
Additive Anticonvulsant Profile and Molecular Docking Analysis of 5,5′-Diphenylhydantoin Schiff Bases and Phenytoin

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Chemicals and Instrumentation
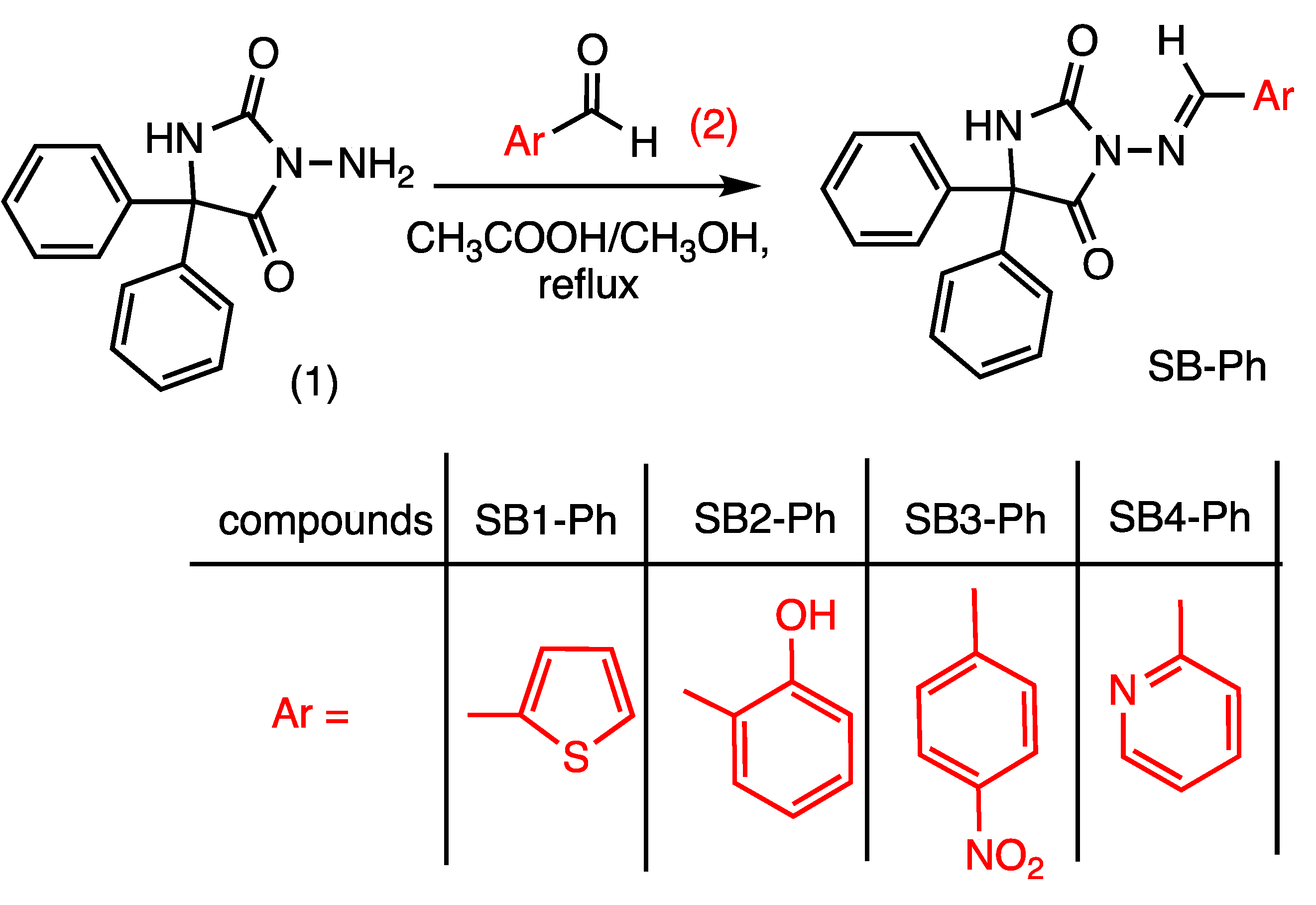
2.2. General Procedure for the Synthesis of 3-Amino-5,5′-diphenylhydantoin Schiff Base Compounds SB1-Ph, SB2-Ph, SB3-Ph, SB4-Ph
2.3. Animals and Experimental Design
2.4. MES Test
2.5. Grip Strength Test
2.6. Rota-Rod Test
2.7. Measurement of Spontaneous Motor Activity
2.8. Docking of Phenytoin Schiff Bases on Opioid Receptors
2.9. Statistical Analysis
3. Results
3.1. Chemistry
3.2. Anticonvulsant Activity
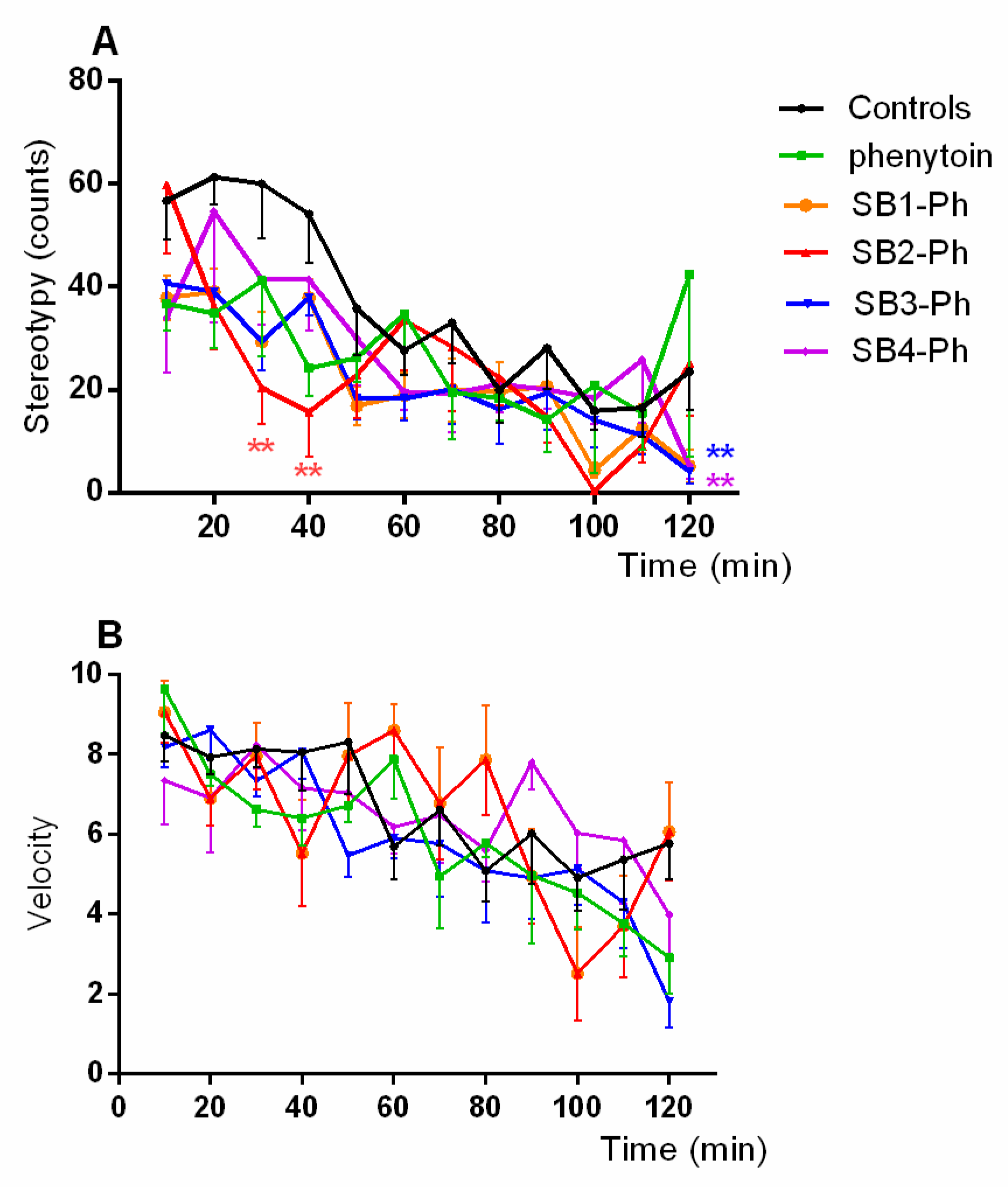
3.3. Muscle Strength and Spontaneous Motor Activity
3.3.1. Muscle Strength
3.3.2. Spontaneous Motor Activity
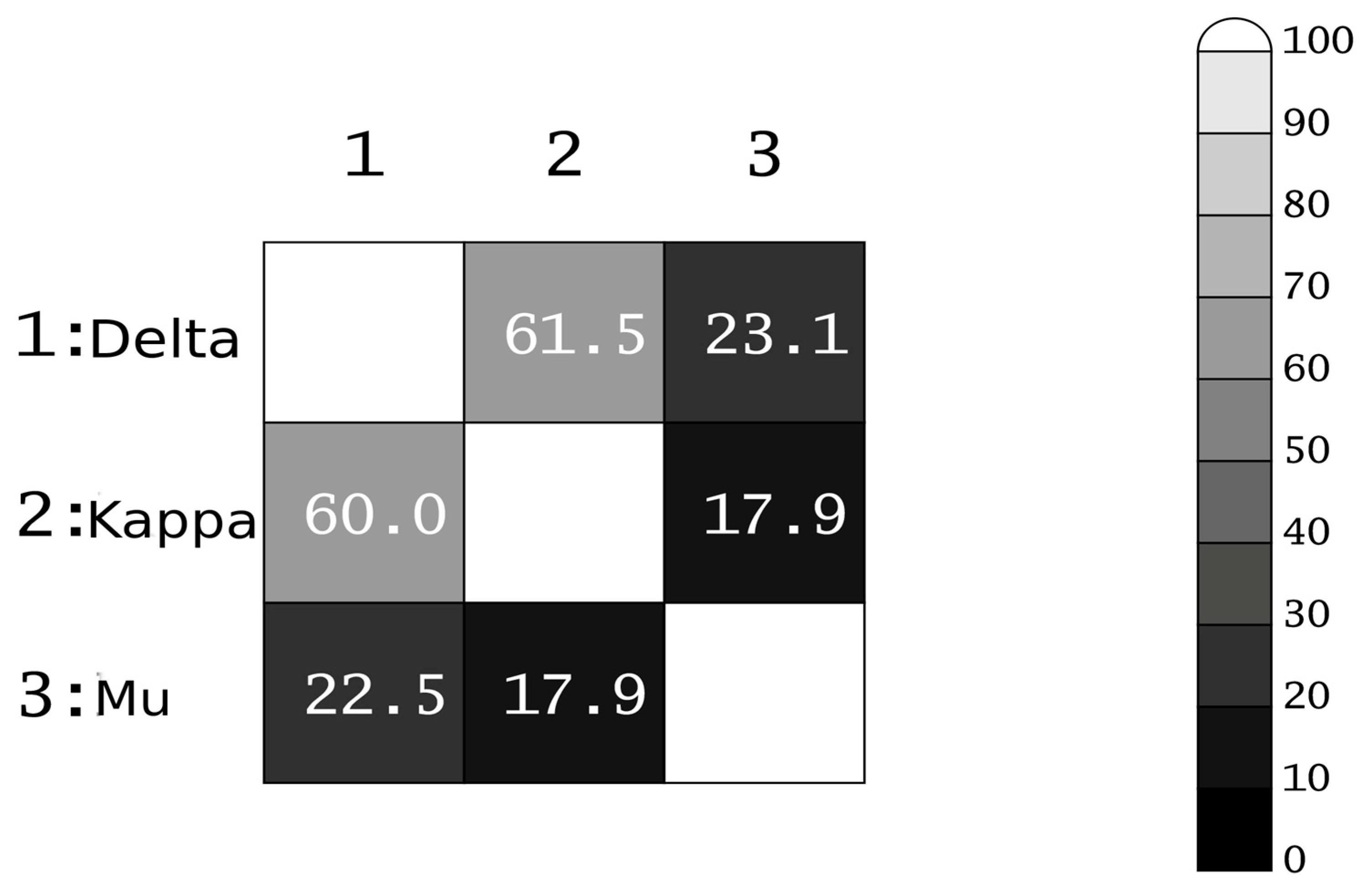
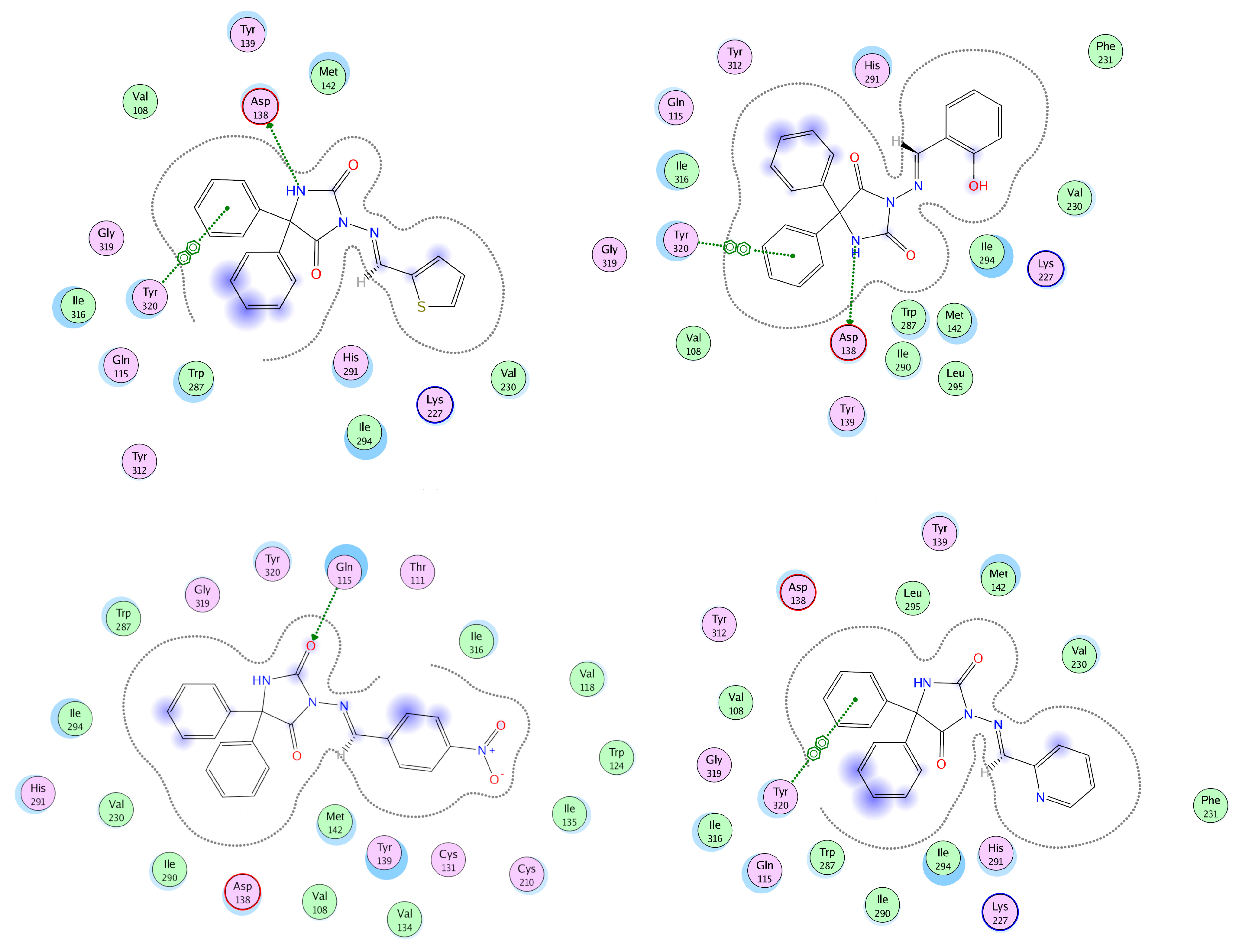
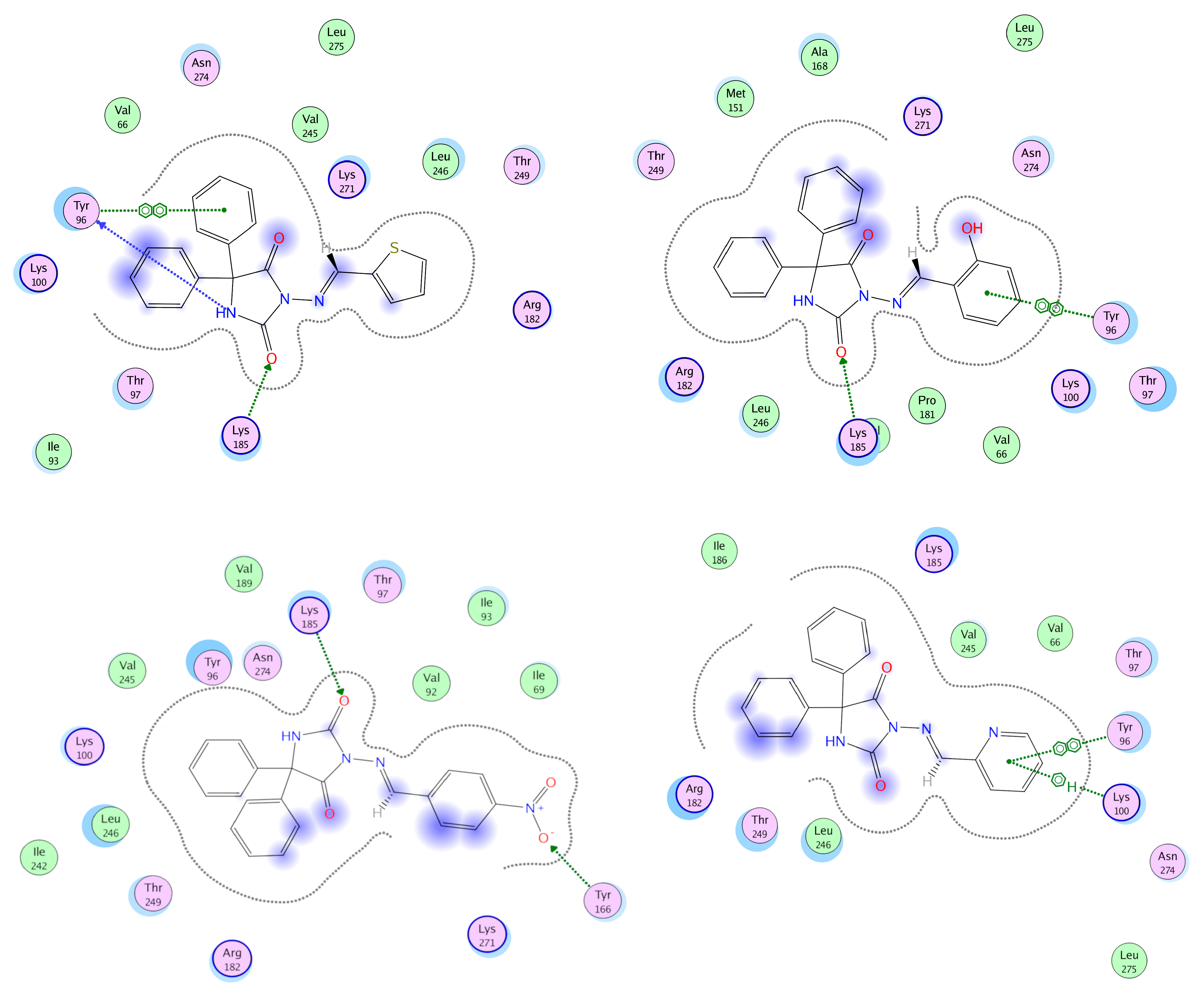
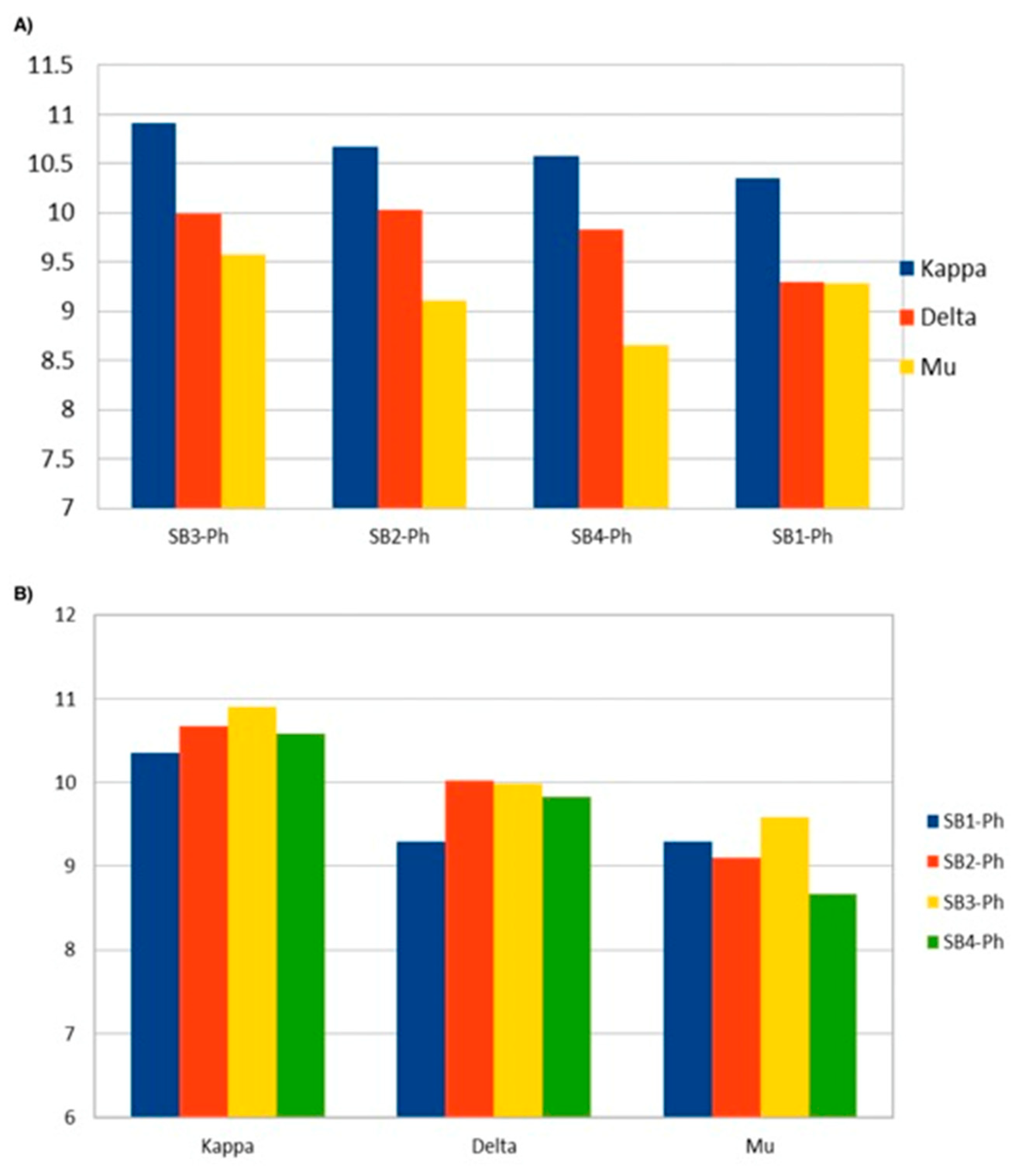
3.4. Docking Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006, 129, 18–35. [Google Scholar] [CrossRef]
- Alanazi, A.M.; El-Azab, A.S.; Al-Swaidan, I.A.; Maarouf, A.R.; El-Bendary, E.R.; Abu El-Enin, M.A.; Abdel-Aziz, A.A.-M. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5,5-diphenylimidazolidine-2,4-dione derivatives. Med. Chem. Res. 2013, 22, 6129–6142. [Google Scholar] [CrossRef]
