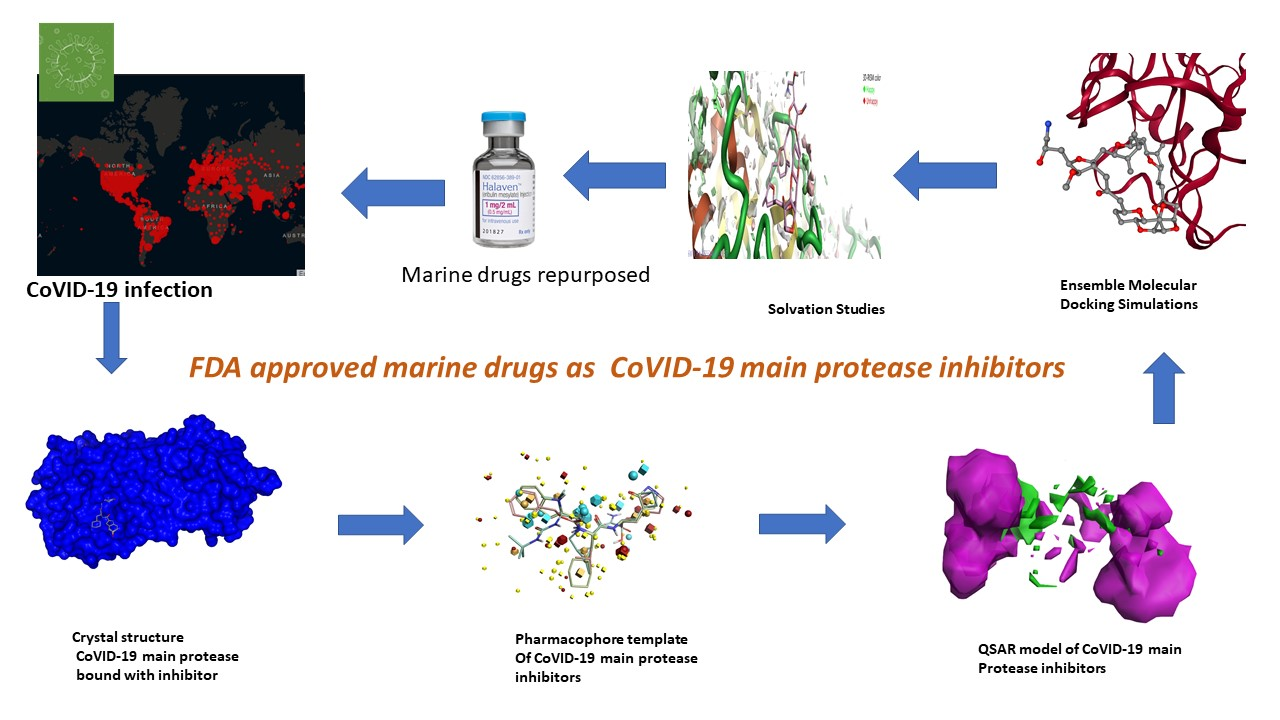
1. Introduction
2. Results and Discussion
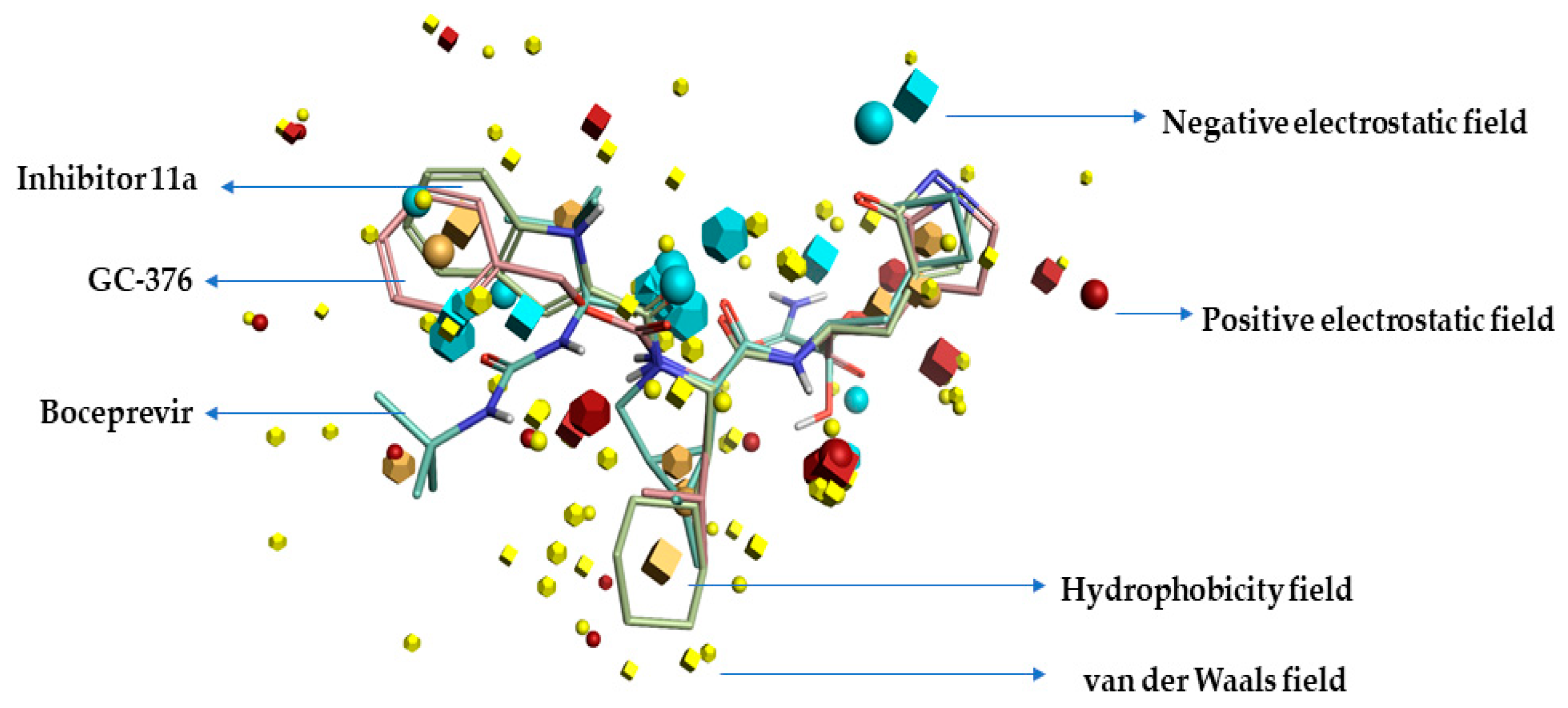
2.1. Pharmacophore Model (Field Template) of SARS-CoVID-19 Main-Protease Inhibitors
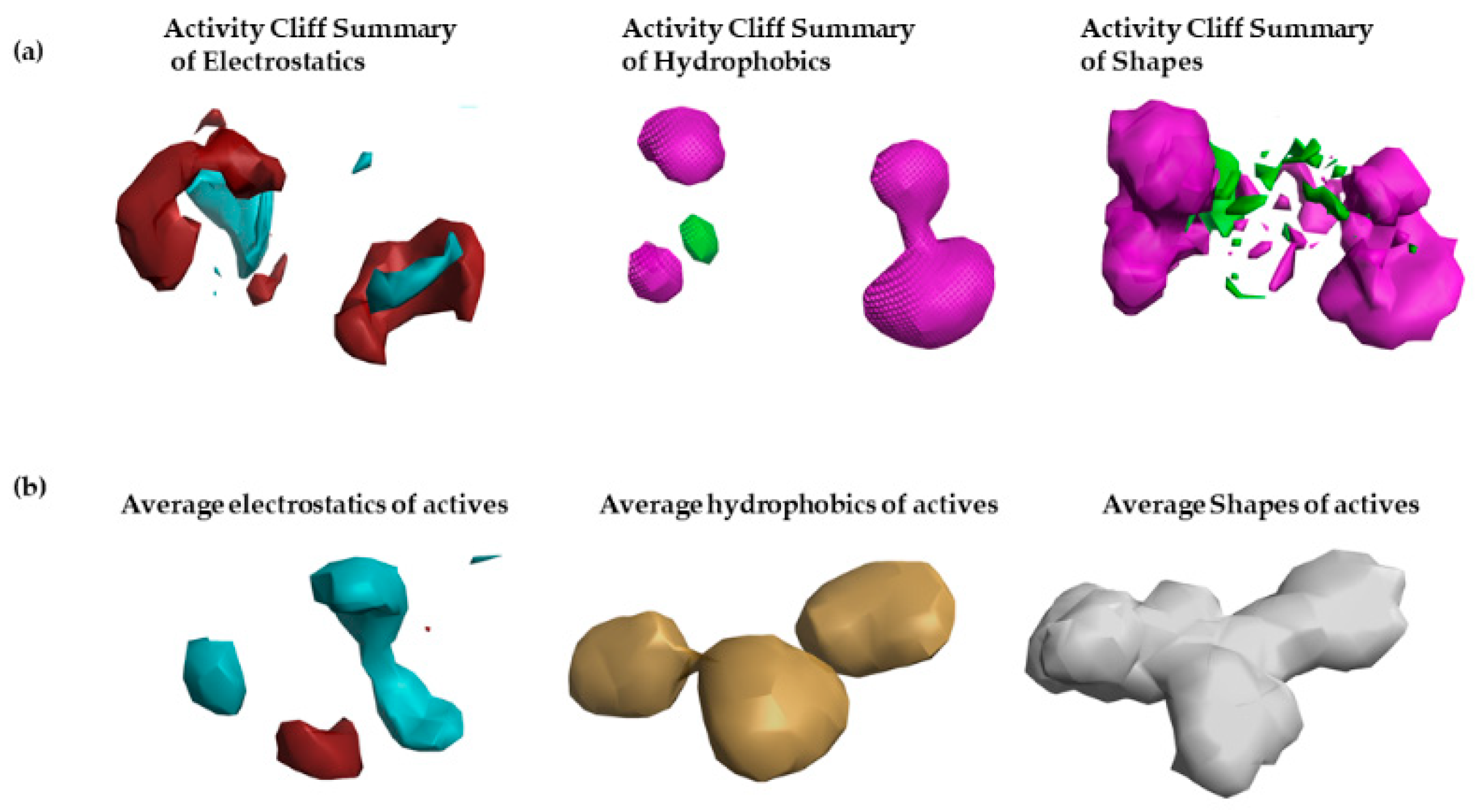
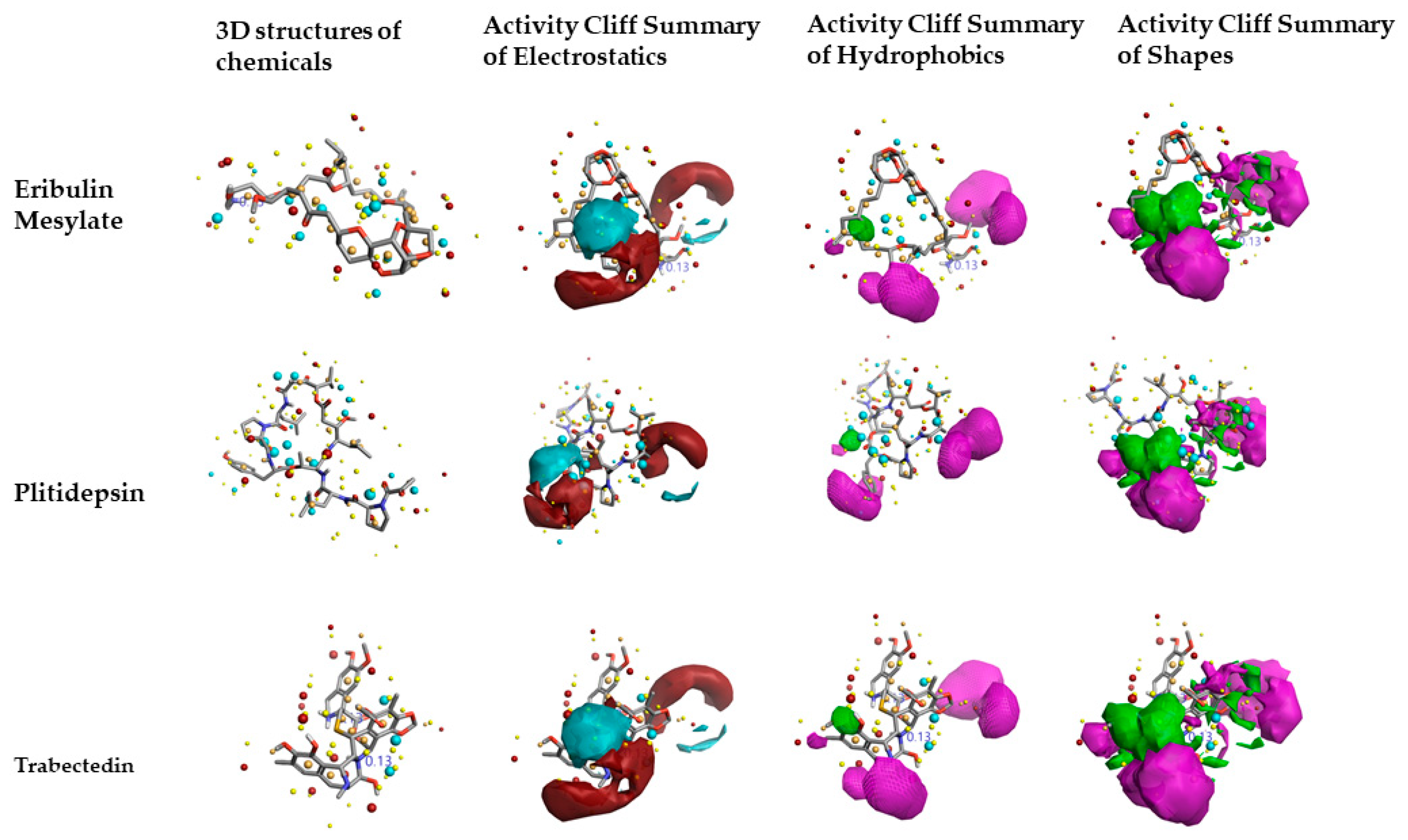
2.2. Structure–Activity Relationship Model of SAR-CoVID-19 Main-Protease Inhibitors
2.3. Molecular Features of FDA-Approved Marine Drugs as CoVID-19 Main-Protease Inhibitors
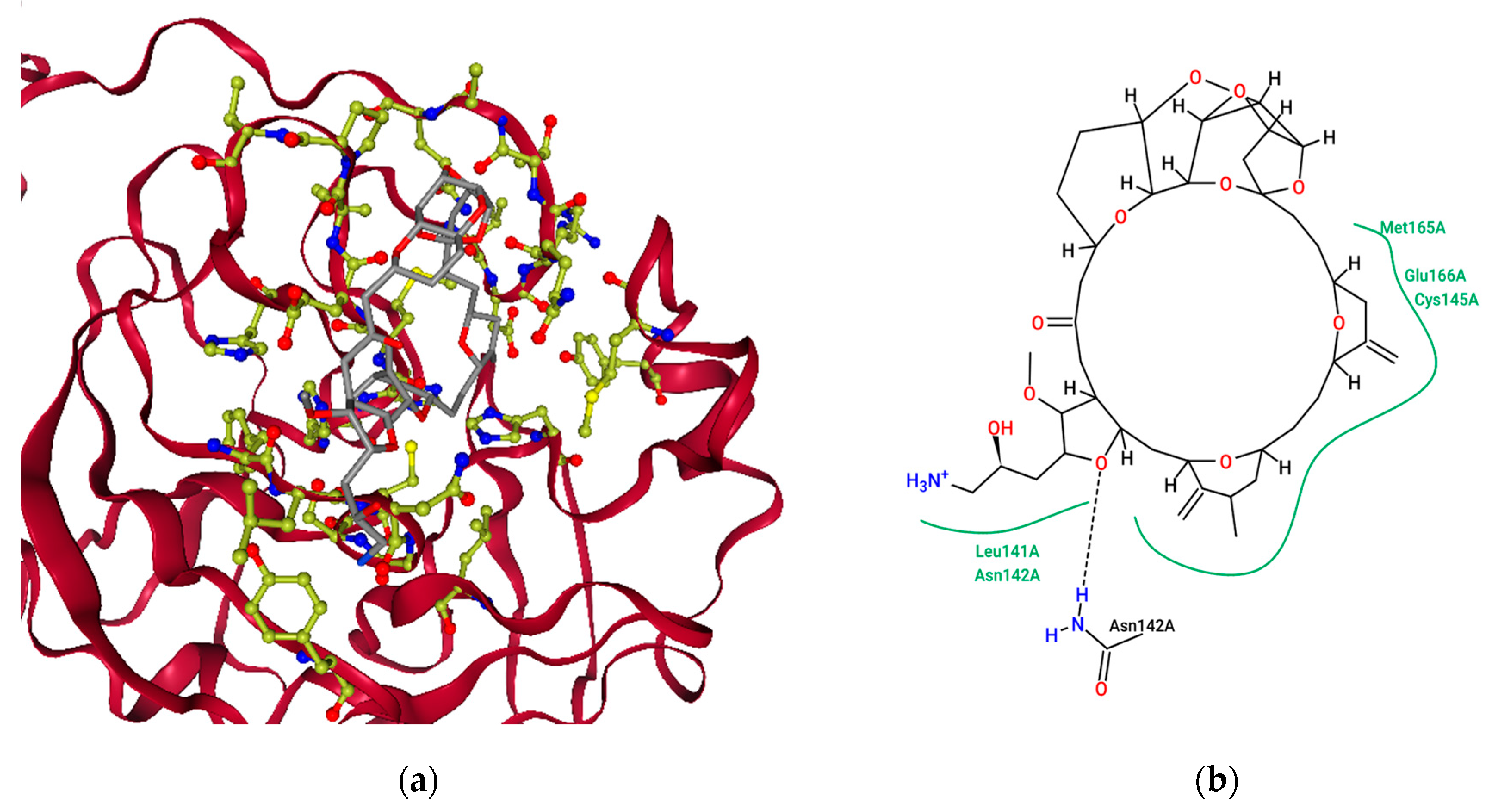
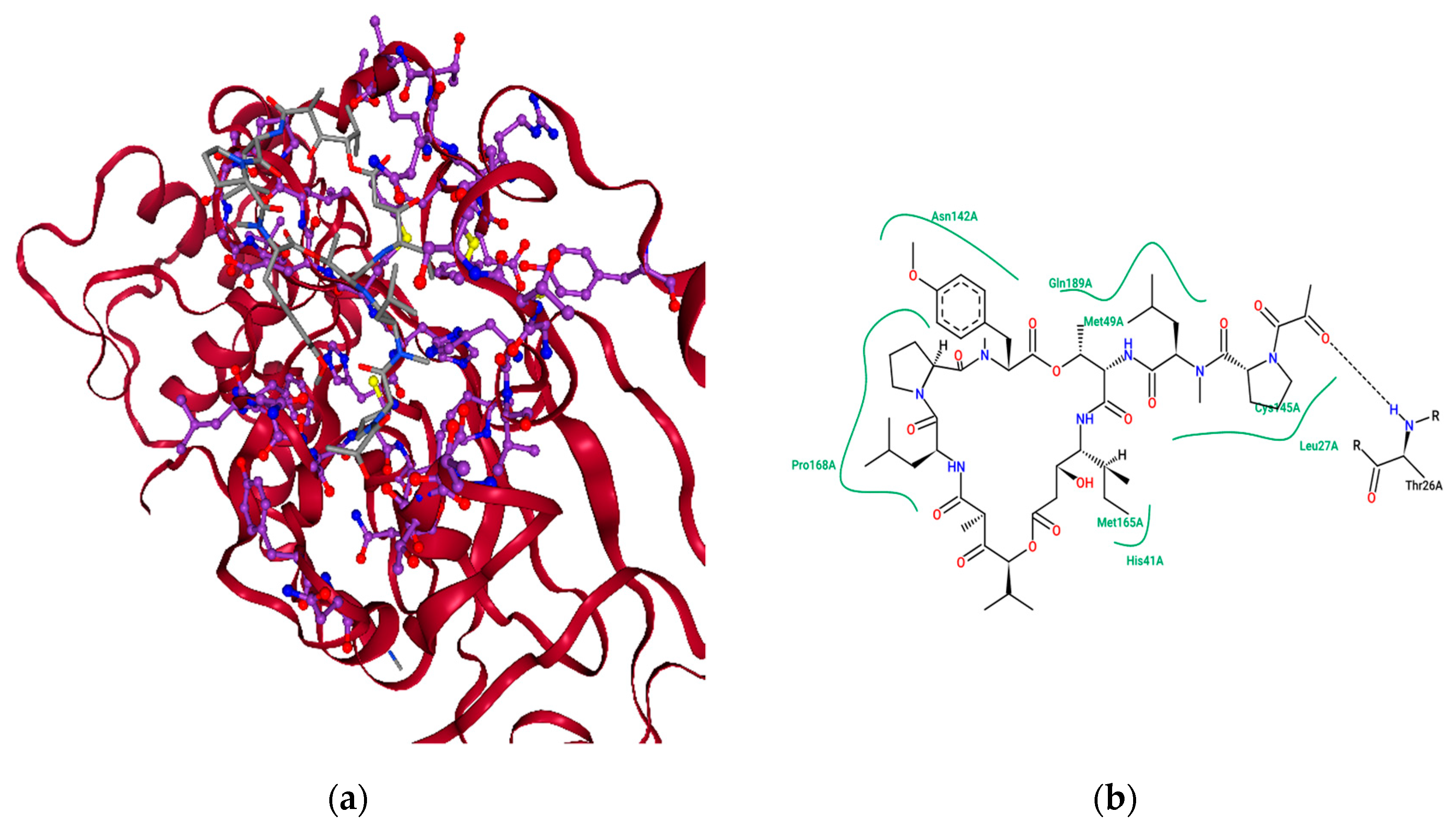
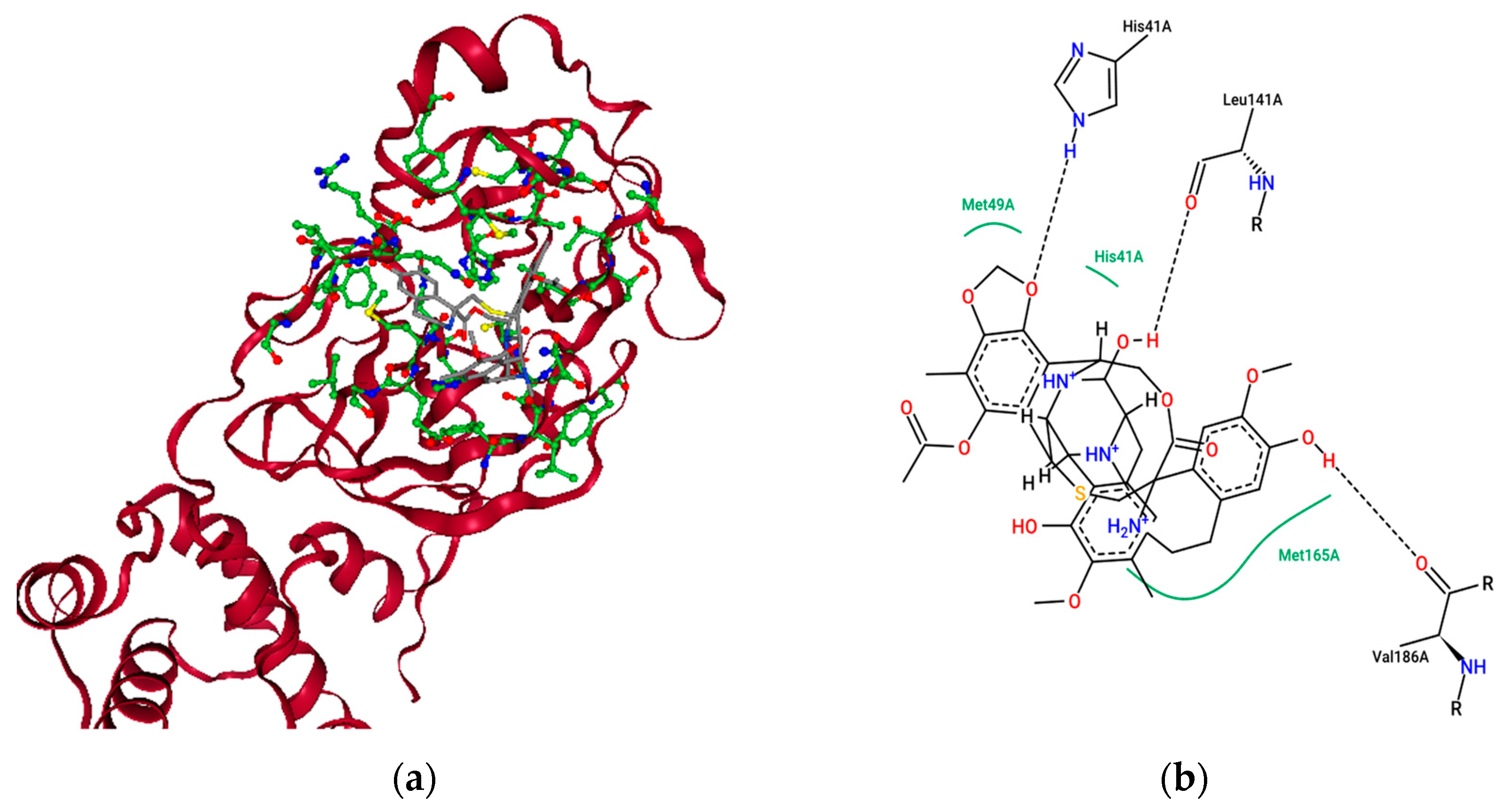
2.4. Ensemble Molecular-Docking Simulations
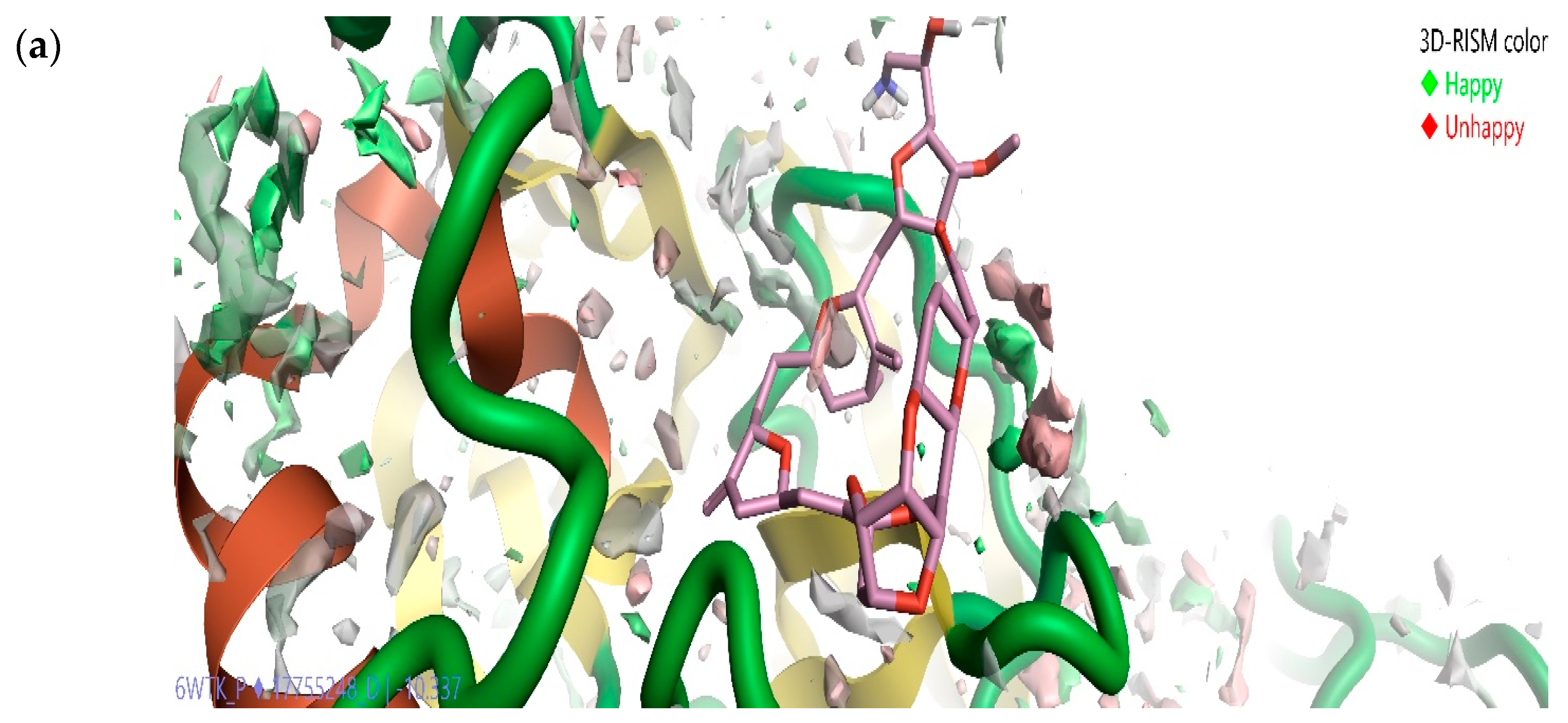
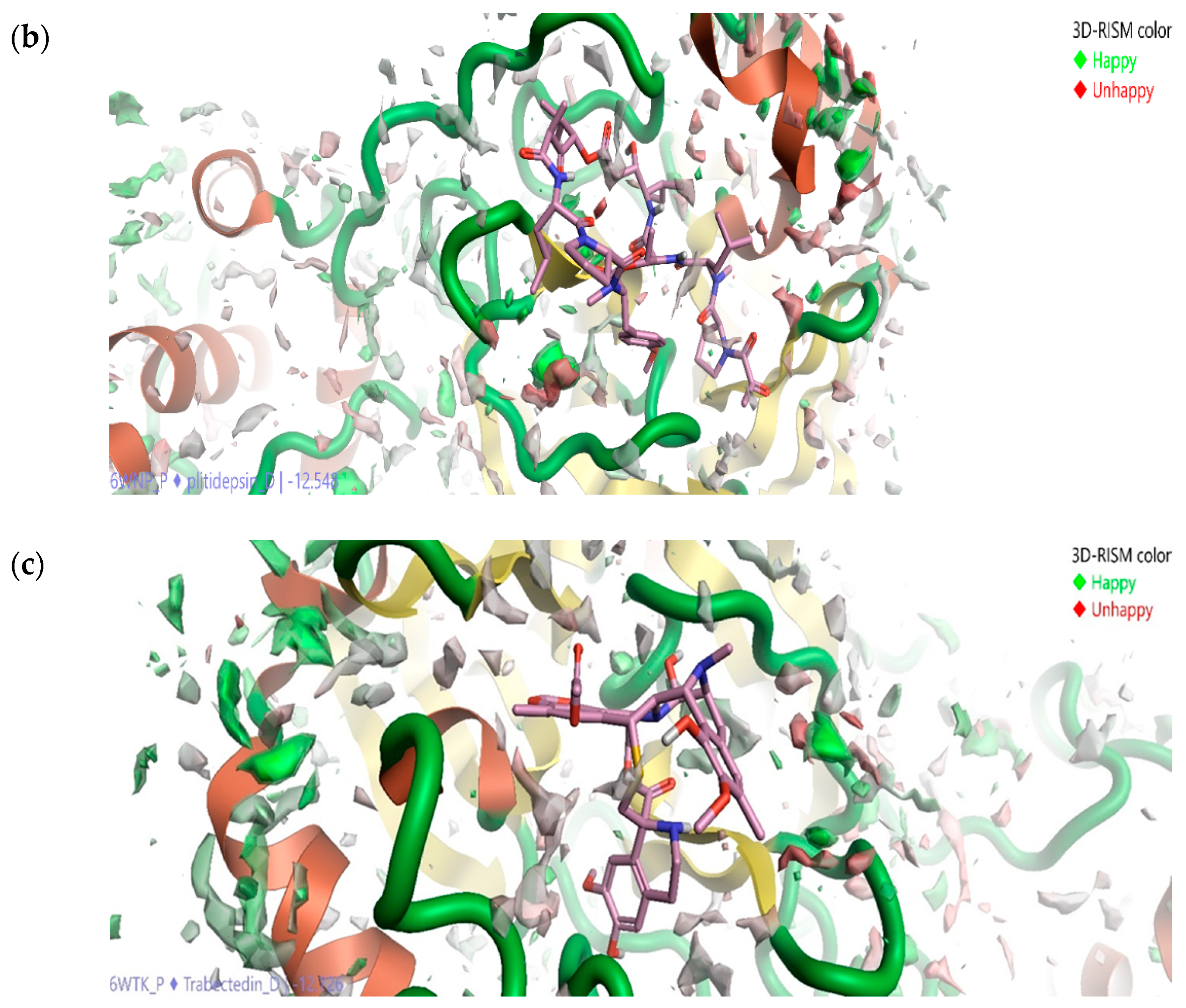
2.5. Solvation Studies on FDA-Approved Marine Drugs Bound with CoVID-19 Main Protease
