
Molecular Modification of Kex2 P1’ Site Enhances Expression and Druggability of Fungal Defensin




Abstract
:1. Introduction
2. Results
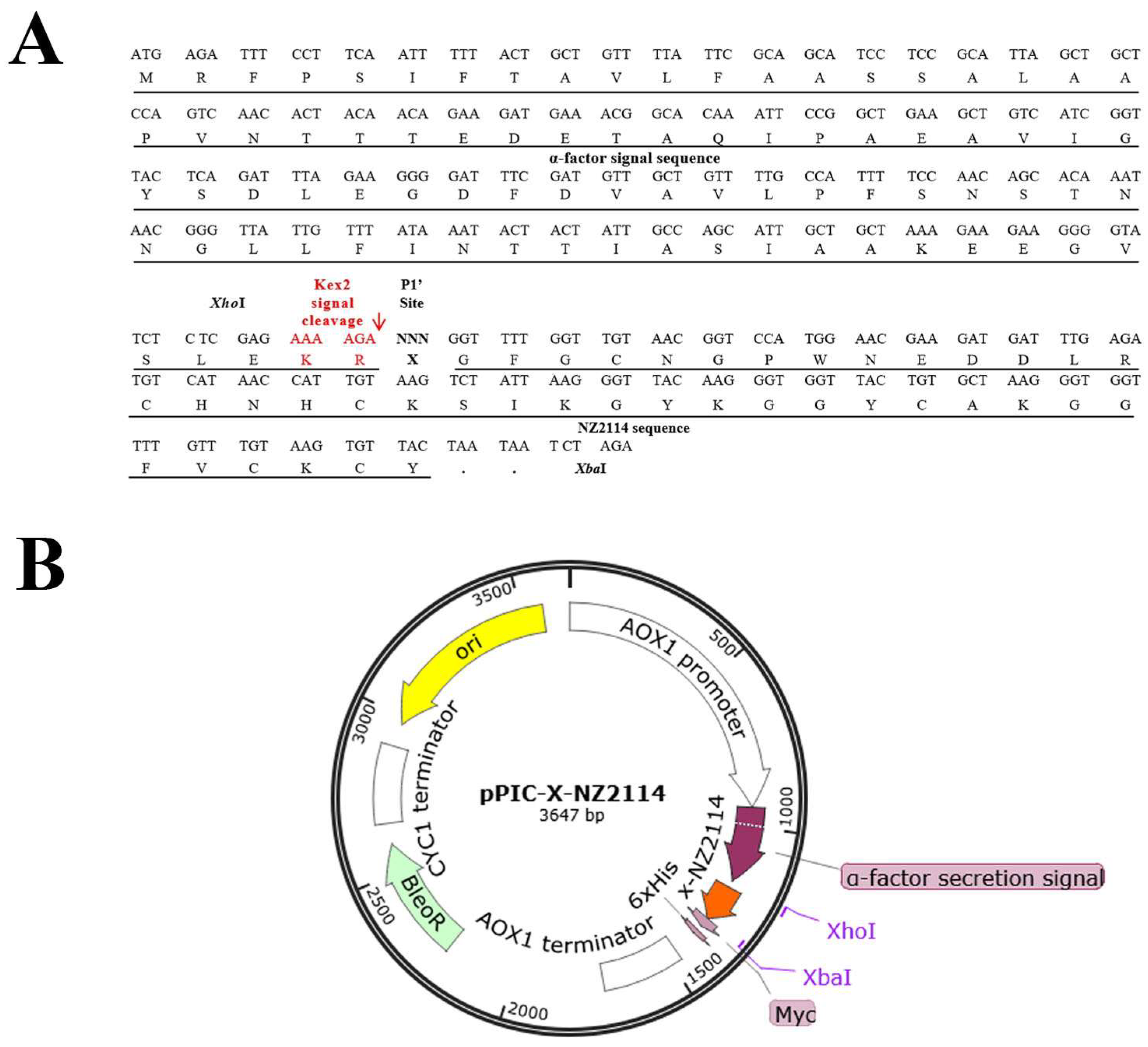
2.1. Construction of Recombinant Plasmids
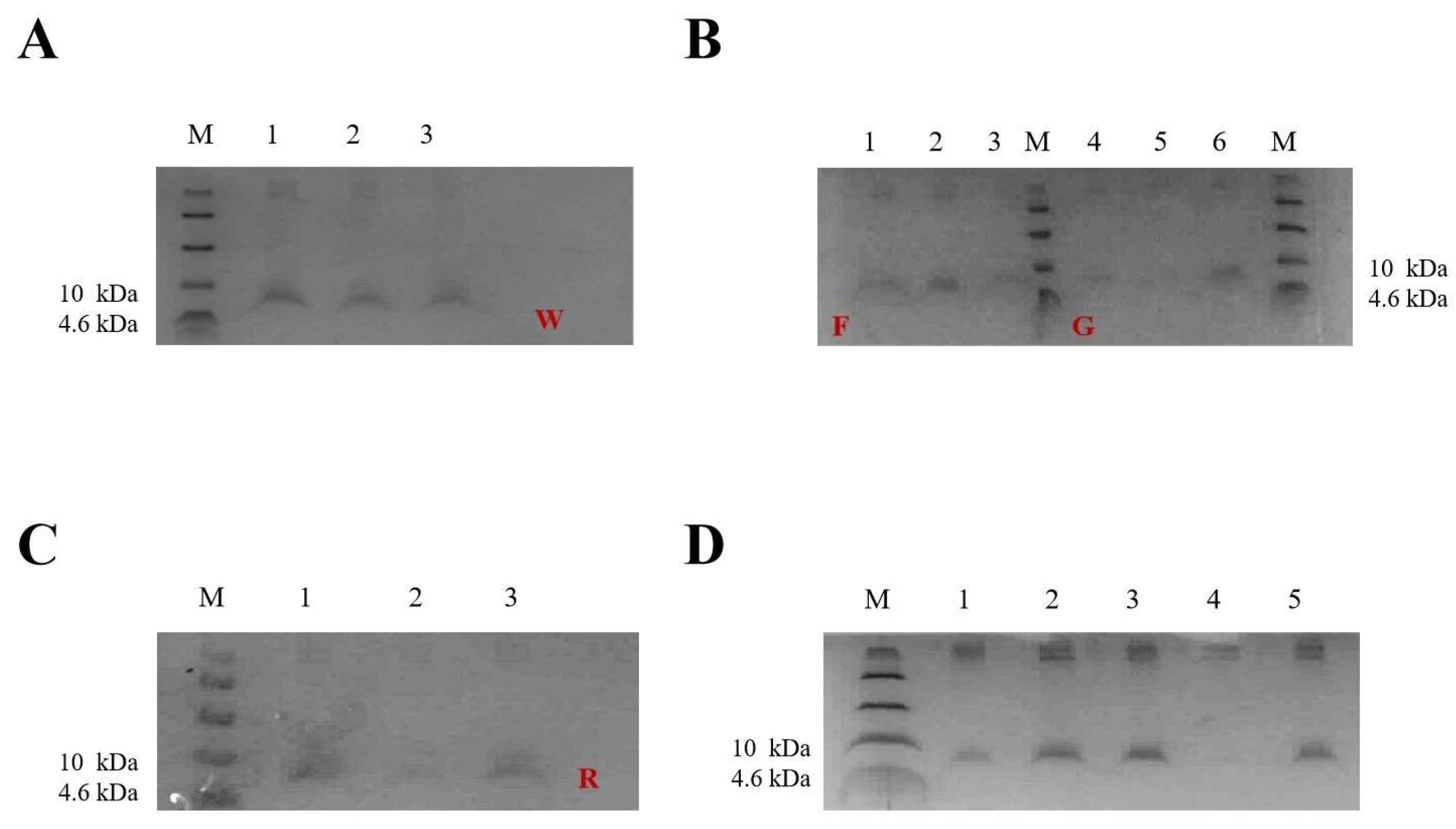
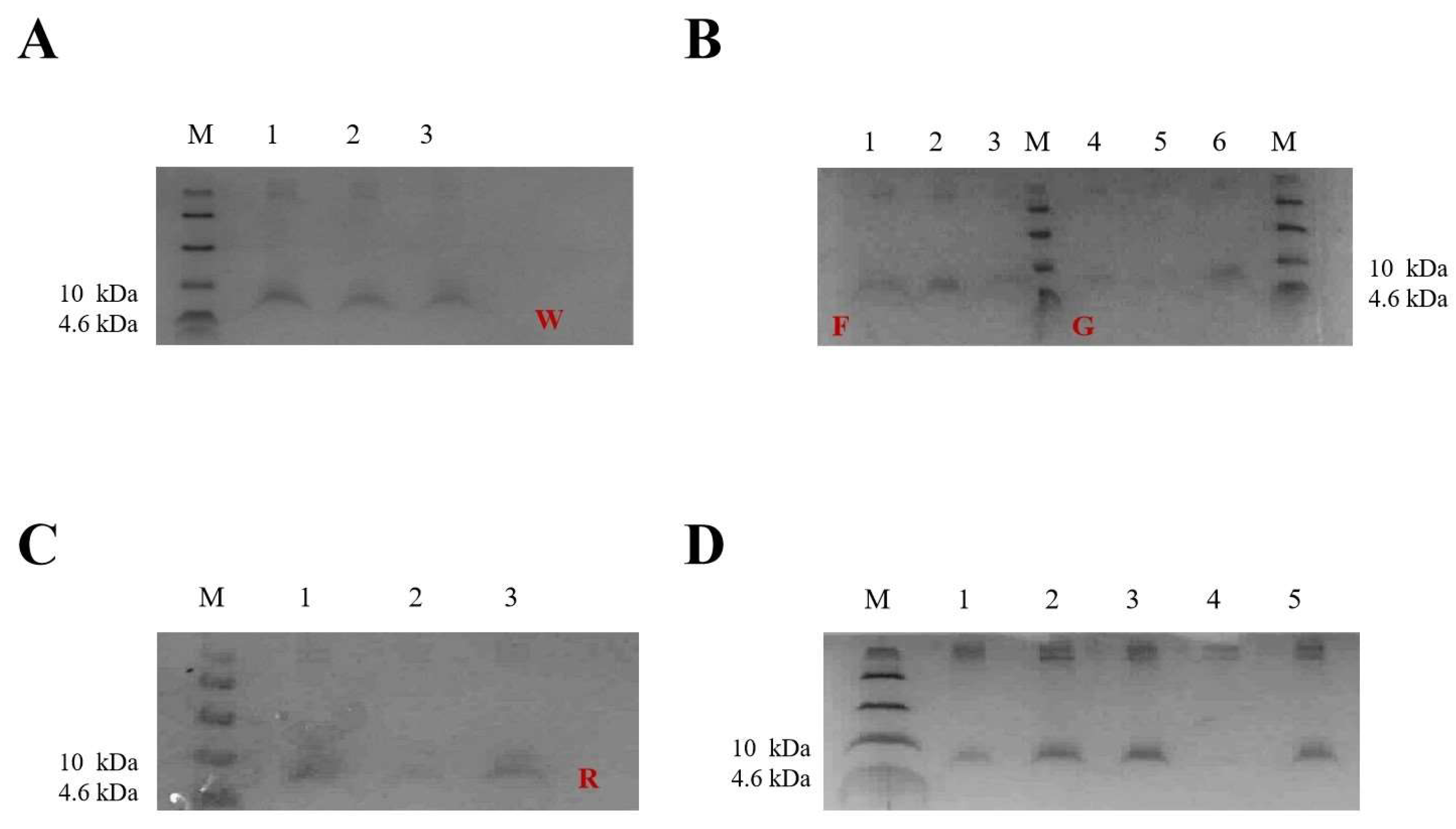
2.2. Selection of Positive Transformants at the Well Plate, Shake Flask Level
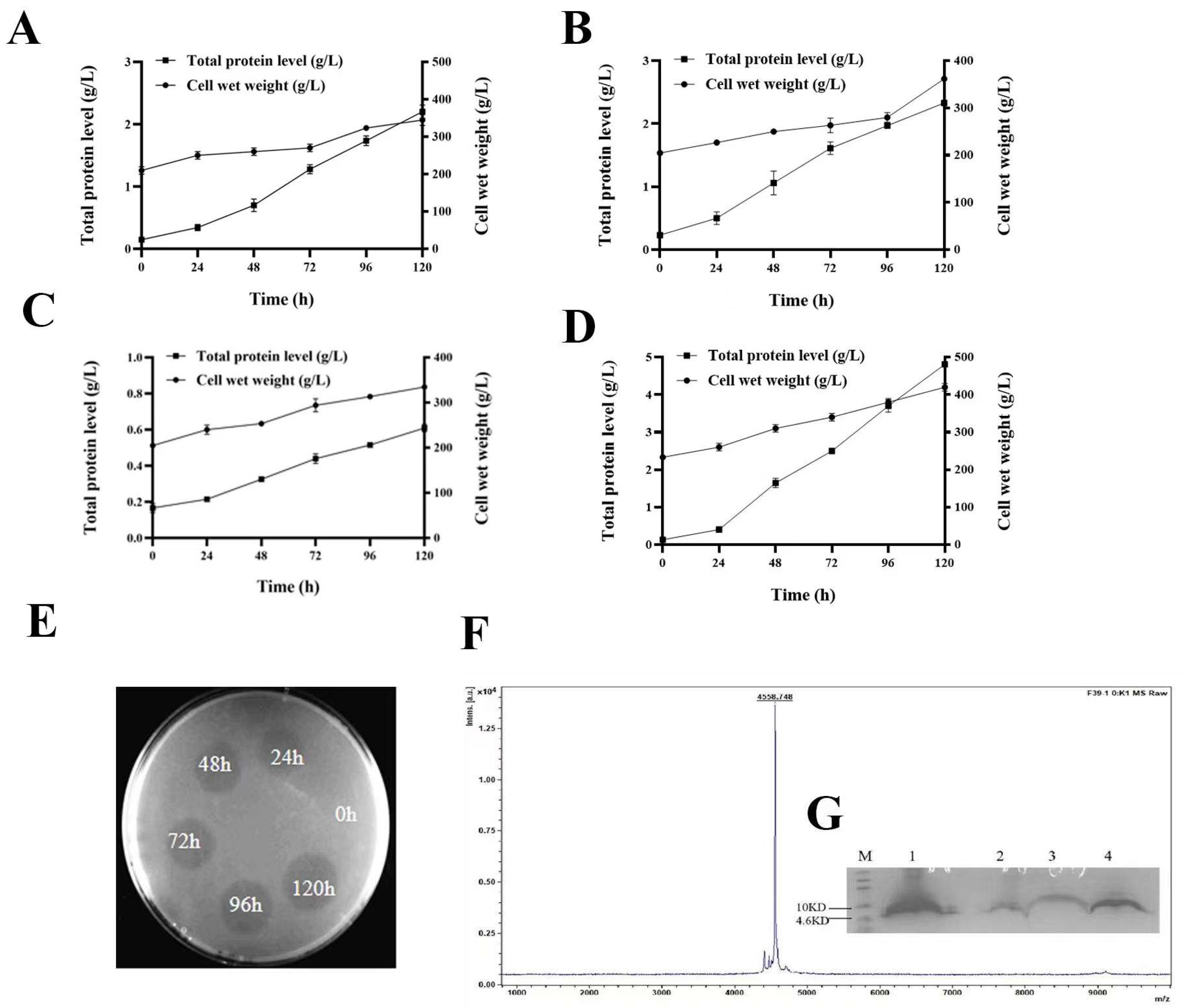
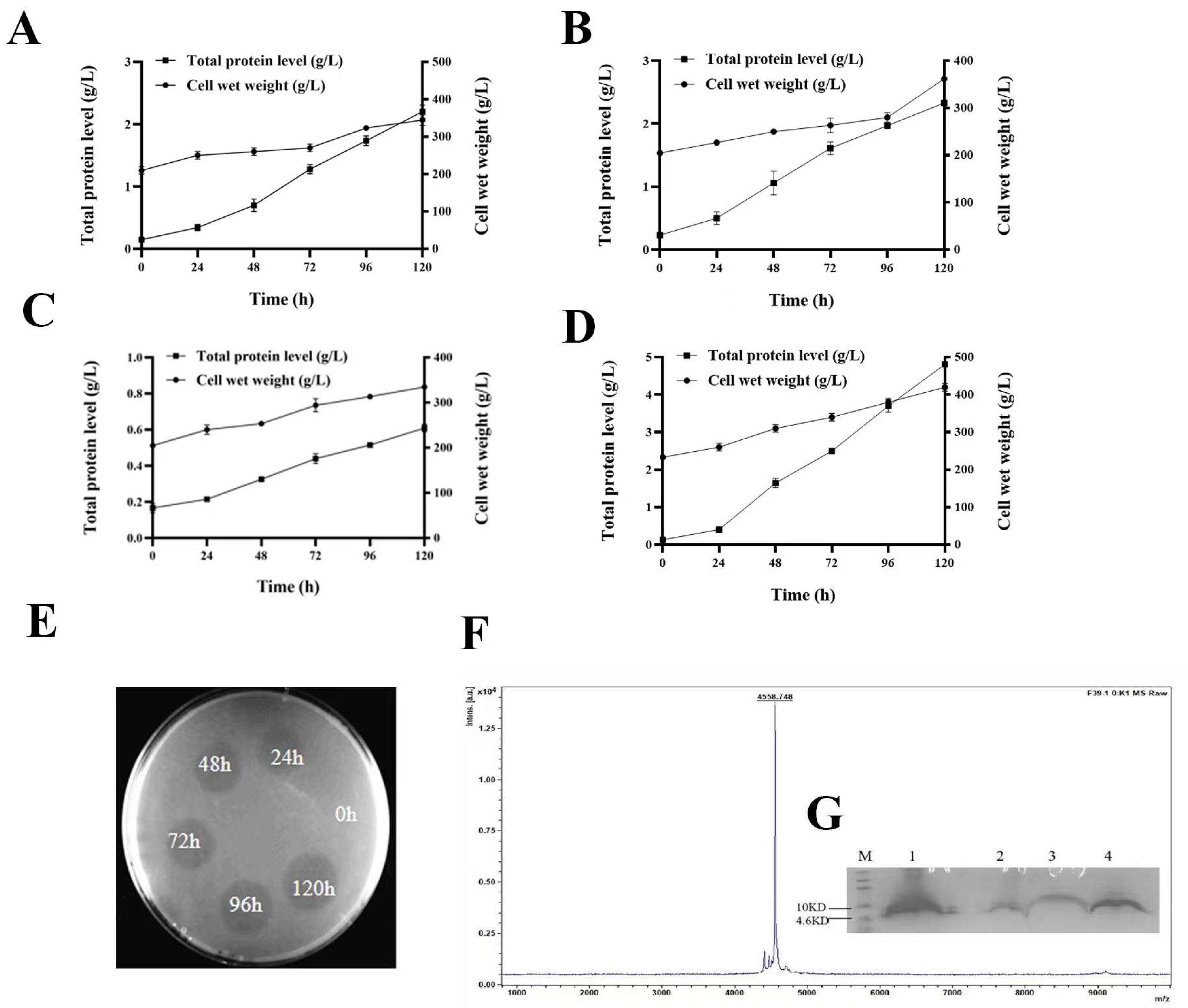
2.3. Expression of W25, R33, G1, and F39 in Fermenter Level
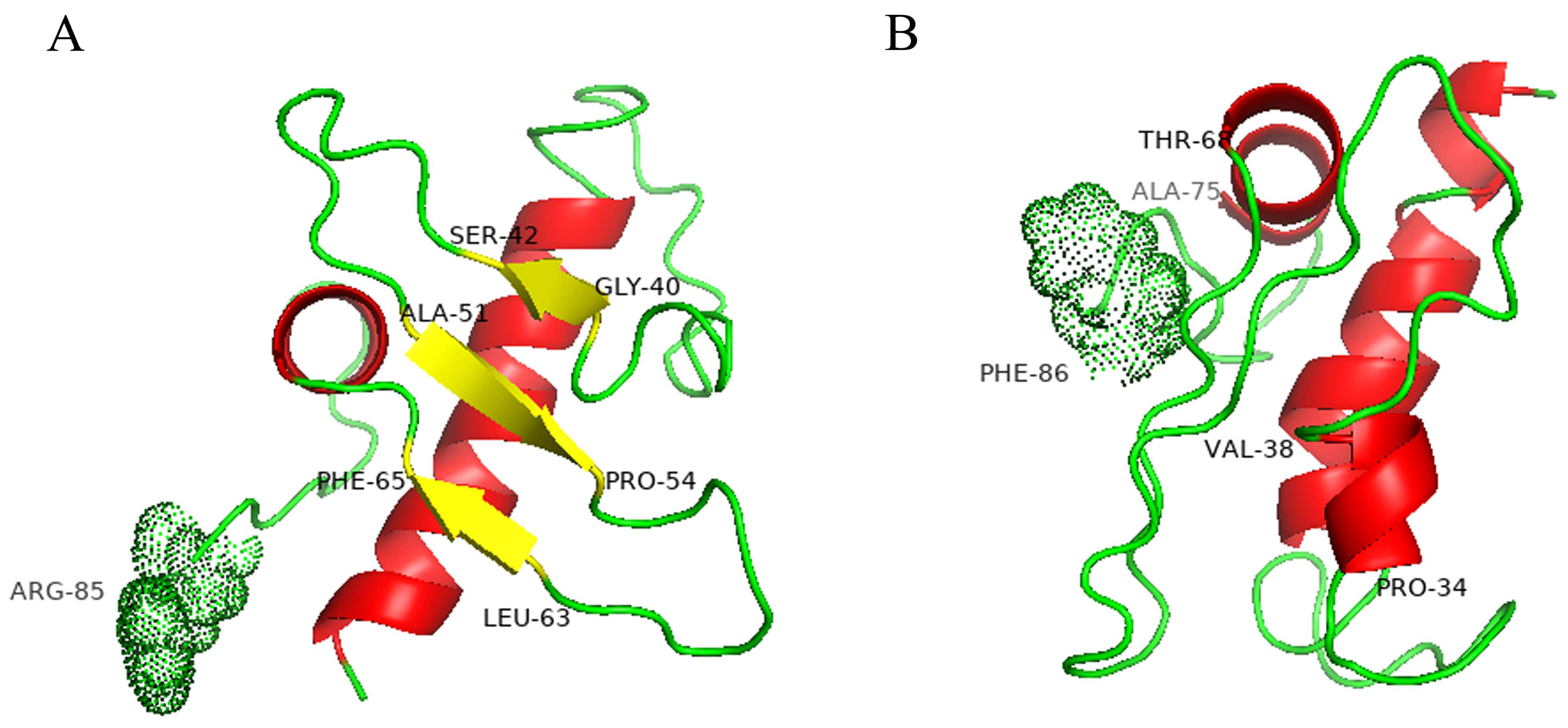
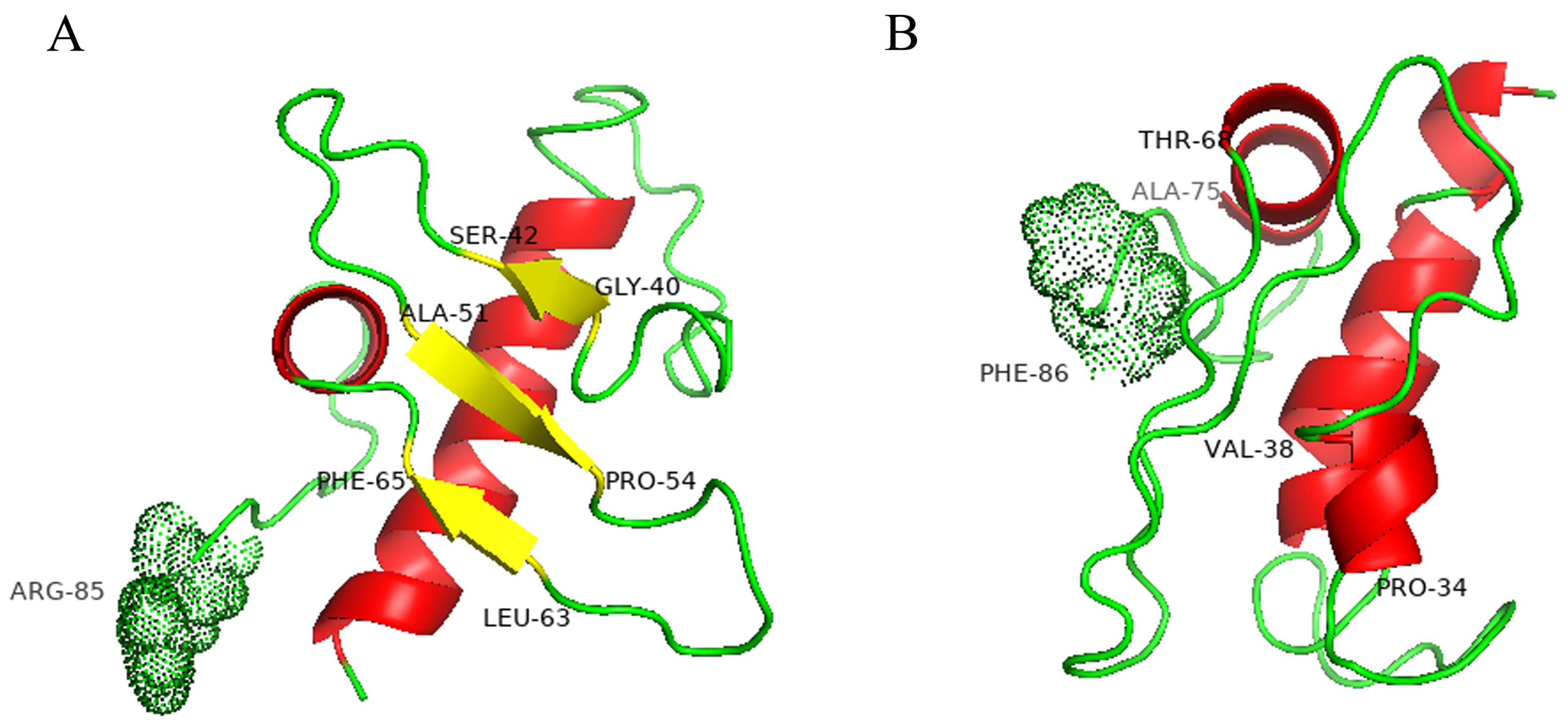
2.4. Model Structures of Wild-Type and Mutant Pro-Signal Peptides
2.5. Purification of FNZ
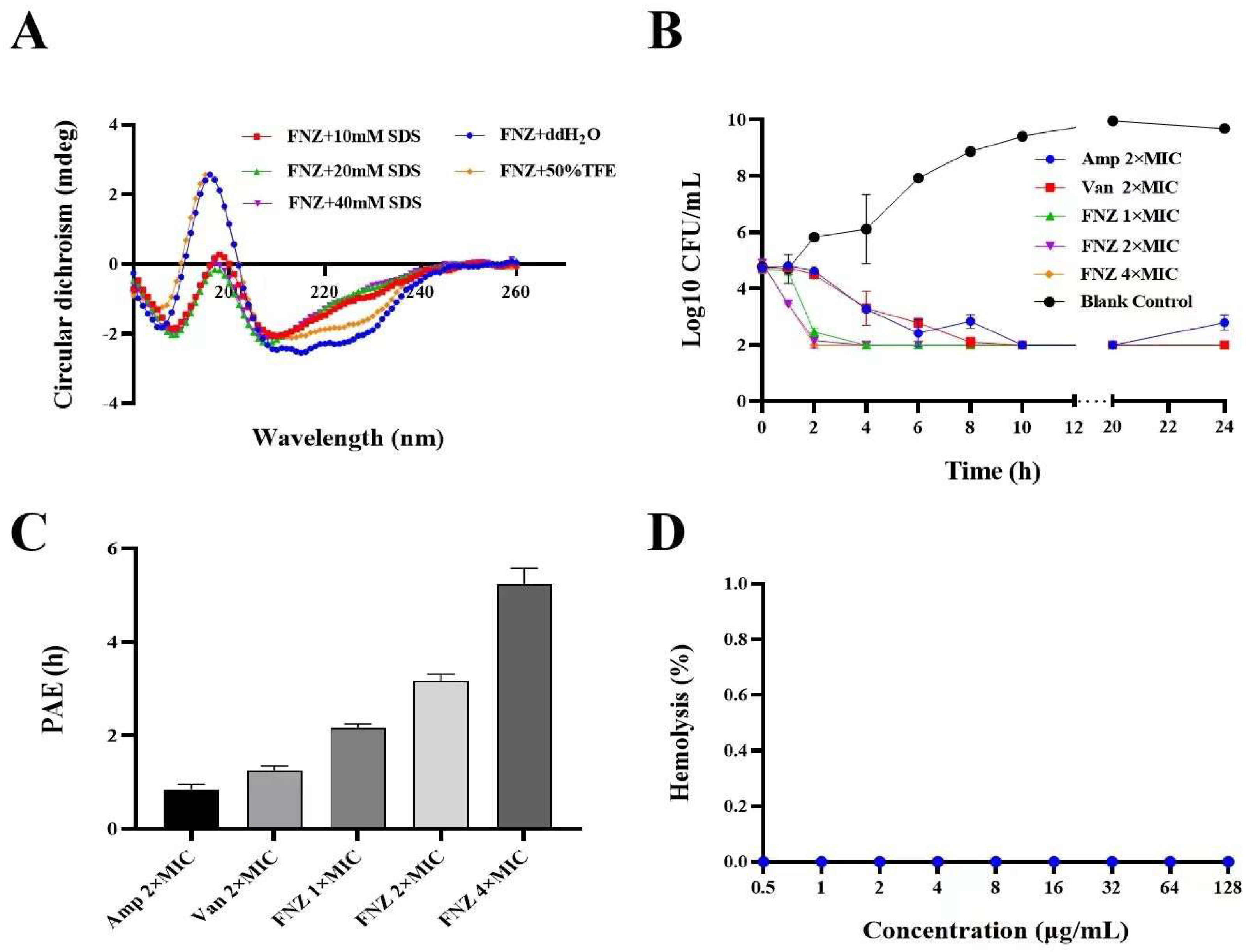
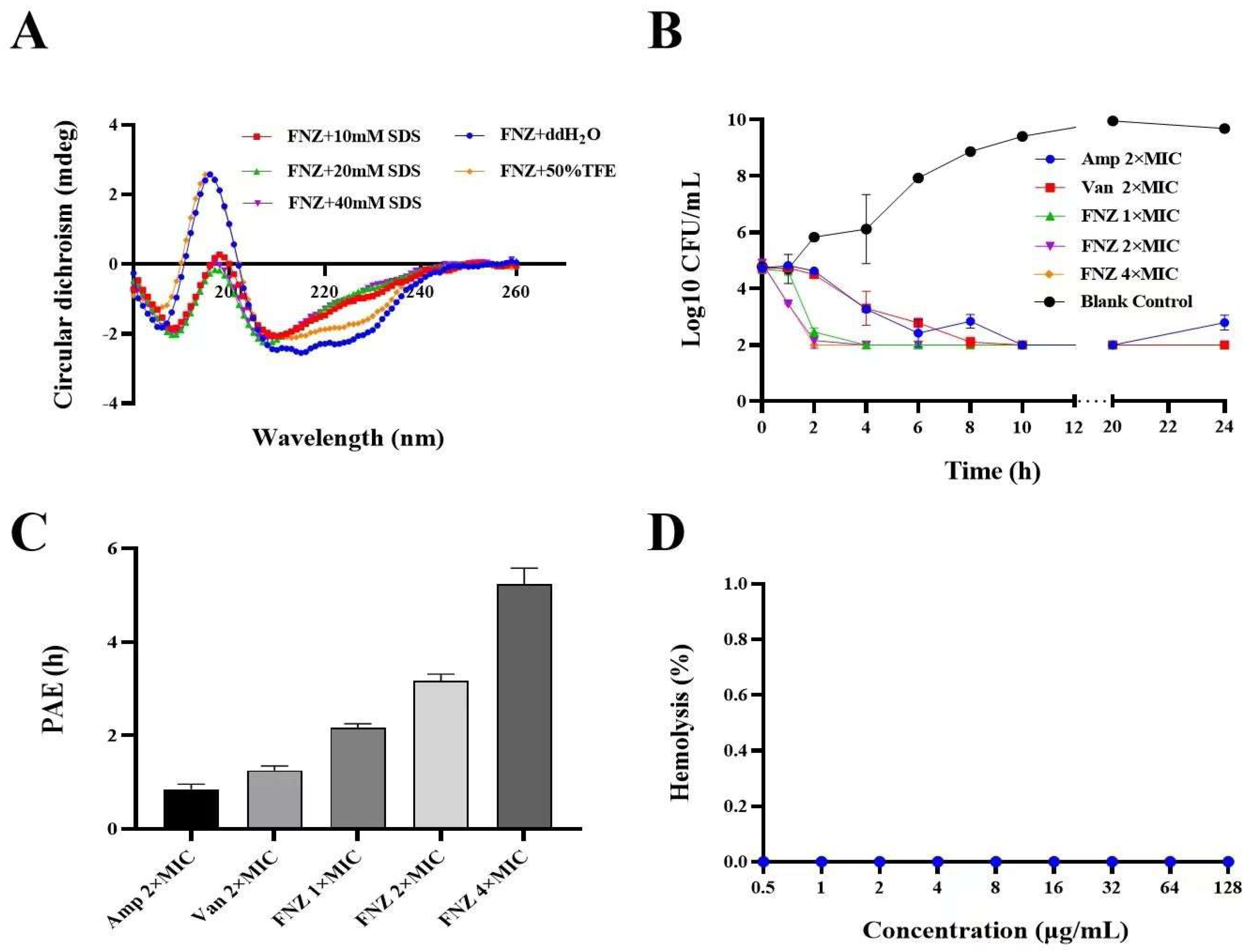
2.6. Secondary Structure Determination of FNZ
2.7. Antimicrobial Activity Assays of FNZ
2.7.1. Minimal Inhibitory Concentrations (MICs) of FNZ
2.7.2. Synergism Assays of FNZ with Traditional Antibiotics
2.7.3. Bactericidal Kinetics Assay
2.7.4. The Post−Antibiotic Effect (PAE) of FNZ against S. aureus
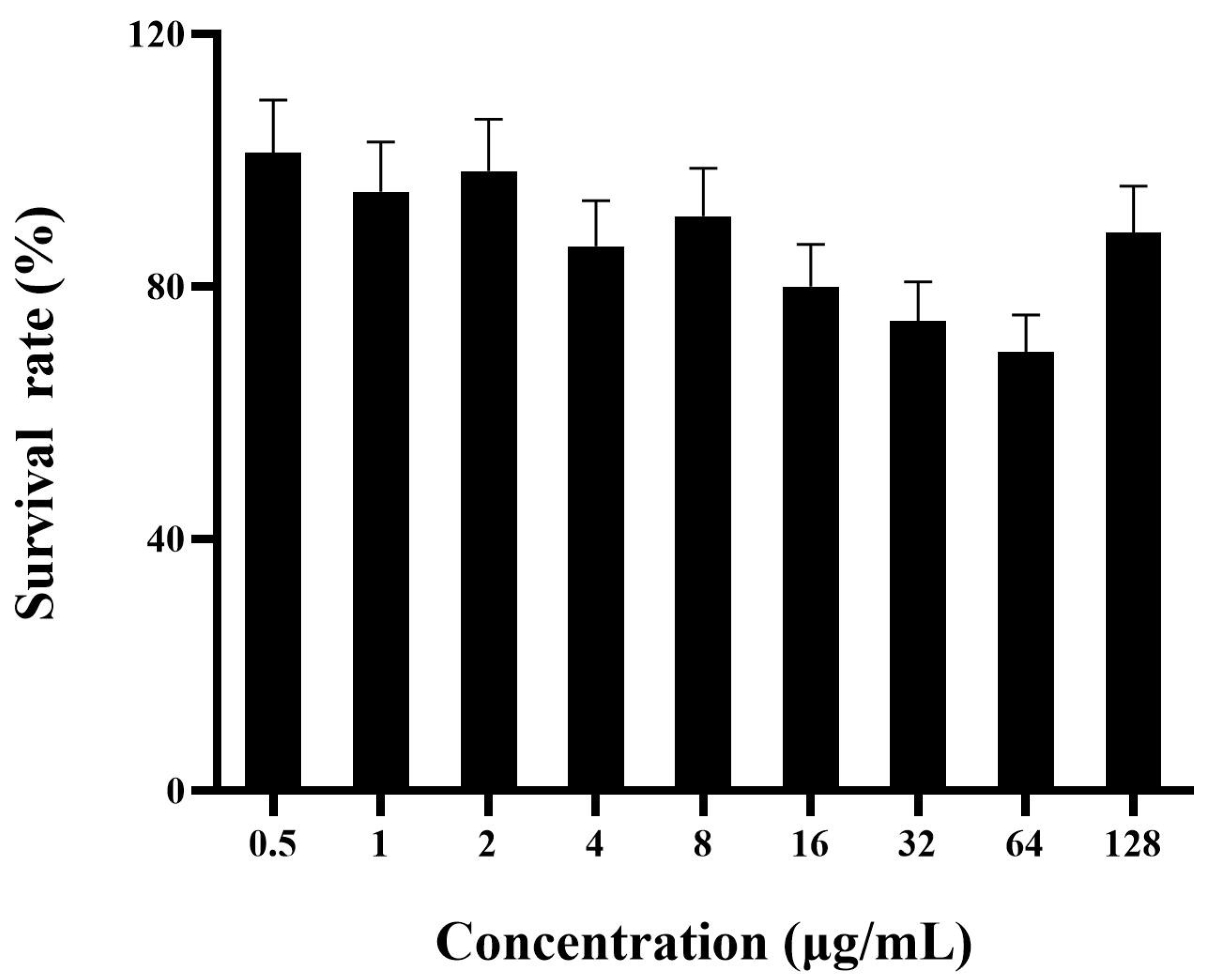
2.8. Hemolysis, Cytotoxicity, and Stability of FNZ
2.8.1. Hemolytic Assay and Cytotoxicity
2.8.2. Thermal, pH, Salts, and Protease Stability of FNZ
3. Discussion
4. Materials and Methods
4.1. Plasmids, Strains, and Reagents
4.2. Construction of the Expression Vector pPICZαA-X-NZ2114
4.3. Expression of the XNZ in P. pastoris in 48-Well Plates and Shake Flasks
4.4. High-Density Cultivation and Purification
4.5. Structure Modeling of Wild-Type and Mutant Pro-Signal Peptides
4.6. Secondary Structure Determination of FNZ
4.7. Antimicrobial Characteristics of FNZ
4.7.1. Minimal Inhibitory Concentration (MIC)
4.7.2. In Vitro Pharmacodynamics of FNZ
4.7.3. The PAE of FNZ
4.7.4. Drug Synergism Assays of FNZ with Traditional Antibiotics
4.8. Hemolysis, Cytotoxicity, and Stability of FNZ
4.8.1. Hemolysis
4.8.2. Cytotoxicity
4.8.3. Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef]
- Costa, C.R.D.M.D.; Feitosa, M.L.T.; Pessoa, G.T.; Bezerra, D.D.O.; Ferraz, M.S.; Carvalho, M.A.M.D. Mastite caprina: Etiologia e epidemiologia: Revisão de literaturea. Pubvet 2013, 7, 1530. [Google Scholar] [CrossRef]
- Shang, C. Identification and diagnosis of avian infectious diseases with arthritis symptoms. China Anim. Husb. 2016, 11, 81–82. (In Chinese) [Google Scholar] [CrossRef]
- Petinaki, E.; Spiliopoulou, I. Methicillin-resistant Staphylococcus aureus among companion and food-chain animals: Impact of human contacts. Clin. Microbiol. Infect. 2012, 18, 626–634. [Google Scholar] [CrossRef]
