
Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia

Abstract
:1. Introduction
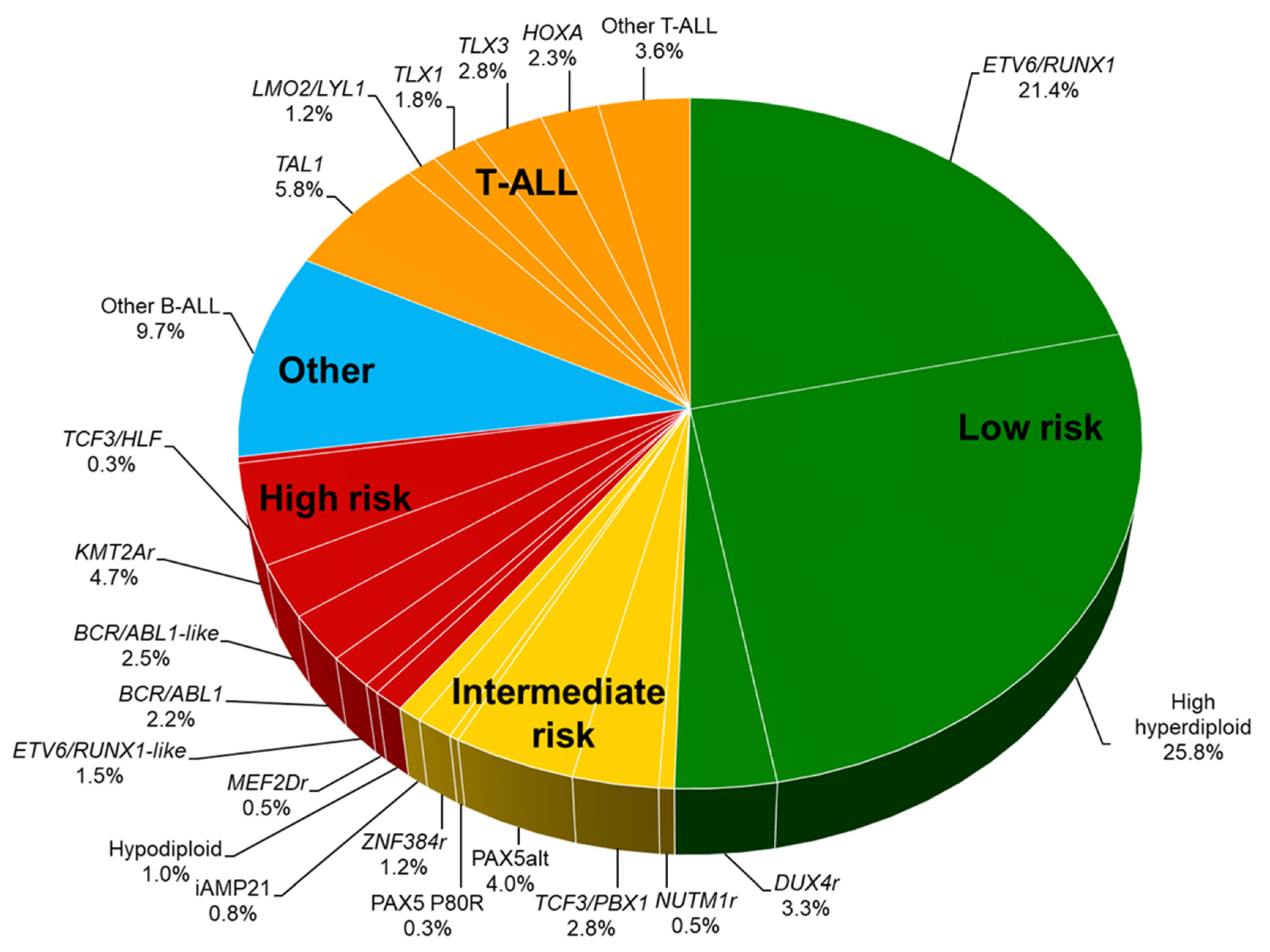
2. Genetic Characterization of Acute Lymphoblastic Leukemia
3. Low-Risk Genetic Subgroups
3.1. ETV6/RUNX1-Rearranged ALL
3.2. Hyperdiploid ALL
3.3. DUX4-Rearranged ALL
4. High-Risk Genetic Subgroups in B-ALL
4.1. Hypodiploid ALL
4.2. BCR/ABL1 (Philadelphia Chromosome)-Positive ALL
4.3. BCR/ABL1 (Philadelphia Chromosome)-Like ALL
4.4. KMT2A-Rearranged ALL
4.5. MEF2D-Rearranged ALL
4.6. TCF3/HLF-Rearranged ALL
