Feature Papers in Mitochondria
A topical collection in Cells (ISSN 2073-4409). This collection belongs to the section "Mitochondria".
Submission Status:
Closed (3 March 2026)
|
Viewed by 31507
Share This Topical Collection
Editor
 Prof. Dr. Paolo Bernardi
Prof. Dr. Paolo Bernardi
Prof. Dr. Paolo Bernardi
E-Mail
Collection Editor
Department of Biomedical Sciences, University of Padova, Via Ugo Bassi 58/B, I-35131 Padova, Italy
Interests: mitochondria; calcium; channels; permeability transition; ATP synthase; cell death
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
This Topical Collection, "Feature Papers in Mitochondria", aims to publish high-quality research articles, review articles and communications from all fields that study the topic of mitochondria, with a focus on cell biological research. The aim of this Topical Collection is to illustrate, through selected works, the state-of-the-art research related to various aspects of mitochondria in diseases. We especially encourage the Editorial Board Members of Cells to contribute papers reflecting the latest progress in their research field. In addition, further experts in relevant research fields are welcome to submit their work to this Topical Collection.
Prof. Dr. Paolo Bernardi
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as communications are invited. For planned papers, a title and short abstract (about 250 words) can be sent to the Editorial Office for assessment.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-anonymized peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Cells is an international peer-reviewed open access semimonthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Published Papers (6 papers)
Open AccessReview
Mitochondrial Metabolic Checkpoints in Human Fertility: Reactive Oxygen Species as Gatekeepers of Gamete Competence
by
Sofoklis Stavros, Nikolaos Thomakos, Efthalia Moustakli, Nikoleta Daponte, Dimos Sioutis, Nikolaos Kathopoulis, Athanasios Zikopoulos, Ismini Anagnostaki, Chrysi Christodoulaki, Themos Grigoriadis, Ekaterini Domali and Anastasios Potiris
Cited by 2 | Viewed by 1492
Abstract
Crucial regulators of gamete metabolism and signaling, mitochondria synchronize energy generation with redox equilibrium and developmental proficiency. Once thought of as hazardous byproducts, reactive oxygen species (ROS) are now understood to be vital signaling molecules that provide a “redox window of competence” that
[...] Read more.
Crucial regulators of gamete metabolism and signaling, mitochondria synchronize energy generation with redox equilibrium and developmental proficiency. Once thought of as hazardous byproducts, reactive oxygen species (ROS) are now understood to be vital signaling molecules that provide a “redox window of competence” that is required for oocyte maturation, sperm capacitation, and early embryo development. This review presents the idea of mitochondrial metabolic checkpoints, which are phases that govern gamete quality and fertilization potential by interacting with cellular signaling, redox balance, and mitochondrial activity. Recent research shows that oocytes may sustain a nearly ROS-free metabolic state by blocking specific respiratory-chain components, highlighting the importance of mitochondrial remodeling in gamete competence. Evidence from in vitro and in vivo studies shows that ROS act as dynamic gatekeepers at critical points in oogenesis, spermatogenesis, fertilization, and early embryogenesis. However, assisted reproductive technologies (ARTs) may inadvertently disrupt this redox–metabolic equilibrium. Potential translational benefits can be obtained via targeted techniques that optimize mitochondrial function, such as modifying oxygen tension, employing mitochondria-directed antioxidants like MitoQ and SS-31, and supplementing with nutraceuticals like melatonin, CoQ10, and resveratrol. Understanding ROS-mediated checkpoints forms the basis for developing biomarkers of gamete competence and precision therapies to improve ART outcomes. By highlighting mitochondria as both metabolic sensors and redox regulators, this review links fundamental mitochondrial biology to clinical reproductive medicine.