- Mendelson, M.; Matsoso, M.P. The world health organization global action plan for antimicrobial resistance. S. Afr. Med. J. 2015, 105, 325. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.W.; Dungan, R.S.; Moore, A.; Leytem, A.B. Occurrence and abundance of antibiotic resistance genes in agricultural soil receiving dairy manure. FEMS Microbiol. Ecol. 2018, 94, 10. [Google Scholar] [CrossRef]
- Esmaeil Amini, M.; Shomali, N.; Bakhshi, A.; Rezaei, S.; Hemmatzadeh, M.; Hosseinzadeh, R.; Eslami, S.; Babaie, F.; Aslani, S.; Torkamandi, S.; et al. Gut microbiome and multiple sclerosis: New insights and perspective. Int. Immunopharmacol. 2020, 88, 107024. [Google Scholar] [CrossRef]
- Cong, Y.; Yang, S.; Rao, X. Vancomycin resistant Staphylococcus aureus infections: A review of case updating and clinical features. J. Adv. Res. 2019, 21, 169–176. [Google Scholar] [CrossRef]
- Smith, T.L.; Pearson, M.L.; Wilcox, K.R.; Cruz, C.; Lancaster, M.V.; Robinson-Dunn, B.; Tenover, F.C.; Zervos, M.J.; Band, J.D.; White, E.; et al. Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-intermediate Staphylococcus aureus working group. N. Engl. J. Med. 1999, 340, 493–501. [Google Scholar] [CrossRef]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with vancomycin-resistant Staphylococcus aureus containing the van A resistance gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef]
- Tenover, F.C.; Weigel, L.M.; Appelbaum, P.C.; McDougal, L.K.; Chaitram, J.; McAllister, S.; Clark, N.; Killgore, G.; Hara, C.M.; Jevitt, L.; et al. Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrob. Agents Chemother. 2004, 48, 275–280. [Google Scholar] [CrossRef] [PubMed]
- WHO publishes list of bacteria for which new antibiotics are urgently needed. Neurosciences 2017, 38, 444–445.
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, J.H.; de la Fuente-Nunez, C.; Franco, O.L. Editorial: Antimicrobial peptides: Molecular design, structure-function relationship, and biosynthesis optimization. Front. Microbiol. 2022, 13, 888540. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Aminov, R.; Franco, O.L.; de la Fuente-Nunez, C.; Wang, J.H. Editorial: Community series in antimicrobial peptides: Molecular design, structure function relationship and biosynthesis optimization. Front. Microbiol. 2023, 14, 1125426. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sönksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Kruse, T.; Wimmer, R.; Wiedemann, I.; Sass, V.; Pag, U.; Jansen, A.; Nielsen, A.K.; Mygind, P.H.; Raventós, D.S.; et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 2010, 328, 1168–1172. [Google Scholar] [CrossRef]
- Zhang, Y.; Teng, D.; Mao, R.; Wang, X.; Xi, D.; Hu, X.; Wang, J. High expression of a plectasin-derived peptide NZ2114 in Pichia pastoris and its pharmacodynamics, postantibiotic and synergy against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2014, 98, 681–694. [Google Scholar] [CrossRef]