5. Intermediate-Risk Genetic Subtypes in B-ALL
5.1. TCF3/PBX1-Rearranged ALL
5.2. Intrachromosomal Amplification of Chromosome 21 (iAMP21)
5.3. PAX5-Driven Subtypes: PAX5alt and PAX5 p.Pro80Arg
5.4. ZNF384-Rearranged ALL
6. Other Newly Identified B-ALL Subtypes
6.1. ETV6/RUNX1-Like ALL
6.2. NUTM1-Rearranged ALL
7. T-Acute Lymphoblastic Leukemia
Early T-Cell Precursor ALL
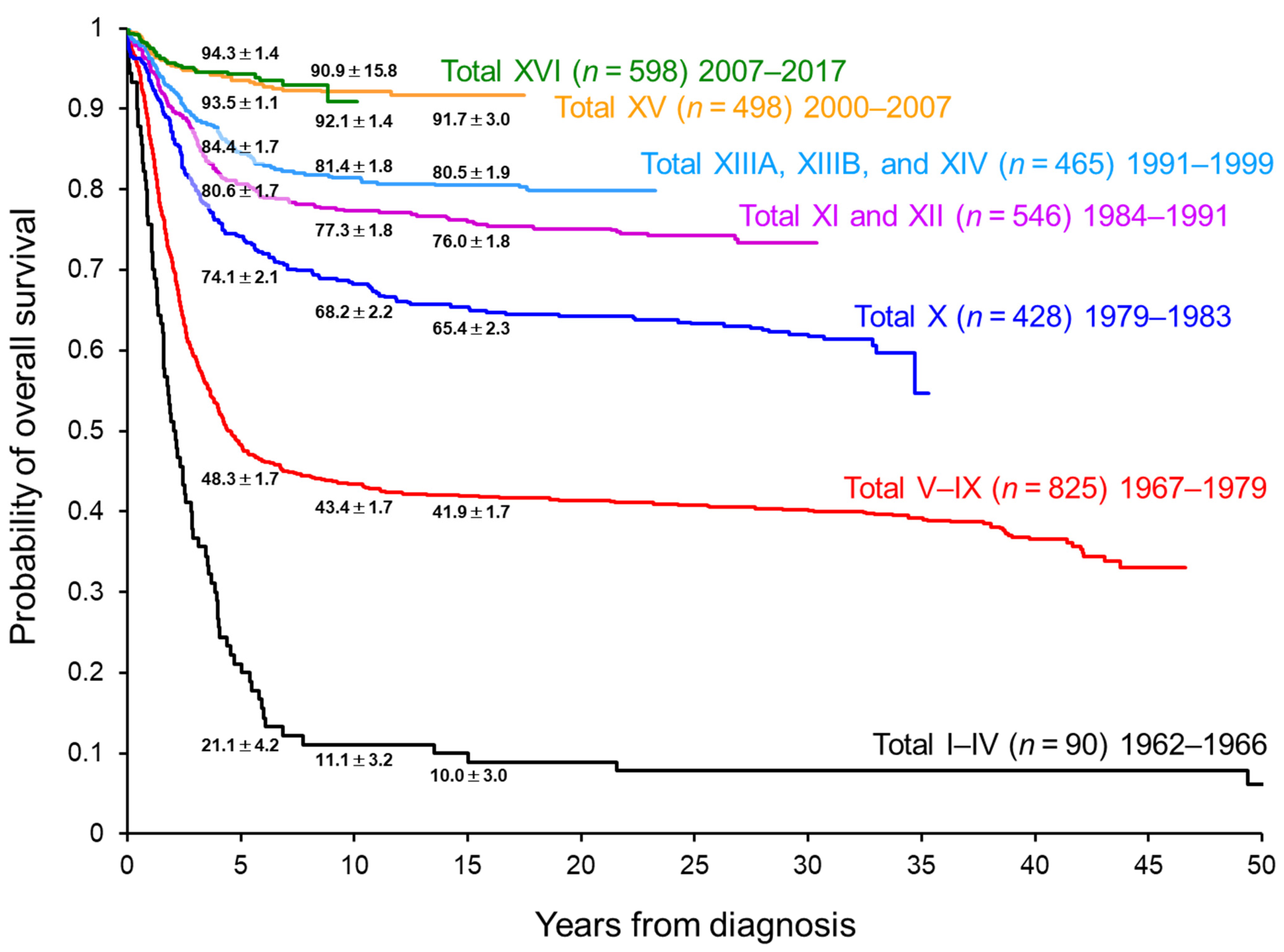
8. Minimal Residual Disease
9. Emerging Therapy: Molecular Targeted Therapy
9.1. Tyrosine Kinase Inhibitors
9.2. BCL-2 and BCL-XL Inhibitors
9.3. Proteasome Inhibitors
