High-Throughput 2020, 9(3), 17; https://doi.org/10.3390/ht9030017 - 29 Jul 2020
Cited by 15
Abstract
Recent advances in microbiome studies have revealed much information about how the gut virome, mycobiome, and gut bacteria influence health and disease. Over the years, many studies have reported associations between the gut microflora under different pathological conditions. However, information about the role
[...] Read more.
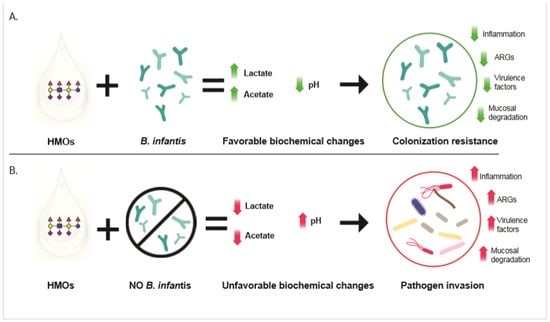
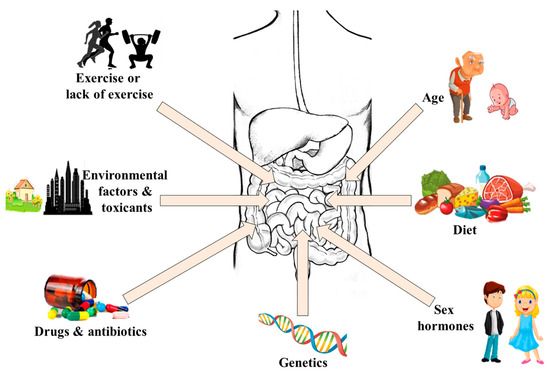
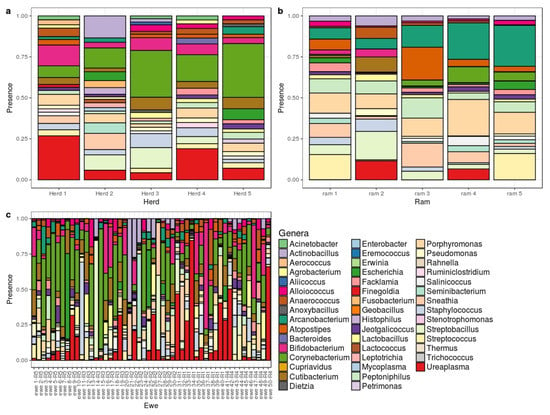
Recent advances in microbiome studies have revealed much information about how the gut virome, mycobiome, and gut bacteria influence health and disease. Over the years, many studies have reported associations between the gut microflora under different pathological conditions. However, information about the role of gut metabolites and the mechanisms by which the gut microbiota affect health and disease does not provide enough