- Yang, N.; Teng, D.; Mao, R.; Hao, Y.; Wang, X.; Wang, Z.; Wang, X.; Wang, J. A recombinant fungal defensin-like peptide-P2 combats multidrug-resistant Staphylococcus aureus and biofilms. Appl. Microbiol. Biotechnol. 2019, 103, 5193–5213. [Google Scholar] [CrossRef]
- Liu, H.; Yang, N.; Mao, R.; Teng, D.; Hao, Y.; Wang, X.; Wang, J. A new high-yielding antimicrobial peptide NZX and its antibacterial activity against Staphylococcus hyicus in vitro/vivo. Appl. Microbiol. Biotechnol. 2020, 104, 1555–1568. [Google Scholar] [CrossRef]
- Zhao, F.; Yang, N.; Wang, X.; Mao, R.; Hao, Y.; Li, Z.; Wang, X.; Teng, D.; Fan, H.; Wang, J. In vitro/vivo mechanism of action of MP1102 with low/nonresistance against Streptococcus suis type 2 strain CVCC 3928. Front. Cell. Infect. Microbiol. 2019, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, N.; Mao, R.; Hao, Y.; Ma, X.; Teng, D.; Fan, H.; Wang, J. A recombinant fungal defensin-like peptide-P2 combats Streptococcus dysgalactiae and biofilms. Appl. Microbiol. Biotechnol. 2021, 105, 1489–1504. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, N.; Mao, R.; Hao, Y.; Teng, D.; Wang, J. In vitro pharmacodynamics and bactericidal mechanism of fungal defensin derived peptides NZX and P2 against Streptococcus agalactiae. Microorganisms 2022, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Teng, D.; Mao, R.; Yang, N.; Hao, Y.; Wang, J. Thinking on the construction of antimicrobial peptide databases: Powerful tools for the molecular design and screening. Int. J. Mol. Sci. 2023, 24, 3134. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Deng, T.; Ge, H.; He, H.; Liu, Y.; Zhai, C.; Feng, L.; Yi, L. The heterologous expression strategies of antimicrobial peptides in microbial systems. Protein Expr. Purif. 2017, 140, 52–59. [Google Scholar] [CrossRef]
- Macauley-Patrick, S.; Fazenda, M.L.; Mcneil, B.; Harvey, L.M. Heterologous protein production using the Pichia pastoris expression system. Yeast 2010, 22, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.; Craig, W.; Nielsen, L.A.; Kristensen, H.H. In vivo pharmacodynamic characterization of a novel plectasin antibiotic, NZ2114, in a murine infection model. Antimicrob. Agents Chemother. 2009, 53, 3003–3009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Teng, D.; Tian, Z.; Wang, S.; Wang, J. Expression of plectasin in Pichia pastoris and its characterization as a new antimicrobial peptide against Staphyloccocus and Streptococcus. Protein Expres. Purif. 2011, 78, 189–196. [Google Scholar] [CrossRef]