- Alaa, A.M.; El-Azab, A.S.; Abou-Zeid, L.A.; ElTahir, K.E.H.; Abdel-Aziz, N.I.; Ayyad, R.R.; Al-Obaid, A.M. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Eur. J. Med. Chem. 2016, 115, 121–131. [Google Scholar]
- Zhang, M.; Liang, Y.R.; Li, H.; Liu, M.M.; Wang, Y. Design, synthesis, and biological evaluation of hydantoin bridged analogues of combretastatin A-4 as potential anticancer agents. Bioorg. Med. Chem. 2017, 25, 6623–6634. [Google Scholar] [CrossRef] [PubMed]
- Alkahtani, H.M.; Alanazi, M.M.; Aleanizy, F.S.; Alqahtani, F.Y.; Alhoshani, A.; Alanazi, F.E.; Almehizia, A.A.; Abdalla, A.N.; Alanazi, M.G.; El-Azab, A.S.; et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5, 5-diphenylimidazolidine-2, 4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019, 27, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.L.; Brown, G.; Brouillette, W.J. Effects of log P and phenyl ring conformation on the binding of 5-phenylhydantoins to the voltage-dependent sodium channel. J. Med. Chem. 1997, 40, 602–607. [Google Scholar] [CrossRef]
- Yaari, Y.; Selzer, M.E.; Pincus, J.H. Phenytoin: Mechanisms of its anticonvulsant action. Ann. Neurol. 1986, 20, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Krall, R.; Penry, J.K.; White, B.G.; Kupferberg, H.J.; Swinyard, E.A. Antiepileptic Drug Development: II. Anticonvulsant Drug Screening. Epilepsia 1978, 19, 404–428. [Google Scholar] [CrossRef]
- Varia, S.A.; Stella, V.J. Phenytoin Prodrugs V: In Vivo Evaluation of Some Water-Soluble Phenytoin Prodrugs in Dogs. J. Pharm. Sci. 1984, 73, 1080–1087. [Google Scholar] [CrossRef]