9.4. Other Molecular Targeted Therapies
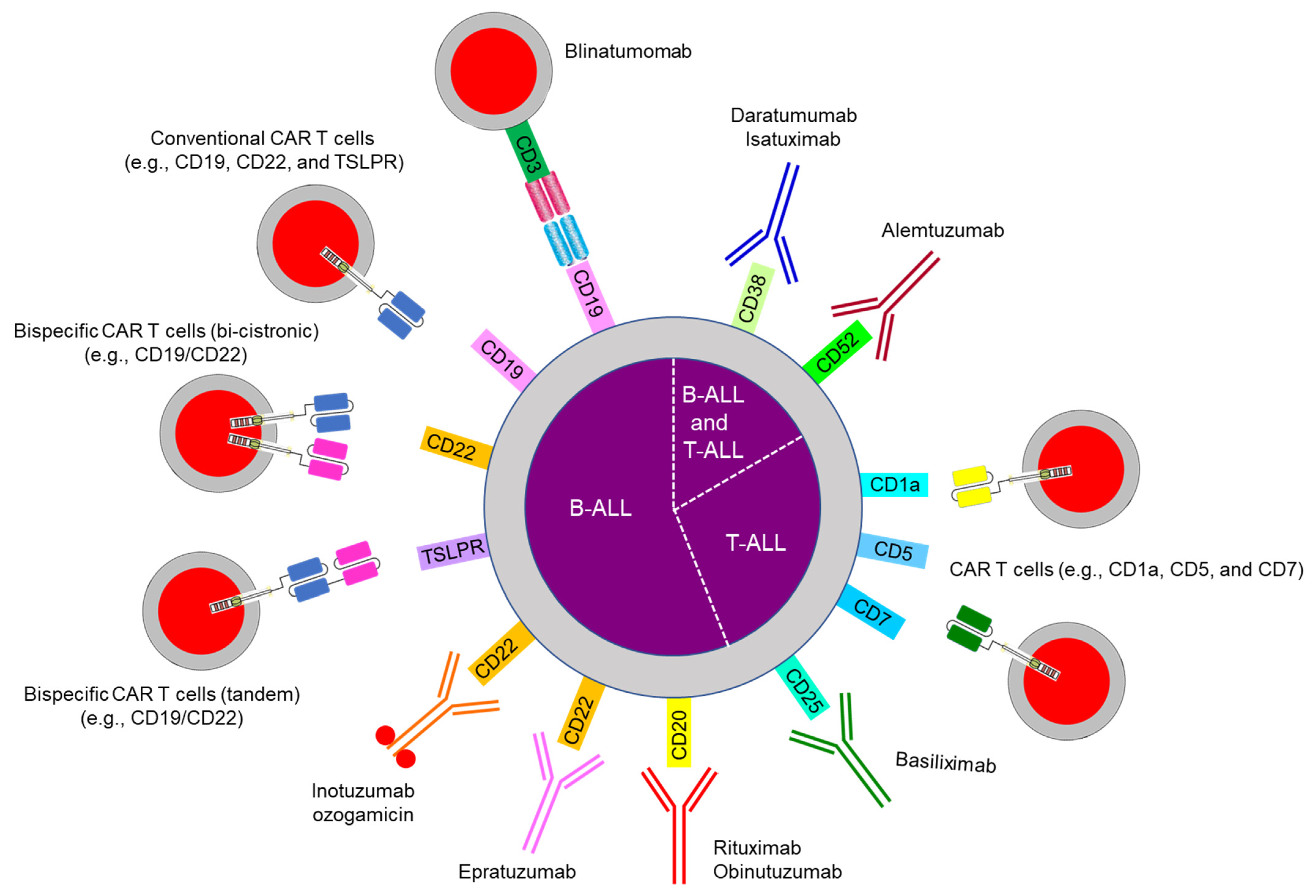
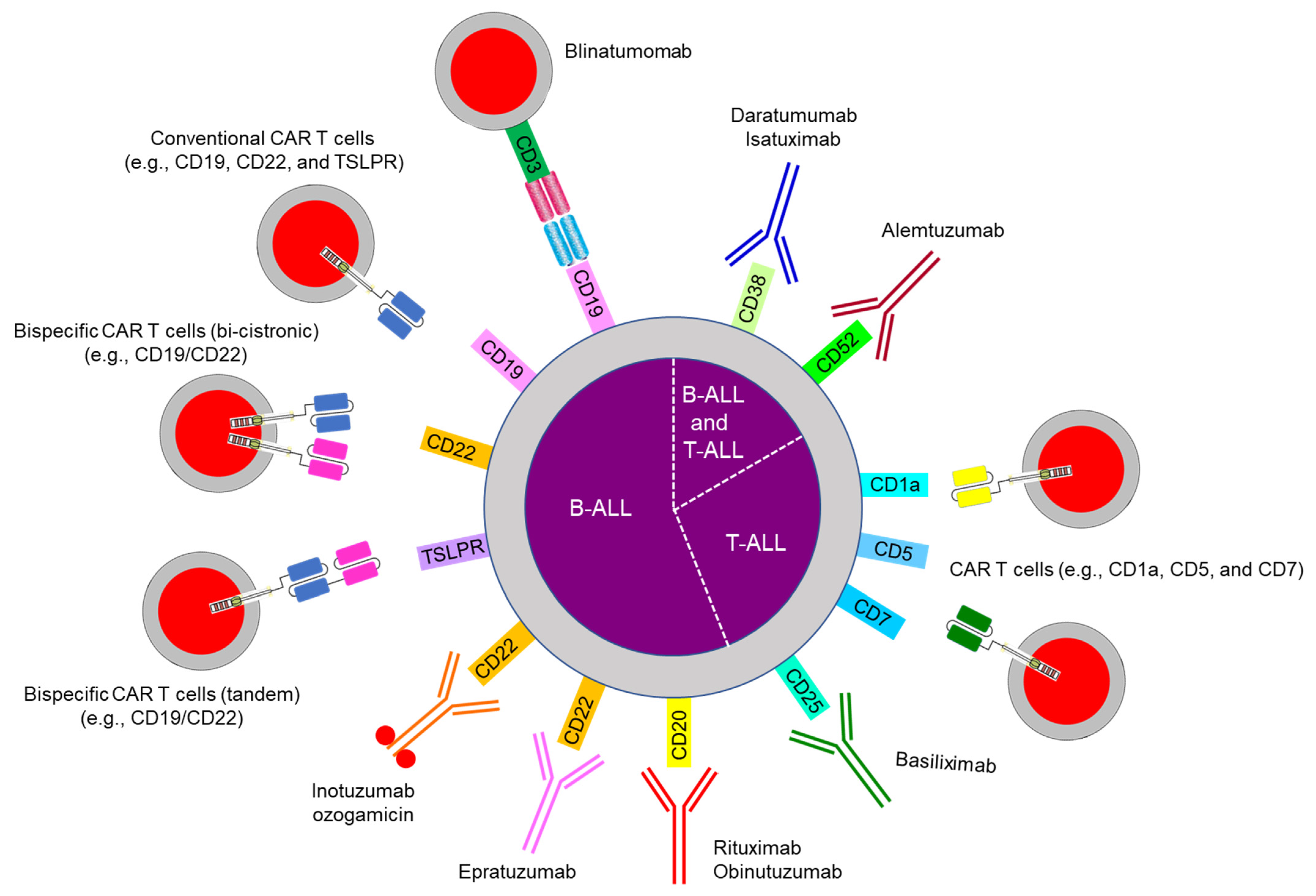
10. Emerging Therapy: Immunotherapy
10.1. Bispecific Antibody Therapy
10.2. Chimeric Antigen Receptor (CAR) T Cells
10.3. Antibody-Drug Conjugates
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- National Cancer Institute. Age-Adjusted and Age-Specific SEER Cancer Incidence Rates, 2014–2018. Available online: https://seer.cancer.gov/csr/1975_2018/results_merged/sect_02_childhood_cancer_iccc.pdf (accessed on 18 April 2021).