- Mizuno, K.; Nakamura, T.; Ohshima, T.; Tanaka, S.; Matsuo, H. Characterization of Kex2-encoded endopeptidase from yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1989, 159, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.S.; Sterne, R.E.; Thorner, J. Enzymes required for yeast prohormone processing. Annu. Rev. Physiol. 1988, 50, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Bevan, A.; Brenner, C.; Fuller, R.S. Quantitative assessment of enzyme specificity in vivo: P2 recognition by Kex2 protease defined in a genetic system. Proc. Natl. Acad. Sci. USA 1998, 95, 10384–10389. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Krysan, D.J.; Komiyama, T.; Fuller, R.S. Precursor processing by kex2/furin proteases. Chem. Rev. 2002, 102, 4525–4548. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, N.C.; Wang, G.T.; Krafft, G.A.; Fuller, R.S. Internally consistent libraries of fluorogenic substrates demonstrate that Kex2 protease specificity is generated by multiple mechanisms. Biochemistry 1997, 36, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kuang, Y.; Li, H.; Liu, Y.; Hui, X.; Li, P.; Jiang, Z.; Zhou, Y.; Wang, Y.; Xu, A.; et al. Enhanced production of recombinant secretory proteins in Pichia pastoris by optimizing Kex2 P1’ site. PLoS ONE 2013, 8, e75347. [Google Scholar] [CrossRef]
- Manfredi, M.A.; Antunes, A.A.; Jesus, L.O.; Juliano, M.A.; Juliano, L.; Judice, W.A. Specificity characterization of the α-mating factor hormone by Kex2 protease. Biochimie 2016, 131, 149–158. [Google Scholar] [CrossRef]
- Liu, W.C.; Inwood, S.; Gong, T.; Sharma, A.; Yu, L.Y.; Zhu, P. Fed-batch high-cell-density fermentation strategies for Pichia pastoris growth and production. Crit. Rev. Biotechnol. 2019, 39, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.M.; Nguyen, T.T.; Nguyen, C.T.; Huynh-Thi, X.M.; Nguyen, C.T.; Trinh, M.T.; Tran, L.T.; Cartwright, S.P.; Bill, R.M.; Tran-Van, H. Pichia pastoris versus Saccharomyces cerevisiae: A case study on the recombinant production of human granulocyte-macrophage colony-stimulating factor. BMC Res. Notes 2017, 10, 148. [Google Scholar] [CrossRef]
- Yang, J.; Rao, X.; Hou, R.; Hu, X.; Chen, Z.; Cong, Y.; Tan, Y.; Zhu, J.; Zhou, Y.; Hu, F. Expression of TG-TPase chimeric gene from Staphylococcus aureus in methanotrophic yeast. J. Immunol. 2009, 25, 145–149. [Google Scholar] [CrossRef]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef]
- Brake, A.J. Alpha-factor leader-directed secretion of heterologous proteins from yeast. Methods Enzymol. 1990, 185, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yi, L.; Hoi, K.H.; Marek, P.; Georgiou, G.; Iverson, B.L. Profiling protease specificity: Combining yeast ER sequestration screening (YESS) with next generation sequencing. ACS Chem. Biol. 2017, 12, 510–518. [Google Scholar] [CrossRef]