- Suzuki, T.; Saitoh, Y.; Nishihara, K. Kinetics of Diphenylhydantoin Disposition in Man. Chem. Pharm. Bull. 1970, 18, 405–411. [Google Scholar] [CrossRef]
- Pandeya, S.N.; Raja, A.S.; Stables, J.P. Synthesis of isatin semicarbazones as novel anticonvulsants—Role of hydrogen bonding. J. Pharm. Pharm. Sci. 2002, 5, 266–271. [Google Scholar] [PubMed]
- Alachkar, A.; Ojha, S.; Sadeq, A.; Adem, A.; Frank, A.; Stark, H.; Sadek, B. Experimental models for the discovery of novel anticonvulsant drugs: Focus on pentylenetetrazole-induced seizures and associated memory deficits. Curr. Pharm. Des. 2020, 26, 1693–1711. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Kumar, Y.; Stables, J.; Sinha, R. Menthone semicarbazides and thiosemicarbazides as anticonvulsant agents. Med. Chem. 2010, 6, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Unverferth, K.; Engel, J.; Höfgen, N.; Rostock, A.; Günther, R.; Lankau, H.J.; Menzer, M.; Rolfs, A.; Liebscher, J.; Muller, B.; et al. Synthesis, Anticonvulsant Activity, and Structure−Activity Relationships of Sodium Channel Blocking 3-Aminopyrroles. J. Med. Chem. 1998, 41, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Patai, S. The Chemistry of the Carbon-Nitrogen Double Bond; John Wiley & Sons Ltd.: London, UK, 1970; p. 794. ISBN 978-0-470-77120-4. [Google Scholar]
- Prakash, A.; Adhikari, D. Application of Schiff Bases and Their Metal Complexes—A Review. Int. J. ChemTech. Res. 2011, 3, 1891–1896. [Google Scholar]