3. Materials and Methods
3.1. Activity Atlas Model of SARS-CoV-2 Main-Protease Inhibitors
3.2. Ensemble Molecular-Docking Simulations
3.3. Solvation Studies Using 3D R.I.S.M.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef]
- Das, S.; Sarmah, S.; Lyndem, S.; Singha Roy, A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Computer-Aided Drug Discovery and Development. Adv. Struct. Saf. Stud. 2011, 716, 23–38. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-Derived Pharmaceuticals—Challenges and Opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field technology applied to virtual screening and finding the bioactive conformation. Expert Opin. Drug Discov. 2007, 2, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extreme as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Chittepu, V.C.S.R.; Kalhotra, P.; Osorio-Gallardo, T.; Gallardo-Velázquez, T.; Osorio-Revilla, G. Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study. Pharmaceutics 2019, 11, 238. [Google Scholar] [CrossRef]
- Kalhotra, P.; Chittepu, V.C.S.R.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Structure–Activity Relationship and Molecular Docking of Natural Product Library Reveal Chrysin as a Novel Dipeptidyl Peptidase-4 (DPP-4) Inhibitor: An Integrated In Silico and In Vitro Study. Molecular 2018, 23, 1368. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins Struct. Funct. Bioinform. 2006, 66, 399–421. [Google Scholar] [CrossRef]
- Bonvin, A.M. Flexible protein-protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef]