- Aggarwal, S.; Mishra, S. Modifications in the Kex2 P1’ cleavage site in the α-MAT secretion signal leads to higher production of human granulocyte colony-stimulating factor in Pichia pastoris. World J. Microbiol. Biotechnol. 2021, 37, 197. [Google Scholar] [CrossRef]
- Mate, D.M.; Garcia-Ruiz, E.; Camarero, S.; Shubin, V.V.; Falk, M.; Shleev, S. Switching from blue to yellow: Altering the spectral properties of a high redox potential laccase by directed evolution. Biocatal. Biotransformation 2013, 31, 8–21. [Google Scholar] [CrossRef]
- Lin-Cereghino, G.P.; Stark, C.M.; Kim, D.; Chang, J.; Shaheen, N.; Poerwanto, H.; Agari, K.; Moua, P.; Low, L.K.; Tran, N. The effect of α-mating factor secretion signal mutations on recombinant protein expression in Pichia pastoris. Gene 2013, 519, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Aza, P.; Molpeceres, G.; de Salas, F.; Camarero, S. Design of an improved universal signal peptide based on the α-factor mating secretion signal for enzyme production in yeast. Cell. Mol. Life Sci. 2021, 78, 3691–3707. [Google Scholar] [CrossRef]
- Barlowe, C.K.; Miller, E.A. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 2013, 193, 383–410. [Google Scholar] [CrossRef]
- Guo, Y.; Teng, D.; Wang, X.; Mao, R.; Hao, Y.; Fan, H.; Wang, J. Effect of antimicrobial peptide NZ2114 on cell membrane, genome and antimicrobial resistance of Staphylococcus aureus isolated from bovine mastitis milk. China Ani. Husban. Vet. Med. 2019, 46, 1181–1190. (In Chinese) [Google Scholar] [CrossRef]
- Hara, S.; Mukae, H.; Sakamoto, N.; Ishimoto, H.; Amenomori, M.; Fujita, H.; Ishimatsu, Y.; Yanagihara, K.; Kohno, S. Plectasin has antibacterial activity and no effect on cell viability or IL-8 production. Biochem. Biophys. Res. Commun. 2008, 374, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Giguère, S.; Lee, E.A.; Guldbech, K.M.; Berghaus, L.J. In vitro synergy, pharmacodynamics, and postantibiotic effect of 11 antimicrobial agents against Rhodococcus equi. Vet. Microbiol. 2012, 160, 207–213. [Google Scholar] [CrossRef]
- Uzogara, E.; Sheehan, D.V.; Manschreck, T.C.; Jones, K.J. A combination drug treatment for acute common migraine. Headache 1986, 26, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Deresinski, S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clin. Infect. Dis. 2009, 49, 1072–1079. [Google Scholar] [CrossRef]
- Eckert, R. Road to clinical efficacy: Challenges and novel strategies for antimicrobial peptide development. Future Microb. 2011, 6, 635–651. [Google Scholar] [CrossRef]
- Leibovici, L.; Paul, M.; Andreassen, S. Balancing the benefits and costs of antibiotic drugs: The TREAT model. Clin. Microbiol. Infect. 2010, 16, 1736–1739. [Google Scholar] [CrossRef]