- Lim, J.Y.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer 2014, 120, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS control of childhood acute lymphoblastic leukemia without cranial irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Li, J.F.; Dai, Y.T.; Lilljebjörn, H.; Shen, S.H.; Cui, B.W.; Bai, L.; Liu, Y.F.; Qian, M.X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [Green Version]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021, in press. [Google Scholar] [CrossRef]
- Teachey, D.T.; Pui, C.H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pei, D.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Shurtleff, S.; Onciu, M.; Cheng, C.; et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: Improved outcome with contemporary therapy. Leukemia 2012, 26, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, D.; Olsen, M.; Lähnemann, D.; Stanulla, M.; Slany, R.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 2018, 131, 821–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpar, D.; Wren, D.; Ermini, L.; Mansur, M.B.; van Delft, F.W.; Bateman, C.M.; Titley, I.; Kearney, L.; Szczepanski, T.; Gonzalez, D.; et al. Clonal origins of ETV6-RUNX1+ acute lymphoblastic leukemia: Studies in monozygotic twins. Leukemia 2015, 29, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, G.; Hauer, J.; Martín-Lorenzo, A.; Scháfer, D.; Bartenhagen, C.; García-Ramírez, I.; Auer, F.; González-Herrero, I.; Ruiz-Roca, L.; Gombert, M.; et al. Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia via impaired H3K4 demethylases. Cancer Res. 2017, 77, 4365–4377. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Pei, D.; Raimondi, S.C.; Coustan-Smith, E.; Jeha, S.; Cheng, C.; Bowman, W.P.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with Response-Adapted therapy. Leukemia 2017, 31, 333–339. [Google Scholar] [CrossRef]
- O’Connor, D.; Enshaei, A.; Bartram, J.; Hancock, J.; Harrison, C.J.; Hough, R.; Samarasinghe, S.; Schwab, C.; Vora, A.; Wade, R.; et al. Genotype-specific minimal residual disease interpretation improves stratification in pediatric acute lymphoblastic leukemia. J. Clin. Oncol. 2018, 36, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Bleckmann, K.; Zimmermann, M.; Biondi, A.; Möricke, A.; Locatelli, F.; Cario, G.; Rizzari, C.; Attarbaschi, A.; Valsecchi, M.G.; et al. Reduced-intensity delayed intensification in standard-risk pediatric acute lymphoblastic leukemia defined by undetectable minimal residual disease: Results of an international randomized trial (AIEOP-BFM ALL 2000). J. Clin. Oncol. 2018, 36, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Ishimaru, S.; Seki, M.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Kakiuchi, N.; Sato, Y.; Ueno, H.; Tanaka, H.; et al. Long-term outcome of 6-month maintenance chemotherapy for acute lymphoblastic leukemia in children. Leukemia 2017, 31, 580–584. [Google Scholar] [CrossRef]
- Look, A.T.; Roberson, P.K.; Williams, D.L.; Rivera, G.; Bowman, W.P.; Pui, C.H.; Ochs, J.; Abromowitch, M.; Kalwinsky, D.; Dahl, G.V.; et al. Prognostic importance of blast cell DNA content in childhood acute lymphoblastic leukemia. Blood 1985, 65, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.L.; Tsiatis, A.; Brodeur, G.M.; Look, A.T.; Melvin, S.L.; Bowman, W.P.; Kalwinsky, D.K.; Rivera, G.; Dahl, G.V. Prognostic importance of chromosome number in 136 untreated children with acute lymphoblastic leukemia. Blood 1982, 60, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.B.; Shuster, J.J.; Carroll, A.; Look, A.T.; Borowitz, M.J.; Crist, W.M.; Nitschke, R.; Pullen, J.; Steuber, C.P.; Land, V.J. Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: A Pediatric Oncology Group study. Blood 1992, 79, 3316–3324. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, V.M.; Vuchich, M.J.; Lauer, S.J.; Mahoney, D.; Carroll, A.J.; Shuster, J.J.; Esseltine, D.W.; Payment, C.; Look, A.T.; Akabutu, J.; et al. Accumulation of high levels of methotrexate polyglutamates in lymphoblasts from children with hyperdiploid (greater than 50 chromosomes) B-lineage acute lymphoblastic leukemia: A Pediatric Oncology Group study. Blood 1992, 80, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kager, L.; Cheok, M.; Yang, W.; Zaza, G.; Cheng, Q.; Panetta, J.C.; Pui, C.H.; Downing, J.R.; Relling, M.V.; Evans, W.E. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J. Clin. Investig. 2005, 115, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lopez, E.; Autry, R.J.; Smith, C.; Yang, W.; Paugh, S.W.; Panetta, J.C.; Crews, K.R.; Bonten, E.J.; Smart, B.; Pei, D.; et al. Pharmacogenomics of intracellular methotrexate polyglutamates in patients’ leukemia cells in vivo. J. Clin. Investig. 2020, 130, 6600–6615. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Hunger, S.P.; Pui, C.H.; Saha, V.; Gaynon, P.S.; Baruchel, A.; Conter, V.; Otten, J.; Ohara, A.; Versluys, A.B.; et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2012, 366, 1371–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016, 48, 569–574. [Google Scholar] [CrossRef]
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 2016, 48, 1481–1489. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Schinnerl, D.; Mejstrikova, E.; Schumich, A.; Zaliova, M.; Fortschegger, K.; Nebral, K.; Attarbaschi, A.; Fiser, K.; Kauer, M.O.; Popitsch, N.; et al. CD371 cell surface expression: A unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019, 104, e352–e355. [Google Scholar] [CrossRef] [Green Version]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cave, H.; Elitzur, S.; Koh, K.; Liu, H.C.; et al. Outcome of children with hypodiploid acute lymphoblastic leukemia: A retrospective multinational study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef] [PubMed]
- McNeer, J.L.; Devidas, M.; Dai, Y.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; Kahwash, S.B.; Borowitz, M.J.; Wood, B.L.; Larsen, E.; et al. Hematopoietic stem-cell transplantation does not improve the poor outcome of children with hypodiploid acute lymphoblastic leukemia: A report from Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 780–789. [Google Scholar] [CrossRef]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T.; et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J. Clin. Oncol. 2018, 36, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safavi, S.; Paulsson, K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: Two distinct subtypes with consistently poor prognosis. Blood 2017, 129, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Flores, E.; Comeaux, E.Q.; Kim, K.L.; Melnik, E.; Beckman, K.; Davis, K.L.; Wu, K.; Akutagawa, J.; Bridges, O.; Marino, R.; et al. Bcl-2 is a therapeutic target for hypodiploid B-lineage acute lymphoblastic leukemia. Cancer Res. 2019, 79, 2339–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talleur, A.C.; Maude, S.L. What is the role for HSCT or immunotherapy in pediatric hypodiploid B-cell acute lymphoblastic leukemia? Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 508–511. [Google Scholar] [CrossRef]