- Weng, Q.; Yi, J.; Chen, X.; Luo, D.; Wang, Y.; Sun, W.; Kang, J.; Han, Z. Controllable Synthesis and Biological Application of Schiff Bases from d-Glucosamine and Terephthalaldehyde. ACS Omega 2020, 5, 24864–24870. [Google Scholar] [CrossRef] [PubMed]
- Cates, A.L.; Rasheed, S.M. Phosphorus GABA Analogues as Potential Prodrugs. Pharm. Res. 1984, 1, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Todorov, P.; Peneva, P.; Georgieva, S.; Tchekalarova, J.; Rangelov, M.; Todorova, N. Synthesis and characterization of new 5,5’-dimethyl- and 5,5’-diphenylhydantoin-conjugated hemorphin derivatives designed as potential anticonvulsant agents. New J. Chem. 2022, 46, 2198–2217. [Google Scholar] [CrossRef]
- Todorov, P.; Georgieva, S.; Peneva, P.; Rusew, R.; Shivachev, B.; Georgiev, A. Experimental and theoretical study of bidirectional photoswitching behavior of the 5,5’-diphenylhydantoin Schiff bases: Synthesis, crystal structure and kinetics approaches. New J. Chem. 2020, 44, 15081–15099. [Google Scholar] [CrossRef]
- Sills, G.J.; Brodie, M.J. Encyclopedia of Basic Epilepsy Research; Schwartzkroin, P.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 97–103. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; Di Berto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the entire human opioid receptor family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Finney, D.J. Probit Analysis; Cambridge University Press: London, UK, 1971; p. 3. [Google Scholar]
