
Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury


Abstract
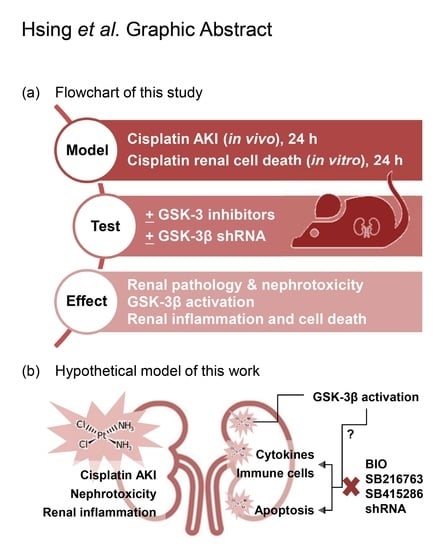
:
{kind=link}
{kind=link}
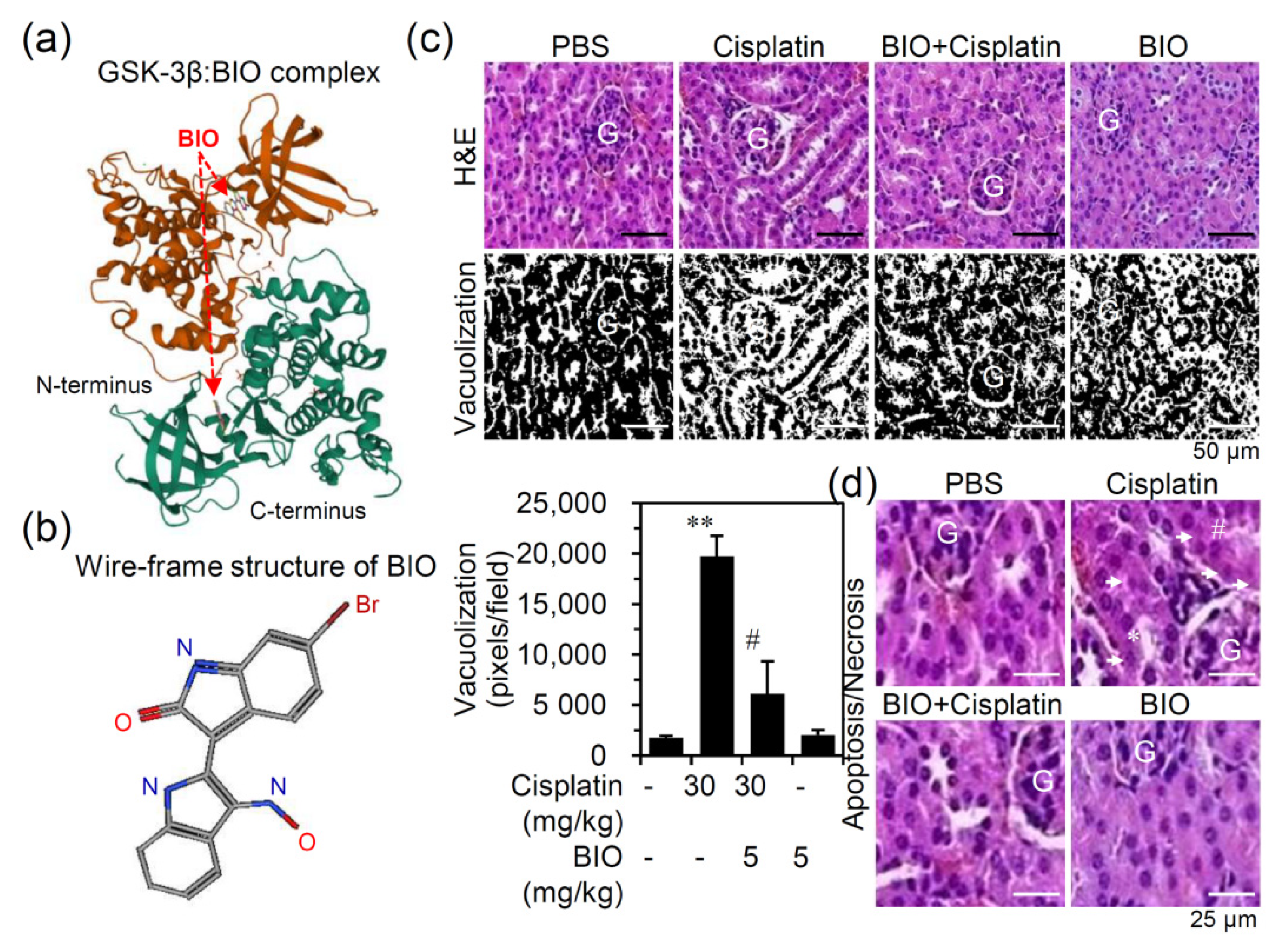{kind=link}
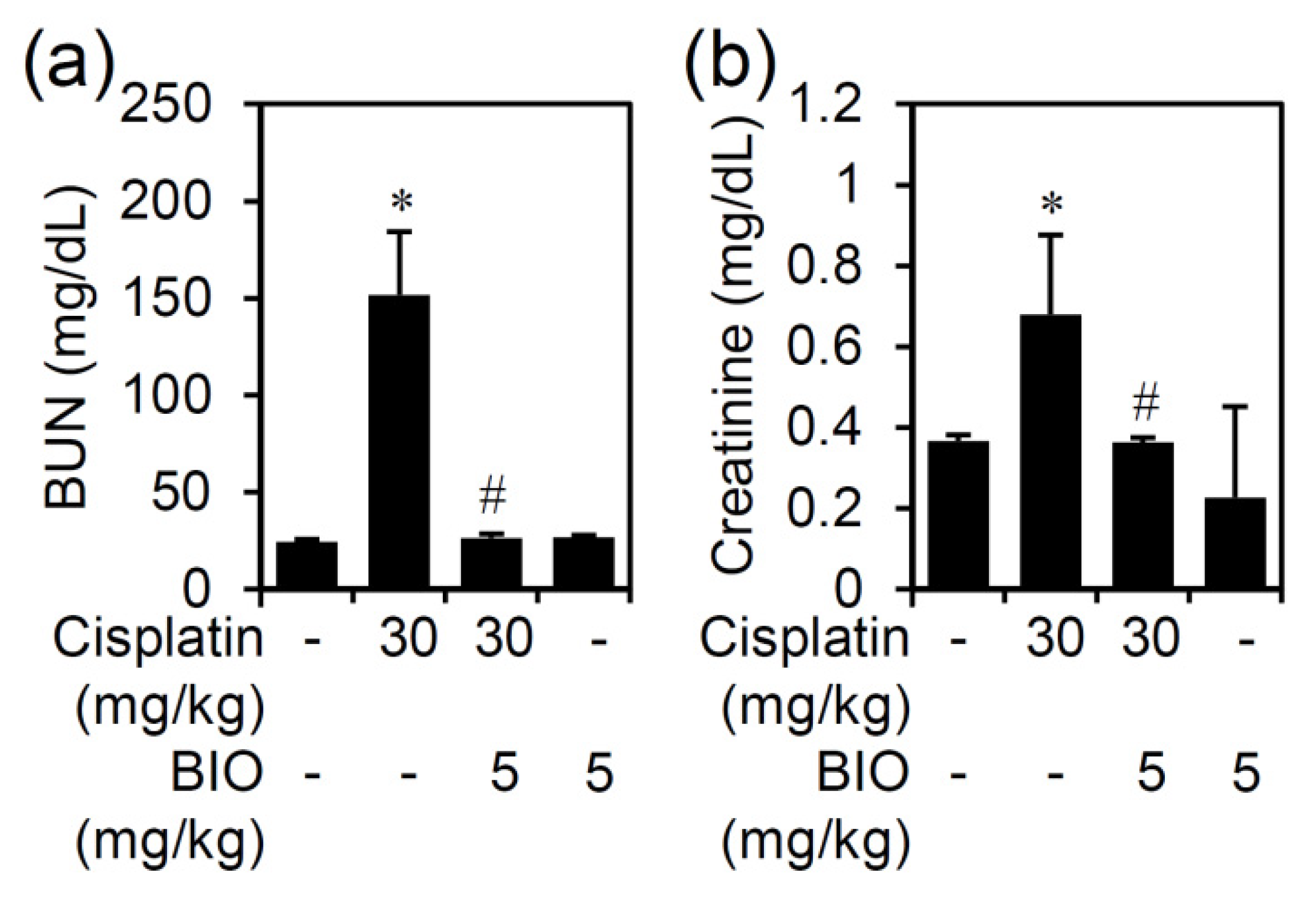{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Animal Treatment