- Slayton, W.B.; Schultz, K.R.; Silverman, L.B.; Hunger, S.P. How we approach Philadelphia chromosome-positive acute lymphoblastic leukemia in children and young adults. Pediatr. Blood Cancer 2020, 67, e28543. [Google Scholar] [CrossRef]
- Aricò, M.; Schrappe, M.; Hunger, S.P.; Carroll, W.L.; Conter, V.; Galimberti, S.; Manabe, A.; Saha, V.; Baruchel, A.; Vettenranta, K.; et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J. Clin. Oncol. 2010, 28, 4755–4761. [Google Scholar] [CrossRef]
- Schultz, K.R.; Carroll, A.; Heerema, N.A.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Zheng, H.W.; Davies, S.M.; et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 2014, 28, 1467–1471. [Google Scholar] [CrossRef] [Green Version]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with Philadelphia chromosome-positive acute lymphoblastic leukemia: Results of Children’s Oncology Group trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of dasatinib vs. imatinib in the treatment of pediatric philadelphia chromosome-positive acute lymphoblastic leukemia: A randomized clinical trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H.; Ravandi, F.; Thomas, D.; Huang, X.; Faderl, S.; Pemmaraju, N.; Daver, N.; Garcia-Manero, G.; Sasaki, K.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: A single-centre, phase 2 study. Lancet Oncol. 2015, 16, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib–blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.T.; Kosaka, Y.; Malla, P.; LaTocha, D.; Lamble, A.; Hayes-Lattin, B.; Byrd, K.; Druker, B.J.; Tyner, J.W.; Chang, B.H.; et al. Concomitant use of a dual Src/ABL kinase inhibitor eliminates the in vitro efficacy of blinatumomab against Ph+ ALL. Blood 2021, 137, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Den Boer, M.L.; van Slegtenhorst, M.; de Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; van Zutven, L.J.; Beverloo, H.B.; van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Pei, D.; Campana, D.; Payne-Turner, D.; Li, Y.; Cheng, C.; Sandlund, J.T.; Jeha, S.; Easton, J.; Becksfort, J.; et al. Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J. Clin. Oncol. 2014, 32, 3012–3020. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Reshmi, S.C.; Harvey, R.C.; Chen, I.M.; Patel, K.; Stonerock, E.; Jenkins, H.; Dai, Y.; Valentine, M.; Gu, Z.; et al. Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: A report from the Children’s Oncology Group. Blood 2018, 132, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Den Boer, M.L.; Cario, G.; Moorman, A.V.; Boer, J.M.; de Groot-Kruseman, H.A.; Fiocco, M.; Escherich, G.; Imamura, T.; Yeoh, A.; Sutton, R.; et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: A multicentre, retrospective, cohort study. Lancet Haematol. 2021, 8, e55–e66. [Google Scholar] [CrossRef]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Tamburini, J.; Maury, S.; et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Pieters, R.; Biondi, A. How I treat infant leukemia. Blood 2019, 133, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the Interfant-06 protocol: Results from an international phase III randomized study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Neff, T.; Armstrong, S.A. Recent progress toward epigenetic therapies: The example of mixed lineage leukemia. Blood 2013, 121, 4847–4853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Okuno, Y.; Kawashima, N.; Muramatsu, H.; Okuno, T.; Wang, X.; Kataoka, S.; Sekiya, Y.; Hamada, M.; Murakami, N.; et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J. Clin. Oncol. 2016, 34, 3451–3459. [Google Scholar] [CrossRef]
- Ohki, K.; Kiyokawa, N.; Saito, Y.; Hirabayashi, S.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yoshimi, A.; Ogata-Kawata, H.; et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2019, 104, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.F.; Wang, B.Y.; Zhang, W.N.; Huang, J.Y.; Li, B.S.; Zhang, M.; Jiang, L.; Li, J.F.; Wang, M.J.; Dai, Y.J.; et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF–positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef]
- Huang, Y.; Mouttet, B.; Warnatz, H.J.; Risch, T.; Rietmann, F.; Frommelt, F.; Ngo, Q.A.; Dobay, M.P.; Marovca, B.; Jenni, S.; et al. The leukemogenic TCF3-HLF complex rewires enhancers driving cellular identity and self-renewal conferring EP300 vulnerability. Cancer Cell 2019, 36, 630–644.e639. [Google Scholar] [CrossRef]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplantation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef] [Green Version]
- Jeha, S.; Pei, D.; Raimondi, S.C.; Onciu, M.; Campana, D.; Cheng, C.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Howard, S.C.; et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 2009, 23, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Hein, D.; Dreisig, K.; Metzler, M.; Izraeli, S.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood 2019, 134, 1355–1358. [Google Scholar] [CrossRef]
- Pui, C.H.; Sandlund, J.T.; Pei, D.; Rivera, G.K.; Howard, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Razzouk, B.I.; Hudson, M.M.; Cheng, C.; et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 2003, 290, 2001–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.R.; Qian, M.; Yang, W.; Diedrich, J.D.; Raetz, E.; Yang, W.; Dong, Q.; Devidas, M.; Pei, D.; Yeoh, A.; et al. Genome-wide association study of susceptibility loci for TCF3-PBX1 acute lymphoblastic leukemia in children. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Harrison, C.J. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood 2015, 125, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Schwab, C.; Ryan, S.; Papaemmanuil, E.; Robinson, H.M.; Jacobs, P.; Moorman, A.V.; Dyer, S.; Borrow, J.; Griffiths, M.; et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014, 508, 98–102. [Google Scholar] [CrossRef]