- Putnam, T.J.; Merritt, H. Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 1937, 85, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016, 201, 157–184. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; White, H.S. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Schwarzer, C. The Opioid system in temporal lobe epilepsy: Functional role and therapeutic potential. Front. Mol. Neurosci. 2017, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Vardy, E.; Mosier, P.D.; Frankowski, K.J.; Wu, H.; Katritch, V.; Westkaemper, R.; Aubé, J.; Stevens, R.C.; Roth, B.L. Chemotype-selective modes of action of κ-opioid receptor agonists. J. Biol. Chem. 2013, 288, 34470–34483. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, J.; Nazarova, A.L. Ligand and G-protein selectivity in the κ-opioid receptor. Nature 2023, 617, 417–425. [Google Scholar] [CrossRef]
- Jonas, H.; Aiello, D.; Schepmann, D.; Diana, P.; Wünsch, B. Synthesis of 8-aminomorphans with high KOR affinity. Eur. J. Med. Chem. 2022, 230, 114079. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Singh, N.; Cheve, G.; Ferguson, D.; McCurdy, C.R. A combined ligand-based and target-based drug design approach for G-protein coupled receptors: Application to salvinorin A, a selective kappa opioid receptor agonist. J. Comput. Aided Mol. Des. 2006, 20, 471–493. [Google Scholar] [CrossRef]