2.3. Immunohistochemistry
2.4. Pathological Examination
2.5. Cell Culture
2.6. Cell Death Assay
2.7. Western Blotting
2.8. RNA Interference
2.9. ELISA
2.10. Statistical Analysis
3. Results
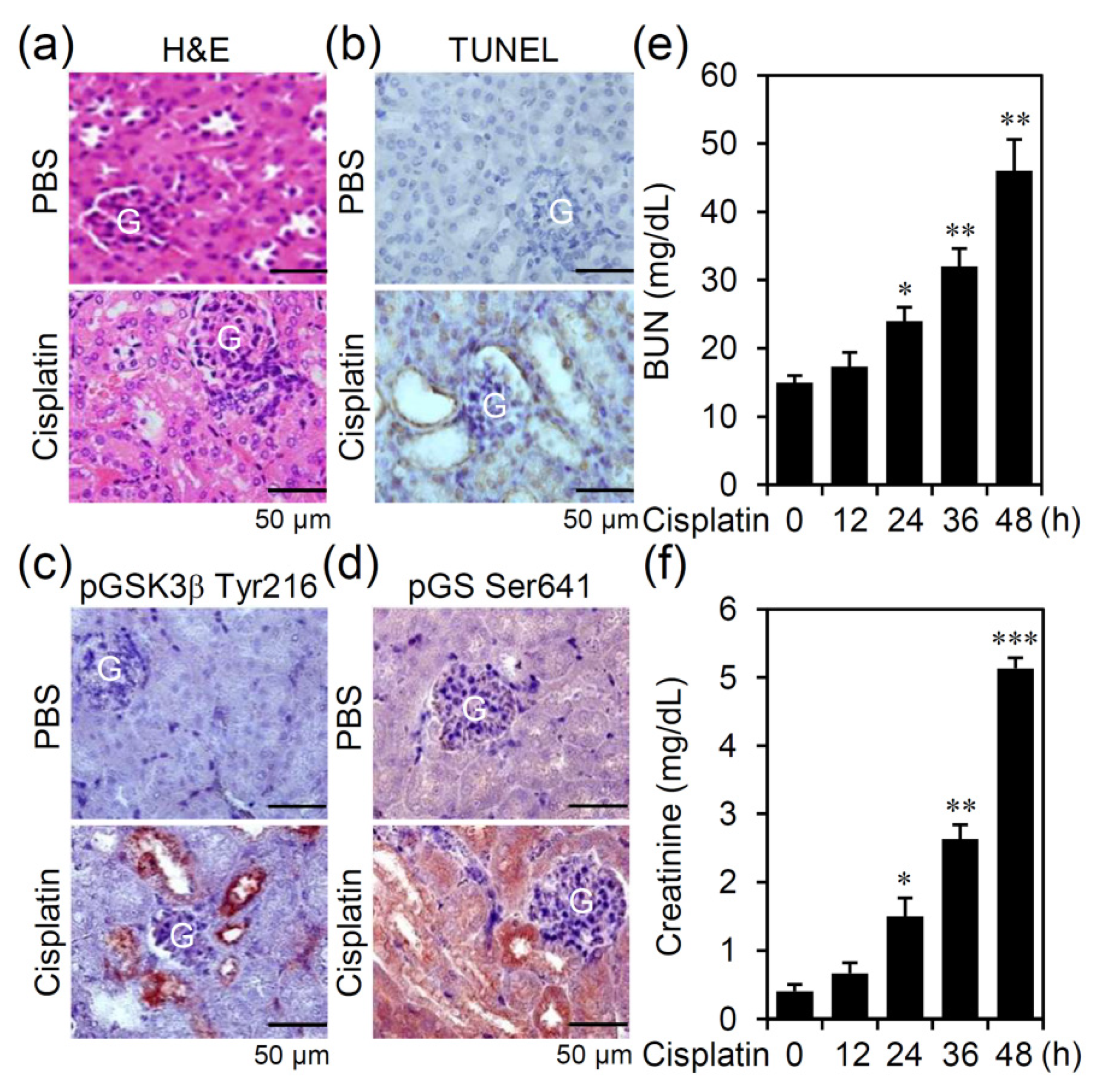
3.1. Cisplatin Treatment Causes GSK-3β Activation Accompanied by Nephrotoxicity in an Experimental Murine Model of AKI