Full article
►▼
Show Figures
Open AccessArticle
Exploring Metabolic Shifts in Kidney Cancer and Non-Cancer Cells Under Pro- and Anti-Apoptotic Treatments Using NMR Metabolomics
by
Lucia Trisolini, Biagia Musio, Beatriz Teixeira, Maria Noemi Sgobba, Anna Lucia Francavilla, Mariateresa Volpicella, Lorenzo Guerra, Anna De Grassi, Vito Gallo, Iola F. Duarte and Ciro Leonardo Pierri
Cited by 3 | Viewed by 2499
Abstract
This study investigates the metabolic responses of cancerous (RCC) and non-cancerous (HK2) kidney cells to treatment with Staurosporine (STAU), which has a pro-apoptotic effect, and Bongkrekic acid (BKA), which has an anti-apoptotic effect, individually and in combination, using
1H NMR metabolomics to
[...] Read more.
This study investigates the metabolic responses of cancerous (RCC) and non-cancerous (HK2) kidney cells to treatment with Staurosporine (STAU), which has a pro-apoptotic effect, and Bongkrekic acid (BKA), which has an anti-apoptotic effect, individually and in combination, using
1H NMR metabolomics to identify metabolite markers linked to mitochondrial apoptotic pathways. BKA had minimal metabolic effects in RCC cells, suggesting its role in preserving mitochondrial function without significantly altering metabolic pathways. In contrast, STAU induced substantial metabolic reprogramming in RCC cells, disrupting energy production, redox balance, and biosynthesis, thereby triggering apoptotic pathways. The combined treatment of BKA and STAU primarily mirrored the effects of STAU alone, with BKA showing little capacity to counteract the pro-apoptotic effects. In non-cancerous HK2 cells, the metabolic alterations were far less pronounced, highlighting key differences in the metabolic responses of cancerous and non-cancerous cells. RCC cells displayed greater metabolic flexibility, while HK2 cells maintained a more regulated metabolic state. These findings emphasize the potential for targeting cancer-specific metabolic vulnerabilities while sparing non-cancerous cells, underscoring the value of metabolomics in understanding apoptotic and anti-apoptotic mechanisms. Future studies should validate these results in vivo and explore their potential for personalized treatment strategies.
Full article
►▼
Show Figures
Open AccessReview
Mitochondrial NME6: A Paradigm Change within the NME/NDP Kinase Protein Family?
by
Bastien Proust, Maja Herak Bosnar, Helena Ćetković, Malgorzata Tokarska-Schlattner and Uwe Schlattner
Cited by 4 | Viewed by 2629
Abstract
Eukaryotic NMEs/NDP kinases are a family of 10 multifunctional proteins that occur in different cellular compartments and interact with various cellular components (proteins, membranes, and DNA). In contrast to the well-studied Group I NMEs (NME1–4), little is known about the more divergent Group
[...] Read more.
Eukaryotic NMEs/NDP kinases are a family of 10 multifunctional proteins that occur in different cellular compartments and interact with various cellular components (proteins, membranes, and DNA). In contrast to the well-studied Group I NMEs (NME1–4), little is known about the more divergent Group II NMEs (NME5–9). Three recent publications now shed new light on NME6. First, NME6 is a third mitochondrial NME, largely localized in the matrix space, associated with the mitochondrial inner membrane. Second, while its monomeric form is inactive, NME6 gains NDP kinase activity through interaction with mitochondrial RCC1L. This challenges the current notion that mammalian NMEs require the formation of hexamers to become active. The formation of complexes between NME6 and RCC1L, likely heterodimers, seemingly obviates the necessity for hexamer formation, stabilizing a NDP kinase-competent conformation. Third, NME6 is involved in mitochondrial gene maintenance and expression by providing (d)NTPs for replication and transcription (in particular the pyrimidine nucleotides) and by a less characterized mechanism that supports mitoribosome function. This review offers an overview of NME evolution and structure and highlights the new insight into NME6. The new findings position NME6 as the most comprehensively studied protein in NME Group II and may even suggest it as a new paradigm for related family members.