evidence. Recent advances in next-generation sequencing and metabolomics coupled with large, randomized clinical trials are helping scientists to understand whether gut dysbiosis precedes pathology or gut dysbiosis is secondary to pathology. In this review, we discuss our current knowledge on the impact of gut bacteria, virome, and mycobiome interactions with the host and how they could be manipulated to promote health.
Full article
(This article belongs to the Special Issue Human Microbiome and Diseases: Implications for Novel Therapies)
►
Show Figures
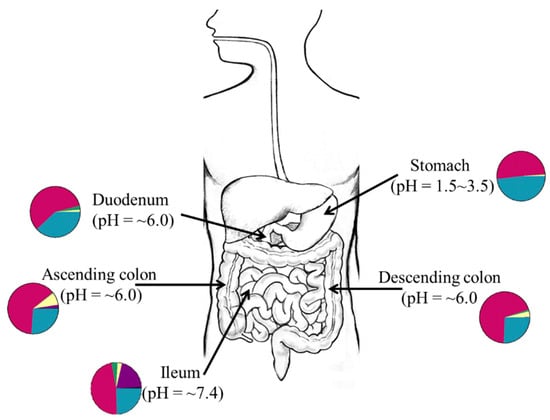
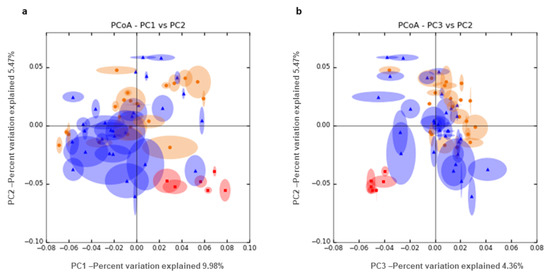
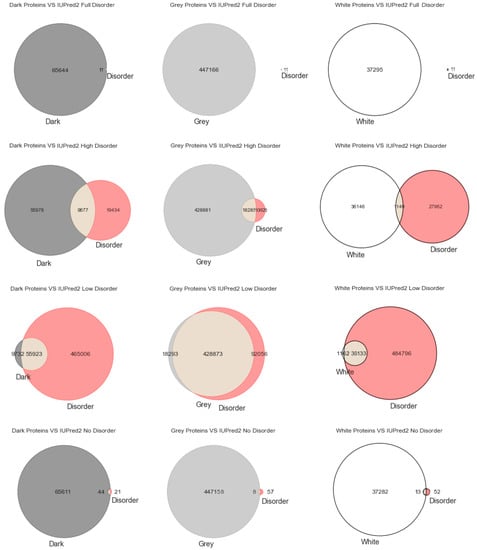
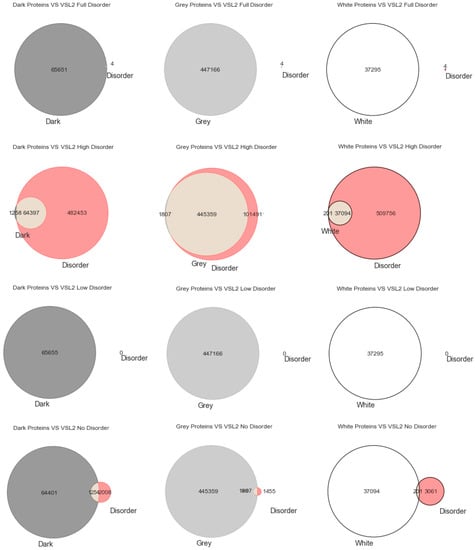
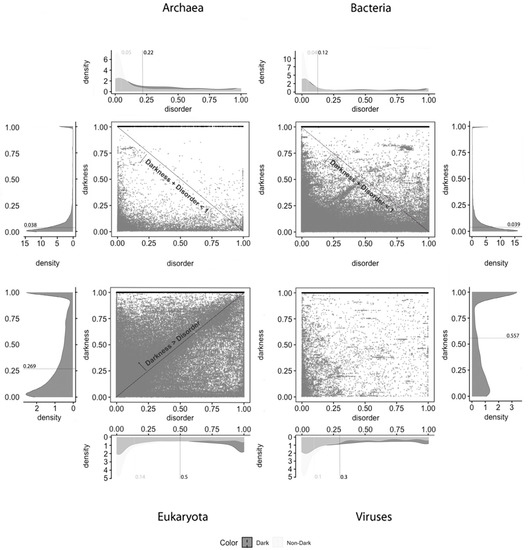
Figure 1
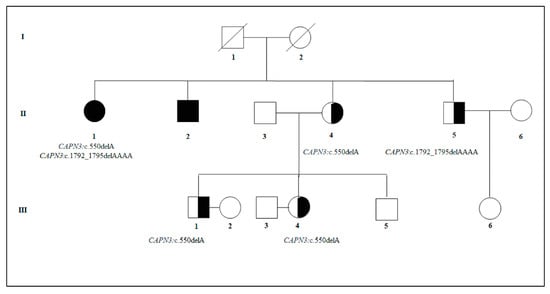


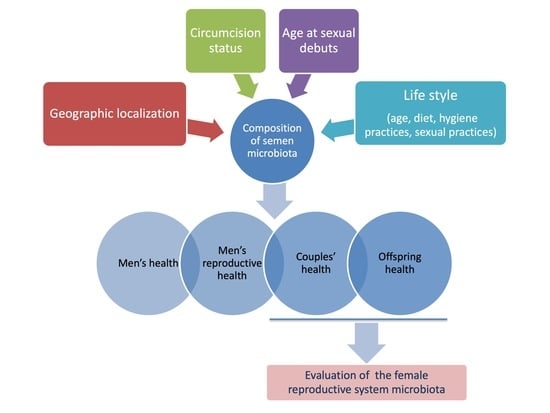
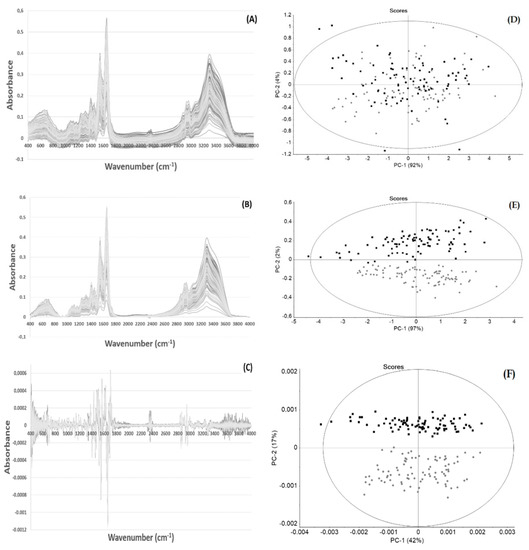
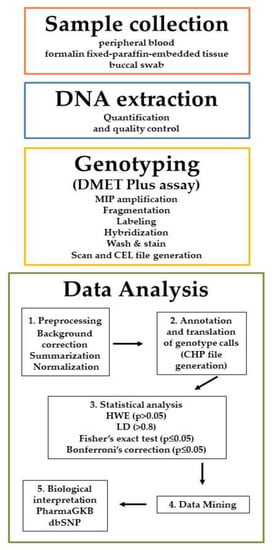



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}