- Hills, R.D.; Brooks, C.L., III. Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 2007, 368, 894–901. [Google Scholar] [CrossRef]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, S.K.; Larson, R.G. Effect of salt on the interactions of antimicrobial peptides with zwitterionic lipid bilayers. BBA 2006, 1758, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Sugiarto, H.; Yu, P.L. Effects of cations on antimicrobial activity of ostricacins-1 and 2 on E. coli O157:H7 and S. aureus 1056MRSA. Curr. Microbiol. 2007, 55, 36–41. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Y.; Mao, R.; Teng, D.; Wang, X.; Wang, J. Design and recombination expression of a novel plectasin-derived peptide MP1106 and its properties against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2015, 99, 2649–2662. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Tan, P.; Lai, Z.; Zhu, Y.; Shao, C.; Akhtar, M.U.; Li, W.; Zheng, X.; Shan, A. Multiple strategy optimization of specifically targeted antimicrobial peptide based on structure-activity relationships to enhance bactericidal efficiency. ACS Biomater. Sci. Eng. 2020, 6, 398–414. [Google Scholar] [CrossRef]
- Saputra, S.K.; Sutantyo, D.; Farmasyanti, C.A.; Alhasyimi, A.A. The effect of the addition of propolis to resin-modified glass ionomer cement bracket adhesive materials on the growth inhibition zone of Streptococcus mutans. F1000Research 2019, 8, 2105. [Google Scholar] [CrossRef]
- Terwee, C.B.; Peipert, J.D.; Chapman, R.; Lai, J.S.; Terluin, B.; Cella, D.; Griffith, P.; Mokkink, L.B. Minimal important change (MIC): A conceptual clarification and systematic review of MIC estimates of PROMIS measures. Qual. Life. Res. 2021, 30, 2729–2754. [Google Scholar] [CrossRef]
- Tessier, P.R.; Nightingale, C.H.; Nicolau, D.P. Postantibiotic effect of trovafloxacin against Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis in cerebrospinal fluid and broth culture media. Diagn. Microbiol. Infect. Dis. 2000, 36, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, B.T.; Rybak, M.J. Etest synergy testing of clinical isolates of Staphylococcus aureus demonstrating heterogeneous resistance to vancomycin. Diagn. Microbiol. Infect. Dis. 2006, 54, 73–77. [Google Scholar] [CrossRef]
- White, R.L.; Burgess, D.S.; Manduru, M.; Bosso, J.A. Comparison of three different in vitro methods of detecting synergy: Time-kill, checkerboard, and E test. Antimicrob. Agents Chemother. 1996, 40, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.M.; Teng, D.; Mao, R.; Hao, Y.; Yang, N.; Li, Z.; Wang, J. Increased intracellular activity of MP1102 and NZ2114 against Staphylococcus aureus in vitro and in vivo. Sci. Rep. 2018, 8, 4204. [Google Scholar] [CrossRef]