- Harrison, C.J.; Moorman, A.V.; Schwab, C.; Carroll, A.J.; Raetz, E.A.; Devidas, M.; Strehl, S.; Nebral, K.; Harbott, J.; Teigler-Schlegel, A.; et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): Cytogenetic characterization and outcome. Leukemia 2014, 28, 1015–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, P.B.; Ryan, S.; Bashton, M.; Hollern, S.; Hanna, R.; Case, M.; Schwalbe, E.C.; Schwab, C.J.; Cranston, R.E.; Young, B.D.; et al. SH2B3 inactivation through CN-LOH 12q is uniquely associated with B-cell precursor ALL with iAMP21 or other chromosome 21 gain. Leukemia 2019, 33, 1881–1894. [Google Scholar] [CrossRef] [Green Version]
- Heerema, N.A.; Carroll, A.J.; Devidas, M.; Loh, M.L.; Borowitz, M.J.; Gastier-Foster, J.M.; Larsen, E.C.; Mattano, L.A., Jr.; Maloney, K.W.; Willman, C.L.; et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk Children’s Oncology Group studies: A report from the Children’s Oncology Group. J. Clin. Oncol. 2013, 31, 3397–3402. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Robinson, H.; Schwab, C.; Richards, S.M.; Hancock, J.; Mitchell, C.D.; Goulden, N.; Vora, A.; Harrison, C.J. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: A comparison of the MRC ALL97/99 and UKALL2003 trials. J. Clin. Oncol. 2013, 31, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Cobaleda, C.; Schebesta, A.; Delogu, A.; Busslinger, M. Pax5: The guardian of B cell identity and function. Nat. Immunol. 2007, 8, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Butler, E.R.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Möricke, A.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; et al. Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: A retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 2021. [Google Scholar] [CrossRef]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ohki, K.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yaguchi, A.; Terada, K.; Saito, Y.; Yoshimi, A.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Bueno, C.; Tejedor, J.R.; Bashford-Rogers, R.; González-Silva, L.; Valdés-Mas, R.; Agraz-Doblás, A.; Díaz de la Guardia, R.; Ribera, J.; Zamora, L.; Bilhou-Nabera, C.; et al. Natural history and cell of origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood 2019, 134, 900–905. [Google Scholar] [CrossRef]
- Boer, J.; Valsecchi, M.; Hormann, F.; Antic, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia: A good prognostic subtype identified in a collaborative international study. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Hormann, F.M.; Hoogkamer, A.Q.; Beverloo, H.B.; Boeree, A.; Dingjan, I.; Wattel, M.M.; Stam, R.W.; Escherich, G.; Pieters, R.; den Boer, M.L.; et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 2019, 104, e455–e459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordo, V.; van der Zwet, J.; Canté-Barrett, K.; Pieters, R.; Meijerink, J. T-cell acute lymphoblastic leukemia: A roadmap to targeted therapies. Blood Cancer Discov. 2021, 2, 19–31. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Palomero, T.; Khiabanian, H.; van der Meulen, J.; Castillo, M.; van Roy, N.; de Moerloose, B.; Philippé, J.; González-García, S.; Toribio, M.L.; et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 338–342. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef]
- Salzer, W.L.; Burke, M.J.; Devidas, M.; Dai, Y.; Hardy, K.K.; Kairalla, J.A.; Gore, L.; Hilden, J.M.; Larsen, E.; Rabin, K.R.; et al. Impact of intrathecal triple therapy versus intrathecal methotrexate on disease-free survival for high-risk B-lymphoblastic leukemia: Children’s Oncology Group Study AALL1131. J. Clin. Oncol. 2020, 38, 2628–2638. [Google Scholar] [CrossRef]
- Teachey, D.T.; Devidas, M.; Wood, B.L.; Chen, Z.; Hayashi, R.J.; Annett, R.D.; Asselin, B.L.; August, K.J.; Cho, S.Y.; Dunsmore, K.P.; et al. Cranial radiation can be eliminated in most children with T-cell acute lymphoblastic leukemia (T-ALL) and bortezomib potentially improves survival in children with T-cell lymphoblastic lymphoma (T-LL): Results of Children’s Oncology Group (COG) trial AALL1231. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Conter, V.; Valsecchi, M.G.; Buldini, B.; Parasole, R.; Locatelli, F.; Colombini, A.; Rizzari, C.; Putti, M.C.; Barisone, E.; Lo Nigro, L.; et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: A retrospective analysis. Lancet Haematol. 2016, 3, e80–e86. [Google Scholar] [CrossRef]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Lamb, A.V.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef]
- Patrick, K.; Wade, R.; Goulden, N.; Mitchell, C.; Moorman, A.V.; Rowntree, C.; Jenkinson, S.; Hough, R.; Vora, A. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br. J. Haematol. 2014, 166, 421–424. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Jabbour, E.; Short, N.J.; Rausch, C.R.; Savoy, J.M.; Bose, P.; Yilmaz, M.; Jain, N.; Borthakur, G.; Ohanian, M.; et al. Clinical experience with venetoclax combined with chemotherapy for relapsed or refractory T-cell acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Campana, D.; Pui, C.H. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood 2017, 129, 1913–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dongen, J.J.; van der Velden, V.H.; Bruggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef] [Green Version]
- Della Starza, I.; Chiaretti, S.; de Propris, M.S.; Elia, L.; Cavalli, M.; de Novi, L.A.; Soscia, R.; Messina, M.; Vitale, A.; Guarini, A.; et al. Minimal residual disease in acute lymphoblastic leukemia: Technical and clinical advances. Front. Oncol. 2019, 9, 726. [Google Scholar] [CrossRef] [Green Version]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Emerson, R.O.; Sherwood, A.; Loh, M.L.; Angiolillo, A.; Howie, B.; Vogt, J.; Rieder, M.; Kirsch, I.; Carlson, C.; et al. Detection of minimal residual disease in B lymphoblastic leukemia by high-throughput sequencing of IGH. Clin. Cancer Res. 2014, 20, 4540–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, M.A.; Carlson, C.; Langholz, B.; Wall, D.A.; Schultz, K.R.; Bunin, N.; Kirsch, I.; Gastier-Foster, J.M.; Borowitz, M.; Desmarais, C.; et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 2015, 125, 3501–3508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Enshaei, A.; O’Connor, D.; Bartram, J.; Hancock, J.; Harrison, C.J.; Hough, R.; Samarasinghe, S.; den Boer, M.L.; Boer, J.M.; de Groot-Kruseman, H.A.; et al. A validated novel continuous prognostic index to deliver stratified medicine in pediatric acute lymphoblastic leukemia. Blood 2020, 135, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.-N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Cancer 2021, 2, 284–299. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef]