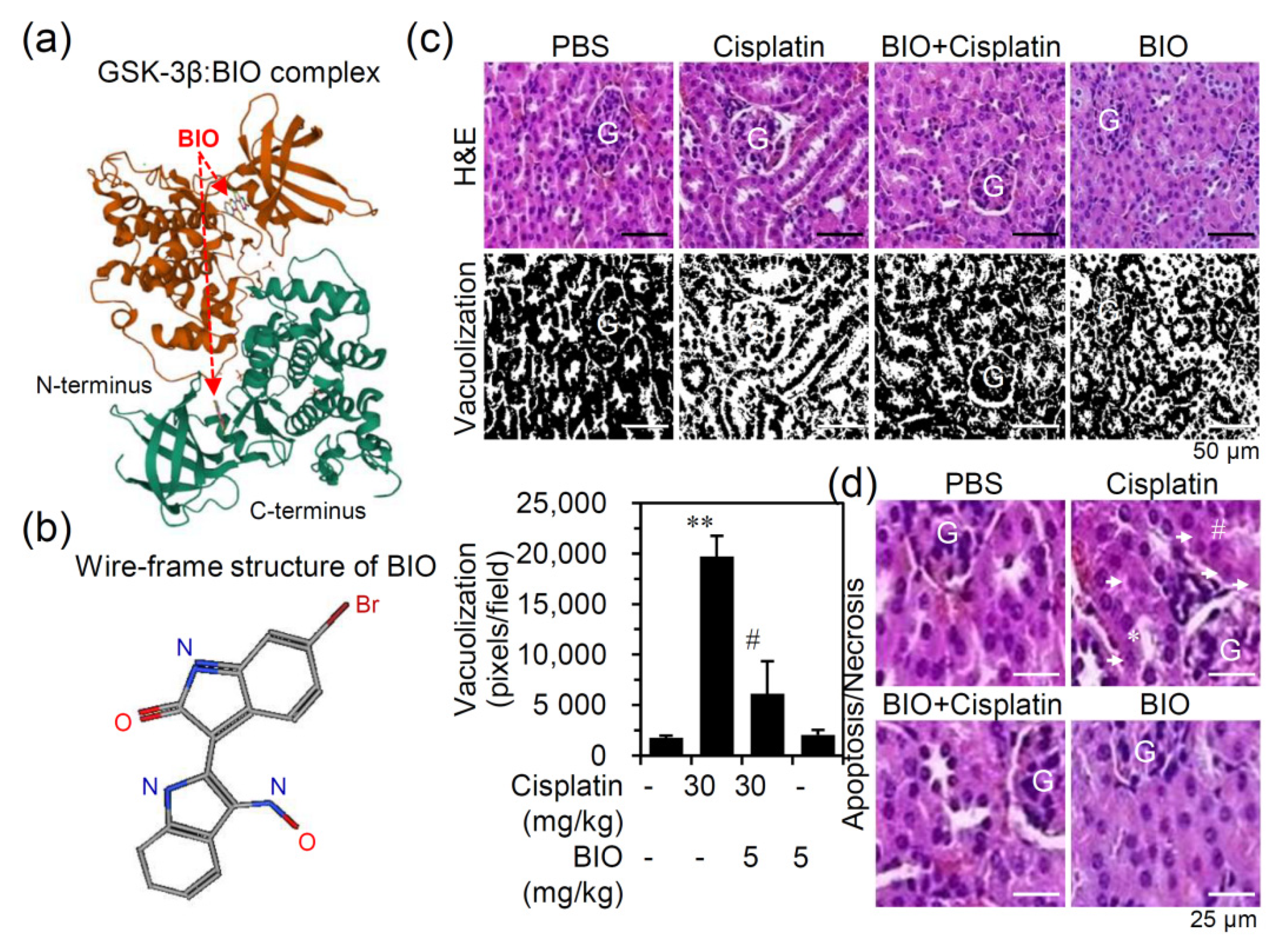
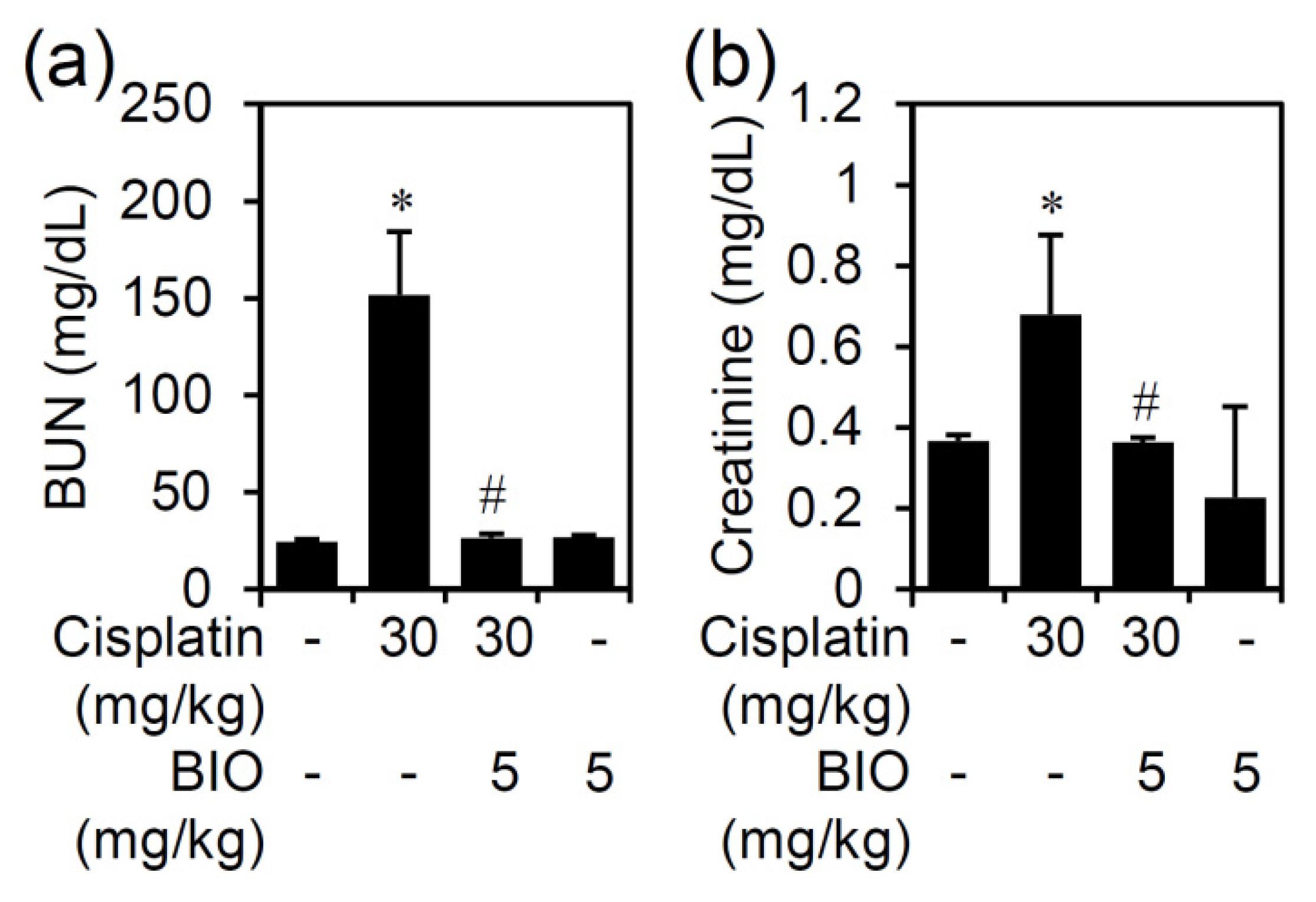
3.2. Pharmacologically Inhibiting GSK-3 Activation Ameliorates Nephrotoxicity and Renal Dysfunction in an Experimental Murine Model of Cisplatin-Induced AKI
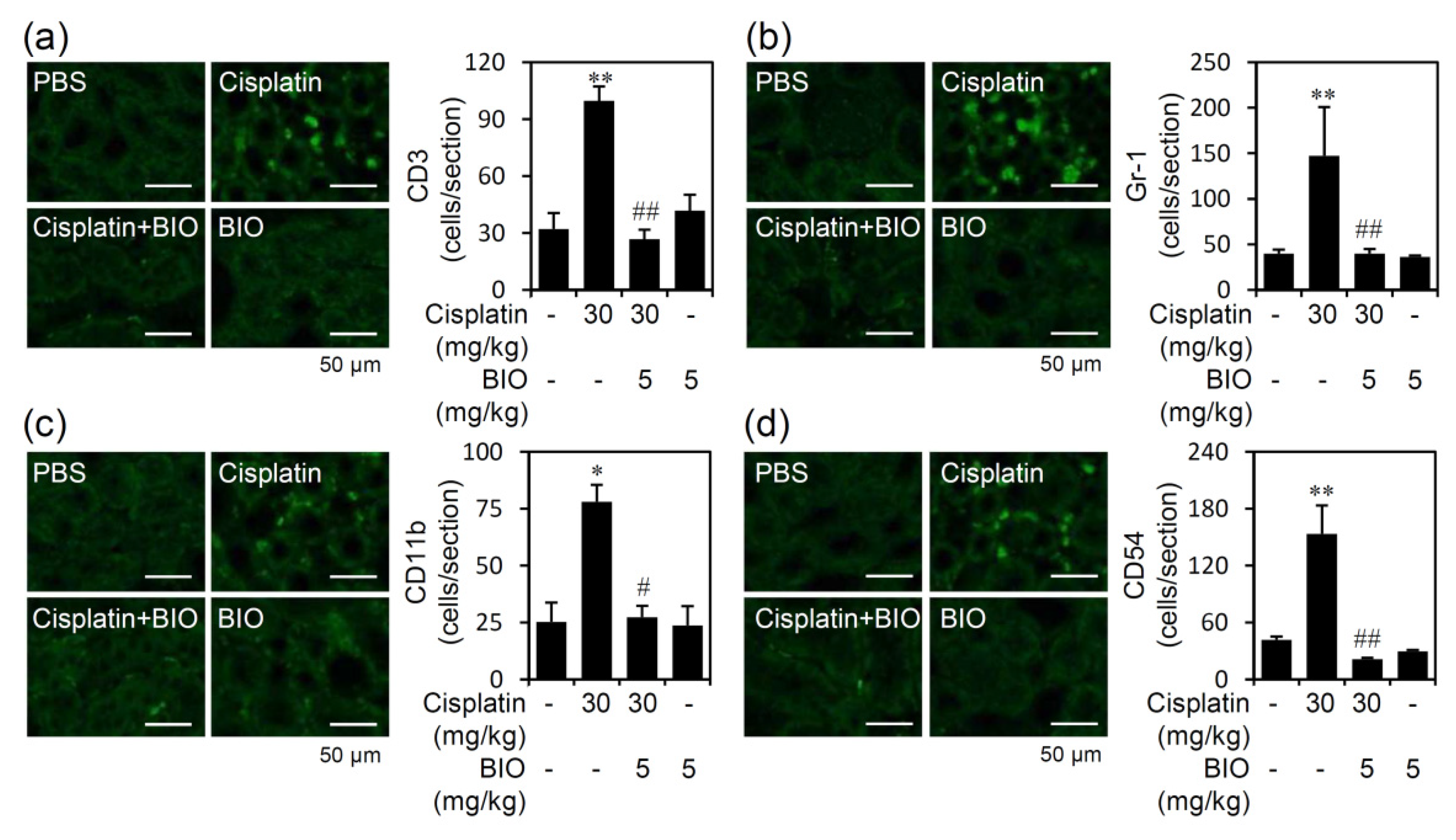