- Wu, G.; Ding, J.; Li, H.; Li, L.; Zhao, R.; Shen, Z.; Fan, X.; Xi, T. Effects of cations and pH on antimicrobial activity of thanatin and s-thanatin against Escherichia coli ATCC25922 and B. subtilis ATCC 21332. Curr. Microbiol. 2008, 57, 552–557. [Google Scholar] [CrossRef]
- Benincasa, M.; Pelillo, C.; Zorzet, S.; Garrovo, C.; Biffi, S.; Gennaro, R.; Scocchi, M. The proline-rich peptide Bac7(1-35) reduces mortality from Salmonella typhimurium in a mouse model of infection. BMC Microbiol. 2010, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Zhu, X.; Wang, J.; Dong, N.; Shan, A. Novel design of heptad amphiphiles to enhance cell selectivity, salt resistance, antibiofilm properties and their membrane-disruptive mechanism. J. Med. Chem. 2017, 60, 2257–2270. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | MIC | |||||||
|---|---|---|---|---|---|---|---|---|
| FNZ | NZ2114 | Ampicillin | Vancomycin | |||||
| μg/mL | μM | μg/mL | μM | μg/mL | μM | μg/mL | μM | |
| Gram-positive bacteria | ||||||||
| Staphylococcus aureus ATCC 43300 | 8 | 1.75 | 4 | 0.91 | 2 | 2.48 | 1 | 0.67 |
| S. aureus ATCC 25923 | 8 | 1.75 | 4 | 0.91 | 0.25 | 0.62 | 4 | 2.69 |
| S. aureus CVCC 546 | 8 | 1.75 | 8 | 1.81 | 0.25 | 0.62 | 1 | 0.67 |
| S. aureus E48 | 4 | 0.88 | NT | NT | 0.25 | 0.62 | 1 | 0.67 |
| S. hyicus ACCC 61734 | 8 | 1.75 | 4 | 0.91 | NT | NT | NT | NT |
| S. epidermidis ATCC 12228 | 8 | 1.75 | NT | NT | 2 | 2.48 | 1 | 0.67 |
| S. agalactiae CAU-FRI 4 | 4 | 0.88 | 2 | 0.45 | NT | NT | NT | NT |
| Gram-negative bacteria | ||||||||
| Escherichia coli ATCC 25922 | >128 | >28.05 | >128 | >28.97 | 1 | 1.24 | 128 | 86 |
| Salmonella enterica ATCC 13076 | >128 | >28.05 | >128 | >28.97 | NT | NT | >128 | >86 |
| Pseudomonas aeruginosa CICC 20625 | >128 | >28.05 | >128 | >28.97 | NT | NT | >128 | >86 |
| Combination | Variety | S. aureus ATCC 43300 | |||
|---|---|---|---|---|---|
| MICa (μg/mL) | MICc (μg/mL) | FIC | FICI | ||
| FNZ + Amp | FNZ | 8 | 0.125 | 0.016 | 0.078 |
| Amp | 2 | 0.125 | 0.063 | ||
| FNZ + Kan | FNZ | 8 | 0.5 | 0.063 | 0.07 |
| Kan | 64 | 0.5 | 0.008 | ||
| FNZ + Nisin | FNZ | 8 | 0.5 | 0.078 | 0.328 |
| Nisin | 4 | 1 | 0.25 | ||
| FNZ + Cip | FNZ | 8 | 0.25 | 0.031 | 1.531 |
| Cip | 0.25 | 0.375 | 1.5 | ||
| FNZ + Van | FNZ | 8 | 1 | 0.125 | 0.25 |
| Van | 1 | 0.125 | 0.125 | ||
| Treatment | MIC | |||
|---|---|---|---|---|
| FNZ | Vancomycin | |||
| μg/mL | μM | μg/mL | μM | |
| Temperature (°C) | ||||
| 20 | 8 | 1.75 | 1 | 0.67 |
| 40 | 8 | 1.75 | 1 | 0.67 |
| 60 | 8 | 1.75 | 1 | 0.67 |
| 80 | 8 | 1.75 | 1 | 0.67 |
| 100 | 16 | 3.5 | 1 | 0.67 |
| pH | ||||
| 2 | 8 | 1.75 | 1 | 0.67 |
| 4 | 8 | 1.75 | 1 | 0.67 |
| 6 | 8 | 1.75 | 1 | 0.67 |
| 8 | 8 | 1.75 | 1 | 0.67 |
| 10 | 16 | 3.5 | 1 | 0.67 |
| Salts | ||||
| 50 mM Na+ | 8 | 1.75 | 1 | 0.67 |
| 100 mM Na+ | 8 | 1.75 | 1 | 0.67 |
| 150 mM Na+ | 8 | 1.75 | 1 | 0.67 |