- Khaw, S.L.; Suryani, S.; Evans, K.; Richmond, J.; Robbins, A.; Kurmasheva, R.T.; Billups, C.A.; Erickson, S.W.; Guo, Y.; Houghton, P.J.; et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 2016, 128, 1382–1395. [Google Scholar] [CrossRef] [Green Version]
- Scherr, M.; Elder, A.; Battmer, K.; Barzan, D.; Bomken, S.; Ricke-Hoch, M.; Schröder, A.; Venturini, L.; Blair, H.J.; Vormoor, J.; et al. Differential expression of miR-17~92 identifies BCL2 as a therapeutic target in BCR-ABL-positive B-lineage acute lymphoblastic leukemia. Leukemia 2014, 28, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Peirs, S.; Matthijssens, F.; Goossens, S.; van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Canté-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014, 124, 3738–3747. [Google Scholar] [CrossRef] [PubMed]
- Chonghaile, T.N.; Roderick, J.E.; Glenfield, C.; Ryan, J.; Sallan, S.E.; Silverman, L.B.; Loh, M.L.; Hunger, S.P.; Wood, B.; DeAngelo, D.J.; et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014, 4, 1074–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autry, R.J.; Paugh, S.W.; Carter, R.; Shi, L.; Liu, J.; Ferguson, D.C.; Lau, C.E.; Bonten, E.J.; Yang, W.; McCorkle, J.R.; et al. Integrative genomic analyses reveal mechanisms of glucocorticoid resistance in acute lymphoblastic leukemia. Nat. Cancer 2020, 1, 329–344. [Google Scholar] [CrossRef]
- Karol, S.E.; Cooper, T.M.; Bittencourt, H.; Gore, L.; O’Brien, M.M.; Fraser, C.; Gambart, M.; Cario, G.; Zwaan, C.M.; Bourquin, J.-P.; et al. Safety, efficacy, and PK of the BCL2 inhibitor venetoclax in combination with chemotherapy in pediatric and young adult patients with relapsed/refractory acute myeloid leukemia and acute lymphoblastic leukemia: Phase 1 study. Blood 2019, 134, 2649. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Horton, T.M.; Gannavarapu, A.; Blaney, S.M.; D’Argenio, D.Z.; Plon, S.E.; Berg, S.L. Bortezomib interactions with chemotherapy agents in acute leukemia in vitro. Cancer Chemother. Pharmacol. 2006, 58, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Messinger, Y.H.; Gaynon, P.S.; Sposto, R.; van der Giessen, J.; Eckroth, E.; Malvar, J.; Bostrom, B.C.; Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Consortium. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 2012, 120, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Obzut, D.A.; Cooperman, J.; Fang, J.; Carroll, M.; Choi, J.K.; Houghton, P.J.; Brown, V.I.; Grupp, S.A. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood 2006, 107, 1149–1155. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crazzolara, R.; Cisterne, A.; Thien, M.; Hewson, J.; Baraz, R.; Bradstock, K.F.; Bendall, L.J. Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood 2009, 113, 3297–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avellino, R.; Romano, S.; Parasole, R.; Bisogni, R.; Lamberti, A.; Poggi, V.; Venuta, S.; Romano, M.F. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood 2005, 106, 1400–1406. [Google Scholar] [CrossRef] [Green Version]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Bueso-Ramos, C.; Daniel, J.; Williamson, J.; Kantarjian, H.M.; Issa, J.P. DNA methylation patterns at relapse in adult acute lymphocytic leukemia. Clin. Cancer Res. 2002, 8, 1897–1903. [Google Scholar] [PubMed]
- Burke, M.J.; Kostadinov, R.; Sposto, R.; Gore, L.; Kelley, S.M.; Rabik, C.; Trepel, J.B.; Lee, M.J.; Yuno, A.; Lee, S.; et al. Decitabine and Vorinostat with Chemotherapy in Relapsed Pediatric Acute Lymphoblastic Leukemia: A TACL Pilot Study. Clin. Cancer Res. 2020, 26, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Pui, C.H. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. 2019, 38, 595–610. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Bruggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of postreinduction therapy consolidation with blinatumomab vs. chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: A randomized clinical trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of blinatumomab vs. chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: A randomized clinical trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Oberley, M.J.; Gaynon, P.S.; Bhojwani, D.; Pulsipher, M.A.; Gardner, R.A.; Hiemenz, M.C.; Ji, J.; Han, J.; O’Gorman, M.R.G.; Wayne, A.S.; et al. Myeloid lineage switch following chimeric antigen receptor T-cell therapy in a patient with TCF3-ZNF384 fusion-positive B-lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27265. [Google Scholar] [CrossRef] [PubMed]
- Braig, F.; Brandt, A.; Goebeler, M.; Tony, H.P.; Kurze, A.K.; Nollau, P.; Bumm, T.; Bottcher, S.; Bargou, R.C.; Binder, M. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017, 129, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Kadauke, S.; Myers, R.M.; Li, Y.; Aplenc, R.; Baniewicz, D.; Barrett, D.M.; Barz Leahy, A.; Callahan, C.; Dolan, J.G.; Fitzgerald, J.C.; et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: A prospective clinical trial. J. Clin. Oncol. 2021, 39, 920–930. [Google Scholar] [CrossRef]
- Hill, J.A.; Seo, S.K. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood 2020, 136, 925–935. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Cho, M.; Haso, W.; Zhang, L.; Tasian, S.K.; Oo, H.Z.; Negri, G.L.; Lin, Y.; Zou, J.; Mallon, B.S.; et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood 2015, 126, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.M.; Muffly, L.S.; Spiegel, J.Y.; Ramakrishna, S.; Hossain, N.; Baggott, C.; Sahaf, B.; Patel, S.; Craig, J.; Yoon, J.; et al. Phase I trial using CD19/CD22 bispecific CAR T cells in pediatric and adult acute lymphoblastic leukemia (ALL). Blood 2019, 134, 744. [Google Scholar] [CrossRef]
- Wang, N.; Hu, X.; Cao, W.; Li, C.; Xiao, Y.; Cao, Y.; Gu, C.; Zhang, S.; Chen, L.; Cheng, J.; et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood 2020, 135, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zuo, S.; Deng, B.; Xu, X.; Li, C.; Zheng, Q.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood 2020, 135, 387–391. [Google Scholar] [CrossRef]