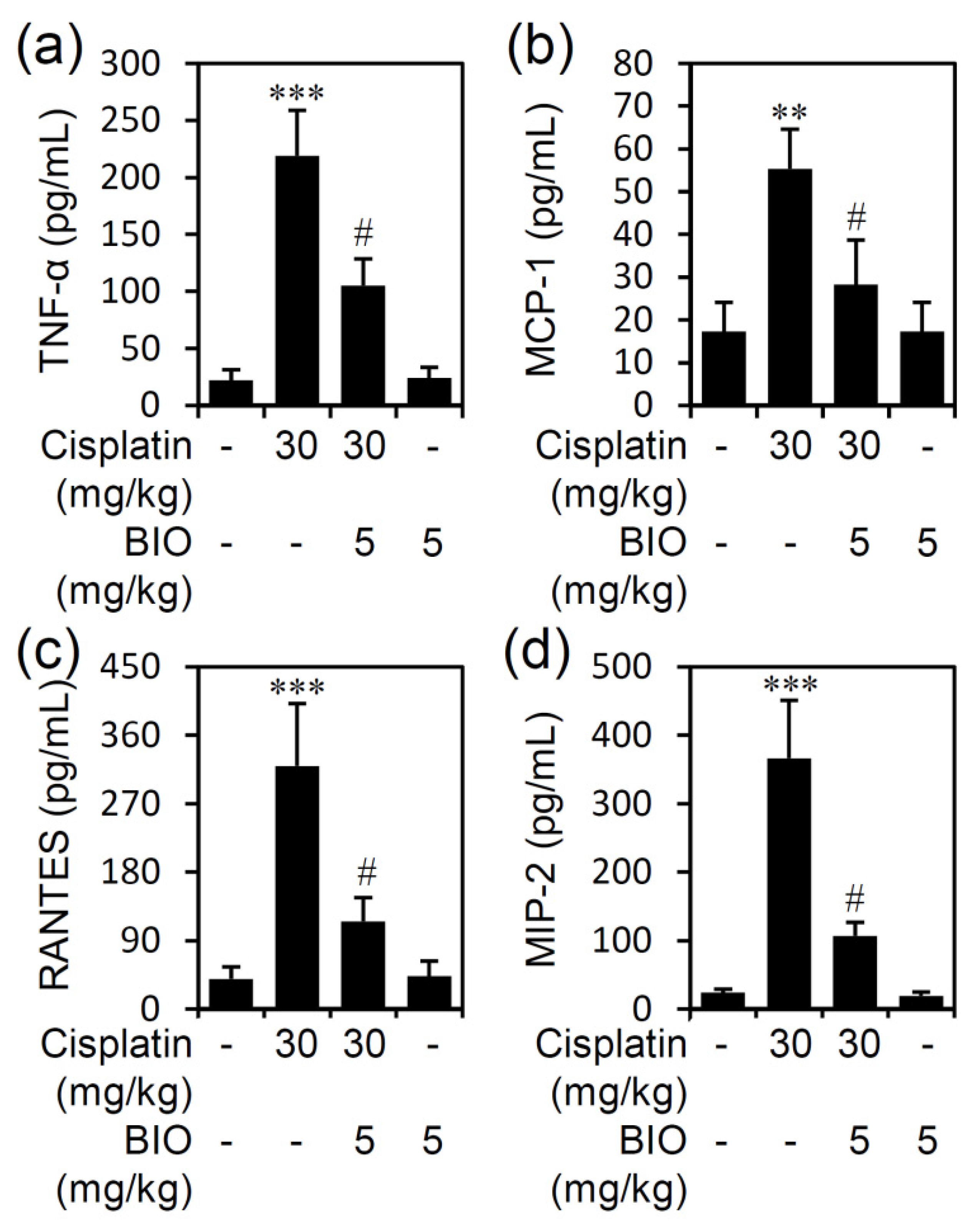
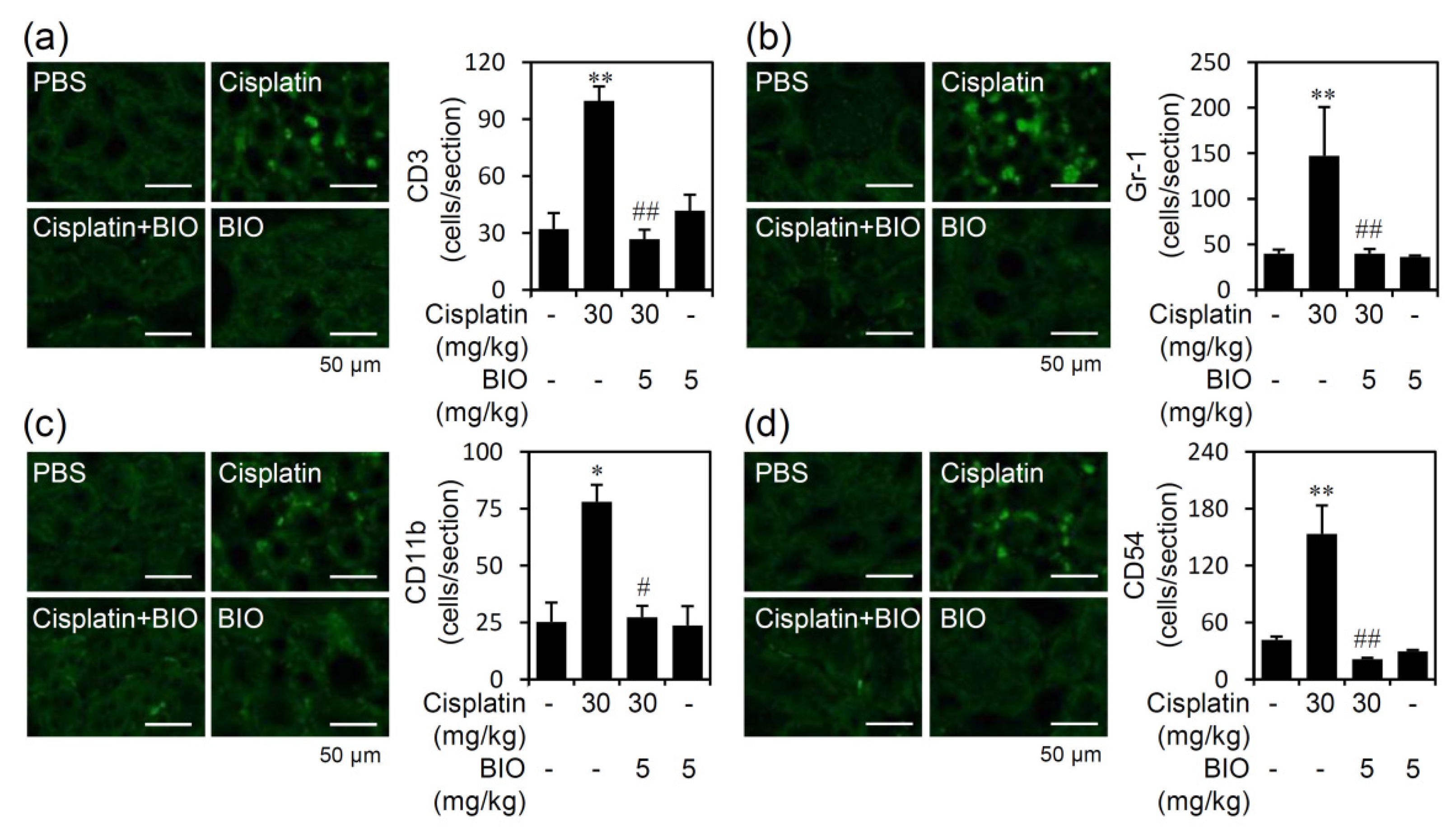
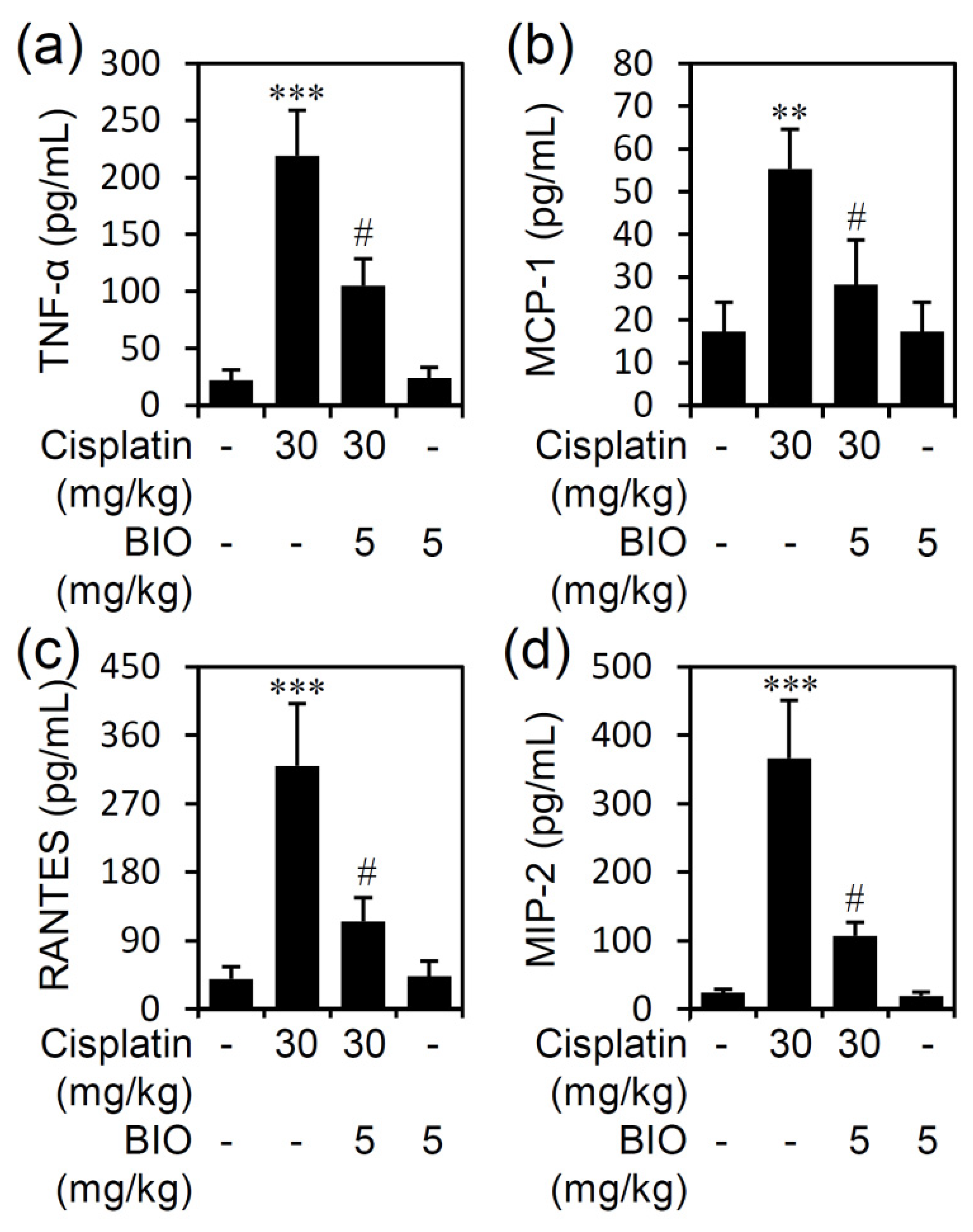
3.3. Pharmacologically Inhibiting GSK-3 Activation Ameliorates Renal Inflammation in an Experimental Murine Model of Cisplatin-Induced AKI
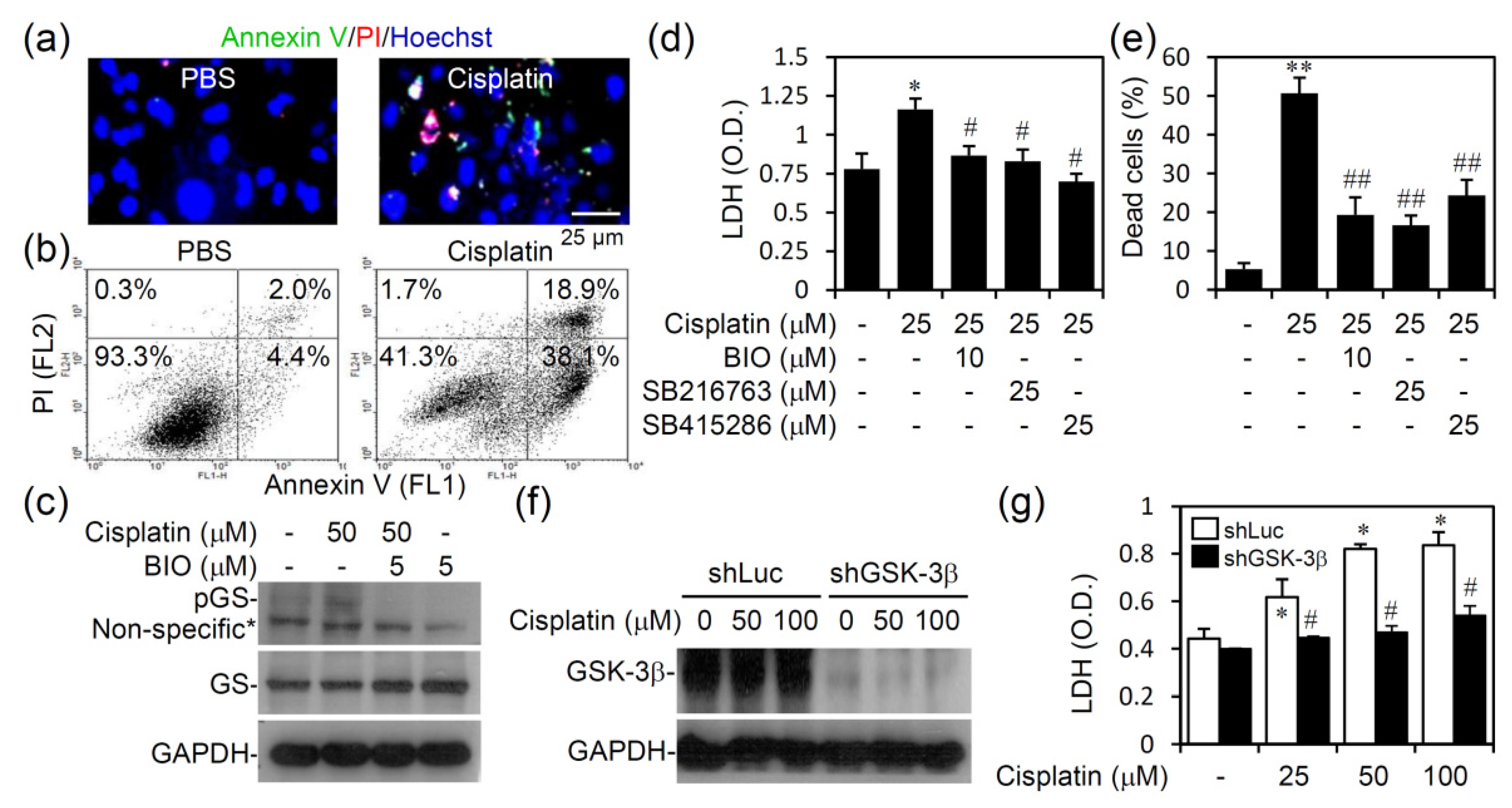