| 300 mM Na+ | 8 | 1.75 | 1 | 0.67 |
| 500 mM Na+ | 16 | 3.5 | 1 | 0.67 |
| 50 mM Mg2+ | 8 | 1.75 | 1 | 0.67 |
| 100 mM Mg2+ | 8 | 1.75 | 1 | 0.67 |
| 150 mM Mg2+ | 8 | 1.75 | 1 | 0.67 |
| 300 mM Mg2+ | 16 | 3.5 | 1 | 0.67 |
| 500 mM Mg2+ | >32 | >7 | 1 | 0.67 |
| Simulated Gastric Fluid (1 h) | 8 | 1.75 | 1 | 0.67 |
| Artificial Intestinal Fluid (10 min) | >16 | >3.5 | 1 | 0.67 |
| Serum (25%) (1–4 h) | 8 | 1.75 | 1 | 0.67 |
| Index | FNZ | NZ2114 |
|---|---|---|
| Expression level | ||
| Yield (g/L) | 4.81 a | 2.31 b |
| Specific production rates (mg/g/h) | 0.095 a | 0.038 b |
| Druggability | ||
| MICs (μg/mL) | S. aureus: 4–8 a S. hyicus: 8 a S. agalactiae: 4 a | S. aureus: 4–8 a S. hyicus: 4 a S. agalactiae: 2 a |
| Bactericidal kinetics | >99% S. aureus were killed a No regrowth within 24 h a | >99% S. aureus were killed b Bacterial regrowth was observed after 6 h of inoculation b |
| PAE (h) | 2.18, 3.13, and 5.27 h at 1× MIC, 2× MIC, and 4× MIC a | 1.7, 2.6, and 3.5 h at 1× MIC, 2× MIC, and 4× MIC b |
| Synergism | Synergistic effect with ampicillin, kanamycin, vancomycin, and nisin a | Synergistic effect with ampicillin, kanamycin, vancomycin, and streptomycin b |
| Hemolysis (%) | 0% a | Less than 0.1% b |
| Cell viability (%) | 88.56% a at 128 μg/mL | 52.13% c at 128 μg/mL |
| Thermal stability | Stable at 20 to 80 °C a 50% activity at 100 °C a | Stable at 20 to 80 °C b 20% activity at 100 °C |
| pH stability | Stable at pH 2–8 a 50% activity at pH 10 a | High activity at pH 8 and 10 b Low activity at pH 2 to 6 b |
| Salts stability | Stable at 50–300 mM Na+ a 50% activity at 500 mM Na+ a Stable at 50–150 mM Mg2+ a 50% activity at 300 mM Mg2+ a Lower than 50% activity at 500 mM Mg2+ a | NT |
| Serum stability | Stablea | NT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Yang, N.; Teng, D.; Hao, Y.; Mao, R.; Wang, J. Molecular Modification of Kex2 P1’ Site Enhances Expression and Druggability of Fungal Defensin. Antibiotics 2023, 12, 786. https://doi.org/10.3390/antibiotics12040786
Jin Y, Yang N, Teng D, Hao Y, Mao R, Wang J. Molecular Modification of Kex2 P1’ Site Enhances Expression and Druggability of Fungal Defensin. Antibiotics. 2023; 12(4):786. https://doi.org/10.3390/antibiotics12040786
Chicago/Turabian StyleJin, Yanjie, Na Yang, Da Teng, Ya Hao, Ruoyu Mao, and Jianhua Wang. 2023. "Molecular Modification of Kex2 P1’ Site Enhances Expression and Druggability of Fungal Defensin" Antibiotics 12, no. 4: 786. https://doi.org/10.3390/antibiotics12040786
APA StyleJin, Y., Yang, N., Teng, D., Hao, Y., Mao, R., & Wang, J. (2023). Molecular Modification of Kex2 P1’ Site Enhances Expression and Druggability of Fungal Defensin. Antibiotics, 12(4), 786. https://doi.org/10.3390/antibiotics12040786