- He, X.; Xiao, X.; Li, Q.; Jiang, Y.; Cao, Y.; Sun, R.; Jin, X.; Yuan, T.; Meng, J.; Ma, L.; et al. Anti-CD19 CAR-T as a feasible and safe treatment against central nervous system leukemia after intrathecal chemotherapy in adults with relapsed or refractory B-ALL. Leukemia 2019, 33, 2102–2104. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Y.; Ruan, M.; Li, J.; Zhong, M.; Li, Z.; Liu, F.; Wang, S.; Chen, Y.; Liu, L.; et al. Treatment of testicular relapse of B-cell acute lymphoblastic leukemia with CD19-specific chimeric antigen receptor T cells. Clin. Lymphoma Myeloma Leuk. 2019, 20, 366–370. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Jabbour, E.; Ravandi, F.; Kebriaei, P.; Huang, X.; Short, N.J.; Thomas, D.; Sasaki, K.; Rytting, M.; Jain, N.; Konopleva, M.; et al. Salvage Chemoimmunotherapy With Inotuzumab Ozogamicin Combined with Mini-Hyper-CVD for Patients With Relapsed or Refractory Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 230–234. [Google Scholar] [CrossRef]
- Kebriaei, P.; Cutler, C.; de Lima, M.; Giralt, S.; Lee, S.J.; Marks, D.; Merchant, A.; Stock, W.; van Besien, K.; Stelljes, M. Management of important adverse events associated with inotuzumab ozogamicin: Expert panel review. Bone Marrow Transpl. 2018, 53, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-de-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.H.J.; Ammerlaan, A.C.; Thano, A.; et al. A Phase I study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drugs | Year Approved in the US * |
|---|---|
| Conventional chemotherapy | |
| Mercaptopurine | 1953 |
| Methotrexate | 1953 |
| Prednisone | 1955 |
| Dexamethasone | 1958 |
| Cyclophosphamide | 1959 |
| Vincristine | 1963 |
| Thioguanine | 1966 |
| Cytarabine | 1969 |
| Doxorubicin | 1974 |
| L-Asparaginase | 1978 |
| Daunorubicin | 1979 |
| New formulations or agents | |
| Pegaspargase | 1994 |
| Nelarabine | 2005 |
| Erwinase | 2011 |
| Vincristine sulfate liposome injection | 2012 |
| Calaspargase | 2018 |
| Molecular targeted therapy | |
| ABL1 inhibitors | |
| Imatinib | 2001 |
| Dasatinib | 2006 |
| Nilotinib | 2007 |
| Ponatinib | 2012 |
| JAK inhibitor | |
| Ruxolitinib | 2011 |
| BCL-2 and BCL-XL inhibitors | |
| Venotoclax | 2016 |
| Navitoclax | NA |
| Proteasome inhibitors | |
| Bortezomib | 2003 |
| Carfilzomib | 2012 |
| Ixazomib | 2015 |
| mTOR inhibitors | |
| Sirolimus | 1999 |
| Temsirolimus | 2007 |
| Everolimus | 2009 |
| DNA methyltransferase inhibitors | |
| Azacitidine | 2004 |
| Decitabine | 2006 |
| Histone deacetylase inhibitors | |
| Vorinostat | 2006 |
| Panobinostat | 2015 |
| Bromodomain inhibitor | |
| JQ1 | NA |
| DOT1 inhibitor | |
| Pinometostat | NA |
| Menin inhibitor | |
| SNDX-5613 | NA |
| Immunotherapy | |
| Unconjugated antibodies | |
| Rituximab (CD20) | 1997 |
| Ofatumumab (CD20) | 2009 |
| Epratuzumab (CD22) | NA |
| Daratumumab (CD38) | 2015 |
| Alemtuzumab (CD52) | 2001 |
| Bispecific antibody | |
| Blinatumomab (CD19) | 2014 |
| Chimeric antigen receptor (CAR) T cells | |
| Tisagenlecleucel (CD19) | 2017 |
| Antibody–drug conjugate | |
| Inotuzumab ozogamicin (CD22) | 2017 |
| Category | Characteristics | Therapeutic Approach |
|---|---|---|
| B-lymphoblastic leukemia | ||
| Low-risk genetics | ||
| ETV6/RUNX1 | Excellent prognosis | Reduction of intensity, MRD based |
| Hyperdiploidy | Excellent prognosis | Reduction of intensity, MRD based |
| DUX4-rearranged | Most have focal ERG deletions and favorable outcome despite IKZF1 alterations | Standard dose intensity, MRD based |
| Intermediate-risk genetics | ||
| TCF3/PBX1 | Higher incidence in African Americans, cytoplasmic μ-chain | Standard dose intensity, MRD based, intensive intrathecal therapy |
| PAX5alt | PAX5 fusions, mutation, or amplifications | Standard dose intensity, MRD based |
| PAX5 p.Pro80Arg | Frequent signaling pathway alterations | Standard dose intensity, MRD based, JAK inhibitors |
| ZNF384-rearranged | Peak age and prognosis vary by fusion partner, expression of myeloid markers | Standard dose intensity, MRD based |
| iAMP21 | Additional copies of chromosome 21, worse outcome with low-intensity therapy | Intensification of therapy |
| NUTM1-rearranged * | Rare; more common in infants, excellent prognosis | Standard dose intensity, MRD based |
| High-risk genetics | ||
| Near-haploid | 24–31 chromosomes, Ras-activating mutations, inactivation of IKZF3 | Intensification of therapy, MRD based, BCL-2 inhibitors |
| Low-hypodiploid | 32–39 chromosomes, TP53 mutations (somatic and germline) | Intensification of therapy, MRD based, BCL-2 inhibitors |
| BCR/ABL1 | Prognosis improved with ABL1 inhibitors, common deletions of IKZF1 | ABL1 inhibitors, BCL-2 inhibitors |
| BCR/ABL1-like; JAK-STAT activating mutation | CRLF2 rearranged (IGH-CRLF2, P2RY8-CRLF2), JAK1/2, EPOR, IL7R, SH2B3 mutation | JAK inhibitors, BCL-2 inhibitors |
| BCR/ABL1-like; ABL1-class | Kinase-activating lesions, potentially amenable to kinase inhibition | ABL1 inhibitors, BCL-2 inhibitors |
| KMT2A (MLL)-rearranged | Common in infant ALL, few cooperating mutations | DOT1L inhibitors, menin inhibitors, proteasome inhibitors, histone deacetylase inhibitors, BCL-2 inhibitors |
| MEF2D-rearranged | Mature B cell leukemia morphology, cytoplasmic μ-chain | Histone deacetylase inhibitors, proteasome inhibitors |
| TCF3-HLF | Rare; dismal prognosis | BCL-2 inhibitors |
| ETV6/RUNX1-like * | Similar gene expression profile to ETV6-RUNX1 but lacks fusion | Intensification of therapy, MRD based |
| T-lymphoblastic leukemia | ||
| Non-early T-cell precursor | Deregulation of TAL1, TAL2, LYL1, LMO1, LMO2, TLX1 (HOX11), TLX3 (HOX11L2), and HOXA; NOTCH1 activating mutation | Standard dose intensity, MRD based, nelarabine, BCL-2 inhibitors |
| JAK-STAT activating mutation | Approximately 25% of patients with T-ALL | Standard dose intensity, MRD based, nelarabine, JAK inhibitors, BCL-2 inhibitors |
| ABL1 fusions (e.g., NUP214-ABL1) | Fusion with BCR and NUP214, potentially amenable to tyrosine kinase inhibition | Standard dose intensity, MRD based, ABL1 inhibitors, nelarabine, BCL-2 inhibitors |
| Early T-cell precursor ALL | Mutations in transcriptional regulators, JAK-STAT and Ras signaling, and epigenetic modifiers | Standard dose intensity, MRD based, JAK inhibitors, BCL-2 inhibitors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. https://doi.org/10.3390/jcm10091926
Inaba H, Pui C-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. Journal of Clinical Medicine. 2021; 10(9):1926. https://doi.org/10.3390/jcm10091926
Chicago/Turabian StyleInaba, Hiroto, and Ching-Hon Pui. 2021. "Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia" Journal of Clinical Medicine 10, no. 9: 1926. https://doi.org/10.3390/jcm10091926
APA StyleInaba, H., & Pui, C.-H. (2021). Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. Journal of Clinical Medicine, 10(9), 1926. https://doi.org/10.3390/jcm10091926