Full article
►▼
Show Figures
Open AccessReview
Cardiomyopathy in Duchenne Muscular Dystrophy and the Potential for Mitochondrial Therapeutics to Improve Treatment Response
by
Shivam Gandhi, H. Lee Sweeney, Cora C. Hart, Renzhi Han and Christopher G. R. Perry
Cited by 14 | Viewed by 10475
Abstract
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease caused by mutations to the dystrophin gene, resulting in deficiency of dystrophin protein, loss of myofiber integrity in skeletal and cardiac muscle, and eventual cell death and replacement with fibrotic tissue. Pathologic cardiac manifestations
[...] Read more.
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disease caused by mutations to the dystrophin gene, resulting in deficiency of dystrophin protein, loss of myofiber integrity in skeletal and cardiac muscle, and eventual cell death and replacement with fibrotic tissue. Pathologic cardiac manifestations occur in nearly every DMD patient, with the development of cardiomyopathy—the leading cause of death—inevitable by adulthood. As early cardiac abnormalities are difficult to detect, timely diagnosis and appropriate treatment modalities remain a challenge. There is no cure for DMD; treatment is aimed at delaying disease progression and alleviating symptoms. A comprehensive understanding of the pathophysiological mechanisms is crucial to the development of targeted treatments. While established hypotheses of underlying mechanisms include sarcolemmal weakening, upregulation of pro-inflammatory cytokines, and perturbed ion homeostasis, mitochondrial dysfunction is thought to be a potential key contributor. Several experimental compounds targeting the skeletal muscle pathology of DMD are in development, but the effects of such agents on cardiac function remain unclear. The synergistic integration of small molecule- and gene-target-based drugs with metabolic-, immune-, or ion balance-enhancing compounds into a combinatorial therapy offers potential for treating dystrophin deficiency-induced cardiomyopathy, making it crucial to understand the underlying mechanisms driving the disorder.
Full article
►▼
Show Figures
Open AccessArticle
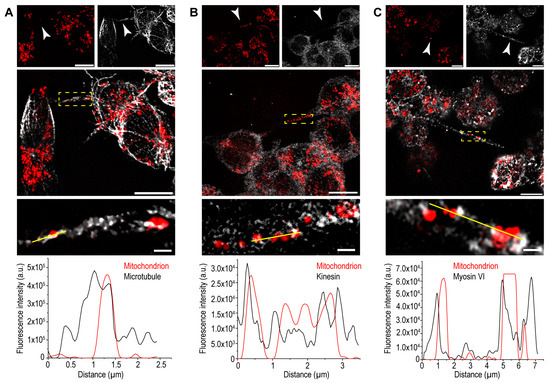
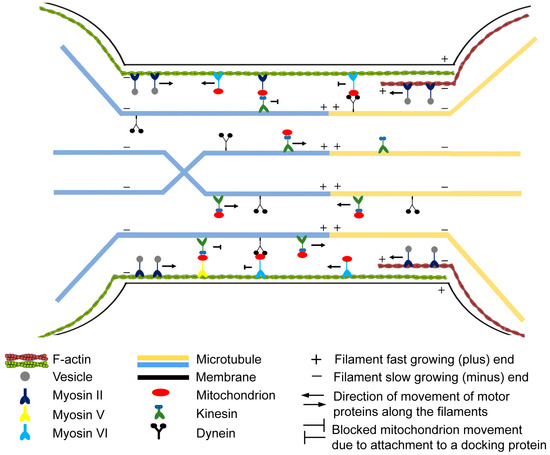
Cooperation of Various Cytoskeletal Components Orchestrates Intercellular Spread of Mitochondria between B-Lymphoma Cells through Tunnelling Nanotubes
by
Henriett Halász, Viktória Tárnai, János Matkó, Miklós Nyitrai and Edina Szabó-Meleg
Cited by 11 | Viewed by 3835
Abstract
Membrane nanotubes (NTs) are dynamic communication channels connecting spatially separated cells even over long distances and promoting the transport of different cellular cargos. NTs are also involved in the intercellular spread of different pathogens and the deterioration of some neurological disorders. Transport processes
[...] Read more.