- Fadhil, I.; Schmidt, R.; Walpole, C.; Carpenter, K.A. Exploring deltorphin II binding to the third extracellular loop of the delta-opioid receptor. J. Biol. Chem. 2004, 279, 21069–21077. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef]
- Pepin, M.C.; Yue, S.Y.; Roberts, E.; Wahlestedt, C.; Walker, P. Novel “restoration of function” mutagenesis strategy to identify amino acids of the delta-opioid receptor involved in ligand binding. J. Biol. Chem. 1997, 272, 9260–9267. [Google Scholar] [CrossRef]
- Clayton, C.C.; Bruchas, M.R.; Lee, M.L.; Chavkin, C. Phosphorylation of the mu-opioid receptor at tyrosine 166 (Tyr3.51) in the DRY motif reduces agonist efficacy. Mol. Pharmacol. 2010, 77, 339–347. [Google Scholar] [CrossRef]
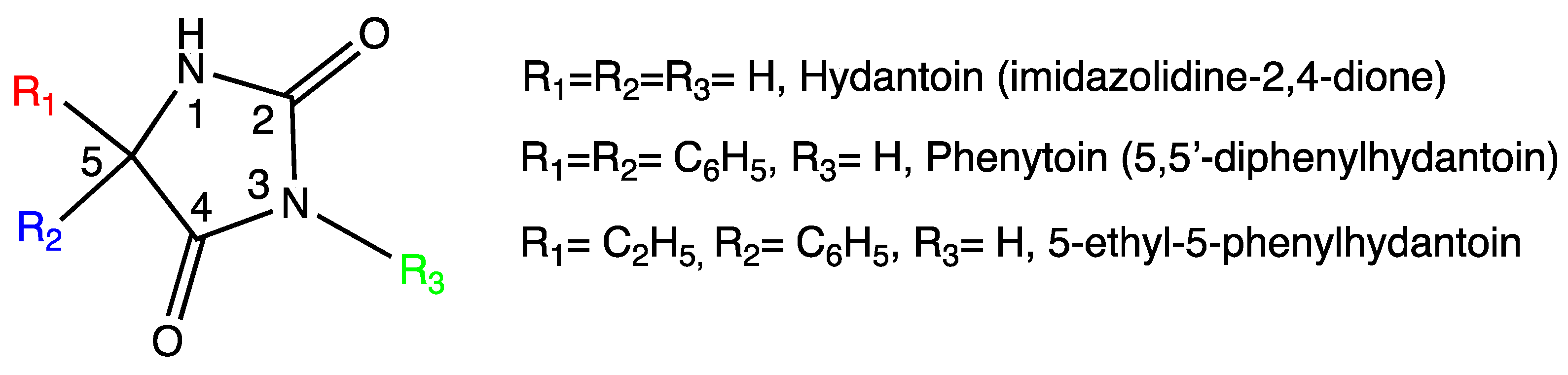





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Compound | Molecular Formula | Molecular Weight |
|---|---|---|---|
| SB1-Ph | 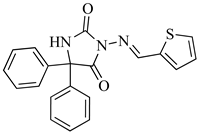 | C20H15N3O2S | 362.0885 |
| SB2-Ph | 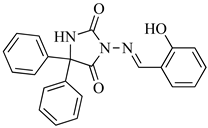 | C22H17N3O3 | 372.1270 |
| SB3-Ph | 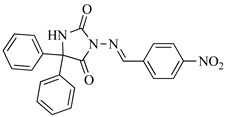 | C22H16N4O4 | 401.1172 |
| SB4-Ph |  | C21H16N4O2 | 357.1273 |
| Drug | a TPE (min) | b ED50 mg·kg−1 | 95% Confidence Interval | c TD50 | d PI |
|---|---|---|---|---|---|
| phenytoin | 60 | 5.96 | (4.65–7.64) | 37.07 | 6.22 |
| SB1-Ph | 30 | 34.09 | (17.66–65.79) | 59.55 | 1.75 |
| phenytoin+SB1-Ph | 60 + 30 | 2.91 | (1.87–4.53) | 112.8 | 38.76 |
| SB2-Ph | 30 | 8.29 | (5.58–12.33) | 54.51 | 6.58 |