- Campbell, A.J.; Lamb, M.L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. J. Chem. Inf. Model. 2014, 54, 2127–2138. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Sindhikara, D.J.; Hirata, F. Analysis of Biomolecular Solvation Sites by 3D-RISM Theory. J. Phys. Chem. B 2013, 117, 6718–6723. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Oda, K.; Kovalenko, A.; Hirata, F.; Kidera, A. Ligand Mapping on Protein Surfaces by the 3D-RISM Theory: Toward Computational Fragment-Based Drug Design. J. Am. Chem. Soc. 2009, 131, 12430–12440. [Google Scholar] [CrossRef]
- Imai, T.; Kovalenko, A.; Hirata, F. Solvation thermodynamics of protein studied by the 3D-RISM theory. Chem. Phys. Lett. 2004, 395, 1–6. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.R.; Rubin, E.H.; Walton, D.C.; Shuster, D.E.; Wong, Y.N.; Fang, F.; Ashworth, S.; Rosen, L.S. Phase I Study of Eribulin Mesylate Administered Once Every 21 Days in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2009, 15, 4213–4219. [Google Scholar] [CrossRef]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.-V.; García-Sancho, A.M. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, eabf4058. [Google Scholar] [CrossRef]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. ChemInform Abstract: Lead Finder: An Approach to Improve Accuracy of Protein-Ligand Docking, Binding Energy Estimation, and Virtual Screening. J. Chem. Inf. Model. 2009, 40. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Fährrolfes, R.; Bietz, S.