Membrane nanotubes (NTs) are dynamic communication channels connecting spatially separated cells even over long distances and promoting the transport of different cellular cargos. NTs are also involved in the intercellular spread of different pathogens and the deterioration of some neurological disorders. Transport processes via NTs may be controlled by cytoskeletal elements. NTs are frequently observed membrane projections in numerous mammalian cell lines, including various immune cells, but their functional significance in the ‘antibody factory’ B cells is poorly elucidated. Here, we report that as active channels, NTs of B-lymphoma cells can mediate bidirectional mitochondrial transport, promoted by the cooperation of two different cytoskeletal motor proteins, kinesin along microtubules and myosin VI along actin, and bidirectional transport processes are also supported by the heterogeneous arrangement of the main cytoskeletal filament systems of the NTs. We revealed that despite NTs and axons being different cell extensions, the mitochondrial transport they mediate may exhibit significant similarities. Furthermore, we found that microtubules may improve the stability and lifespan of B-lymphoma-cell NTs, while F-actin strengthens NTs by providing a structural framework for them. Our results may contribute to a better understanding of the regulation of the major cells of humoral immune response to infections.
Full article
►▼
Show Figures
Open AccessReview
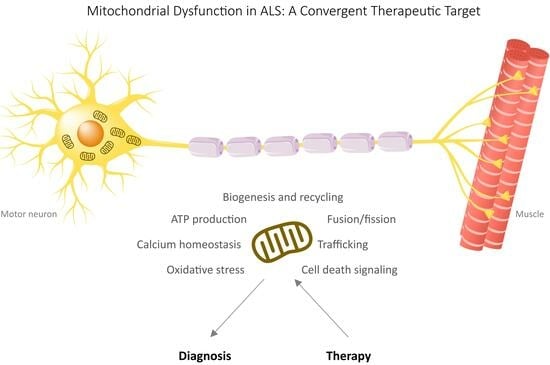
Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis
by
Teresa Cunha-Oliveira, Liliana Montezinho, Rui F. Simões, Marcelo Carvalho, Elisabete Ferreiro and Filomena S. G. Silva
Cited by 28 | Viewed by 9504
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the progressive loss of motor neurons, for which current treatment options are limited. Recent studies have shed light on the role of mitochondria in ALS pathogenesis, making them an attractive therapeutic intervention
[...] Read more.
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the progressive loss of motor neurons, for which current treatment options are limited. Recent studies have shed light on the role of mitochondria in ALS pathogenesis, making them an attractive therapeutic intervention target. This review contains a very comprehensive critical description of the involvement of mitochondria and mitochondria-mediated mechanisms in ALS. The review covers several key areas related to mitochondria in ALS, including impaired mitochondrial function, mitochondrial bioenergetics, reactive oxygen species, metabolic processes and energy metabolism, mitochondrial dynamics, turnover, autophagy and mitophagy, impaired mitochondrial transport, and apoptosis. This review also highlights preclinical and clinical studies that have investigated various mitochondria-targeted therapies for ALS treatment. These include strategies to improve mitochondrial function, such as the use of dichloroacetate, ketogenic and high-fat diets, acetyl-carnitine, and mitochondria-targeted antioxidants. Additionally, antiapoptotic agents, like the mPTP-targeting agents minocycline and rasagiline, are discussed. The paper aims to contribute to the identification of effective mitochondria-targeted therapies for ALS treatment by synthesizing the current understanding of the role of mitochondria in ALS pathogenesis and reviewing potential convergent therapeutic interventions. The complex interplay between mitochondria and the pathogenic mechanisms of ALS holds promise for the development of novel treatment strategies to combat this devastating disease.
Full article
►▼
Show Figures








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}