| phenytoin+SB2-Ph | 60 + 30 | 3.10 | (1.20–8.02) | >150 | 48.39 |
| SB3-Ph | 30 | 15.29 | (6.73–34.75) | 65.62 | 4.29 |
| phenytoin+SB3-Ph | 60 + 30 | 2.67 | (1.24–5.72) | 64.13 | 24.02 |
| SB4-Ph | 30 | 10.71 | (5.92–19.38) | 42.37 | 3.96 |
| phenytoin+SB4-Ph | 60 + 30 | 1.77 | (0.97–3.2) | 79.20 | 44.75 |
| Group/Treatment | Dose (mg/kg). i.p. | Neuromuscular Strength (N) | Rotarod Test N/F |
|---|---|---|---|
| Control (saline) | 0 | 2.08 ± 0.48 | 0/8 |
| phenytoin | 2.5 | 1.98 ± 0.13 | 1/6 |
| 5 | 2.20 ± 0.38 | 0/6 | |
| 10 | 1.84 ± 0.18 | 2/6 | |
| 20 | 2.06 ± 0.49 | 0/6 | |
| SB1-Ph | 5 | 2.25 ± 0.35 | 1/6 |
| 10 | 2.07 ± 0.67 | 2/6 | |
| 20 | 1.82 ± 0.36 | 2/6 | |
| 40 | 2.52 ± 0.32 | 1/6 | |
| phenytoin+SB1-Ph | 2.5 + 10 | 2.22 ± 0.33 | 1/6 |
| 5 + 10 | 2.38 ± 0.34 | 1/6 | |
| 10 + 10 | 2.38 ± 0.21 | 0/6 | |
| 20 + 10 | 1.51 ± 0.22 | 0/6 | |
| SB2-Ph | 2.5 | 2.08 ± 0.34 | 1/6 |
| 5 | 2.39 ± 0.35 | 1/6 | |
| 10 | 1.56 ± 0.28 | 1/6 | |
| 20 | 1.62 ± 0.39 | 2/6 | |
| 40 | 1.74 ± 0.29 | 2/6 | |
| phenytoin+SB2-Ph | 3.5 + 10 | 1.54 ± 0.21 | 0/6 |
| 5 + 10 | 1.55 ± 0.25 | 0/6 | |
| 10 + 10 | 1.97 ± 0.39 | 0/6 | |
| 20 + 10 | 1.70 ± 0.19 | 0/6 | |
| SB3-Ph | 5 | 1.91 ± 0.28 | 1/6 |
| 10 | 1.98 ± 0.53 | 2/6 | |
| 20 | 1.85 ± 0.39 | 2/6 | |
| 40 | 1.69 ± 0.30 | 0/6 | |
| phenytoin+SB3-Ph | 2.5 + 10 | 2.27 ± 0.39 | 2/6 |
| 5 + 10 | 2.27 ± 0.38 | 0/6 | |
| 10 + 10 | 1.76 ± 0.44 | 1/6 | |
| 20 + 10 | 2.22 ± 0.23 | 1/6 | |