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Otto, T.; Volkamer, A.; Rarey, M. ProteinsPlus: A web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017, 45, 337–343. [Google Scholar] [CrossRef]
- Kovalenko, A. Molecular theory of solvation: Methodology summary and illustrations. arXiv 2015, arXiv:1510.06520. [Google Scholar] [CrossRef]
- Skylaris, C.-K.; Haynes, P.D.; Mostofi, A.A.; Payne, M.C. IntroducingONETEP: Linear-scaling density functional simulations on parallel computers. J. Chem. Phys. 2005, 122, 084119. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Name | IC 50 (μM) |
|---|---|
| Tideglusib | 5.81 |
| PX-12 | 4.67 |
| Simeprevir | 4.86 |
| Boceprevir | 5.38 |
| Narlaprevir | 3.33 |
| MG-132 | 5.41 |
| Calpain Inhibitor 11 (ALLM) | 6.01 |
| Calapain Inhibitor X11 | 5.41 |
| Calpetin | 4.97 |
| Calapin Inhibitor 1 | 5.07 |
| GC376 | 7.52 |
| Ebselen | 6.17 |
| Disulfiram | 5.03 |
| Carmofur | 5.74 |
| Shikonin | 4.8 |
| MG-115 | 5.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalhotra, P.; Chittepu, V.C.S.R.; Osorio-Revilla, G.; Gallardo-Velazquez, T. Field-Template, QSAR, Ensemble Molecular Docking, and 3D-RISM Solvation Studies Expose Potential of FDA-Approved Marine Drugs as SARS-CoVID-2 Main Protease Inhibitors. Molecules 2021, 26, 936. https://doi.org/10.3390/molecules26040936
Kalhotra P, Chittepu VCSR, Osorio-Revilla G, Gallardo-Velazquez T. Field-Template, QSAR, Ensemble Molecular Docking, and 3D-RISM Solvation Studies Expose Potential of FDA-Approved Marine Drugs as SARS-CoVID-2 Main Protease Inhibitors. Molecules. 2021; 26(4):936. https://doi.org/10.3390/molecules26040936
Chicago/Turabian StyleKalhotra, Poonam, Veera C. S. R. Chittepu, Guillermo Osorio-Revilla, and Tzayhri Gallardo-Velazquez. 2021. "Field-Template, QSAR, Ensemble Molecular Docking, and 3D-RISM Solvation Studies Expose Potential of FDA-Approved Marine Drugs as SARS-CoVID-2 Main Protease Inhibitors" Molecules 26, no. 4: 936. https://doi.org/10.3390/molecules26040936
APA StyleKalhotra, P., Chittepu, V. C. S. R., Osorio-Revilla, G., & Gallardo-Velazquez, T. (2021). Field-Template, QSAR, Ensemble Molecular Docking, and 3D-RISM Solvation Studies Expose Potential of FDA-Approved Marine Drugs as SARS-CoVID-2 Main Protease Inhibitors. Molecules, 26(4), 936. https://doi.org/10.3390/molecules26040936