| SB4-Ph | 1 | 2.06 ± 0.28 | 0/6 |
| 5 | 2.18 ± 0.43 | 0/6 | |
| 10 | 2.0 ± 0.41 | 0/6 | |
| 20 | 1.94 ± 0.4 | 1/6 | |
| 40 | 1.93 ± 0.32 | 3/6 | |
| phenytoin+SB4-Ph | 2.5 + 10 | 1.89 ± 0.26 | 1/6 |
| 5 + 10 | 2.15 ± 0.27 | 2/6 | |
| 10 + 10 | 2.14 ± 0.23 | 0/6 | |
| 20 + 10 | 2.09 ± 0.38 | 2/6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchekalarova, J.; Todorov, P.; Rangelov, M.; Stoyanova, T.; Todorova, N. Additive Anticonvulsant Profile and Molecular Docking Analysis of 5,5′-Diphenylhydantoin Schiff Bases and Phenytoin. Biomedicines 2023, 11, 2912. https://doi.org/10.3390/biomedicines11112912
Tchekalarova J, Todorov P, Rangelov M, Stoyanova T, Todorova N. Additive Anticonvulsant Profile and Molecular Docking Analysis of 5,5′-Diphenylhydantoin Schiff Bases and Phenytoin. Biomedicines. 2023; 11(11):2912. https://doi.org/10.3390/biomedicines11112912
Chicago/Turabian StyleTchekalarova, Jana, Petar Todorov, Miroslav Rangelov, Tsveta Stoyanova, and Nadezhda Todorova. 2023. "Additive Anticonvulsant Profile and Molecular Docking Analysis of 5,5′-Diphenylhydantoin Schiff Bases and Phenytoin" Biomedicines 11, no. 11: 2912. https://doi.org/10.3390/biomedicines11112912
APA StyleTchekalarova, J., Todorov, P., Rangelov, M., Stoyanova, T., & Todorova, N. (2023). Additive Anticonvulsant Profile and Molecular Docking Analysis of 5,5′-Diphenylhydantoin Schiff Bases and Phenytoin. Biomedicines, 11(11), 2912. https://doi.org/10.3390/biomedicines11112912