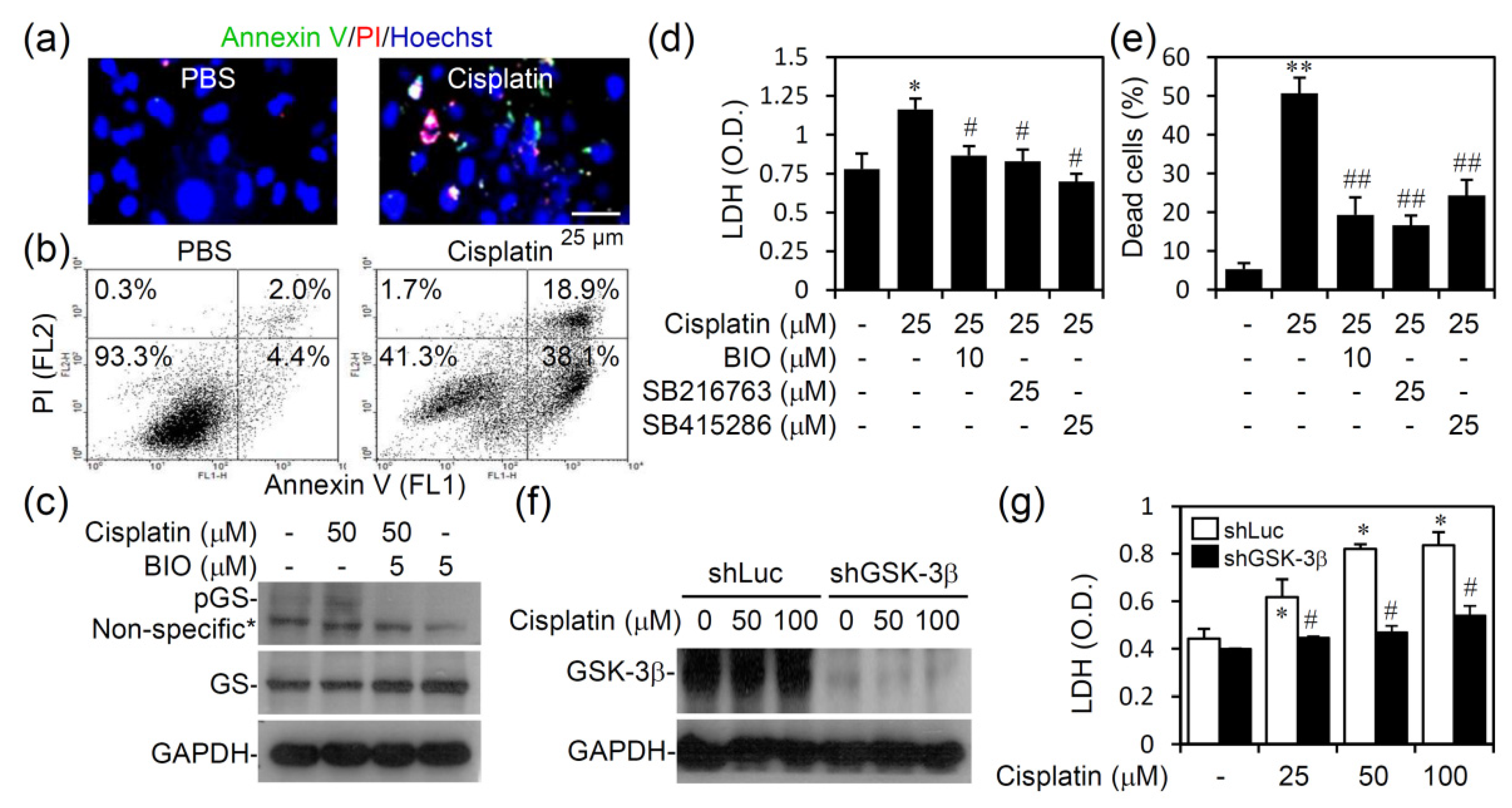
3.4. Pharmacologically Inhibiting GSK-3 Activation Promotes Cell Survival from Cisplatin-Induced Renal Cell Cytotoxicity In Vitro
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Taguchi, T.; Nazneen, A.; Abid, M.R.; Razzaque, M.S. Cisplatin-associated nephrotoxicity and pathological events. Contrib. Nephrol. 2005, 148, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Cornelison, T.L.; Reed, E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol. Oncol. 1993, 50, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Hayati, F.; Hossainzadeh, M.; Shayanpour, S.; Abedi-Gheshlaghi, Z.; Beladi Mousavi, S.S. Prevention of cisplatin nephrotoxicity. J. Nephropharmacol. 2016, 5, 57–60. [Google Scholar]
- Wang, J.; Pabla, N.; Wang, C.Y.; Wang, W.; Schoenlein, P.V.; Dong, Z. Caspase-mediated cleavage of ATM during cisplatin-induced tubular cell apoptosis: Inactivation of its kinase activity toward p53. Am. J. Physiol. Ren. Physiol. 2006, 291, F1300–F1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashed, L.A.; Hashem, R.M.; Soliman, H.M. Oxytocin inhibits NADPH oxidase and P38 MAPK in cisplatin-induced nephrotoxicity. Biomed. Pharm. 2011, 65, 474–480. [Google Scholar] [CrossRef]
- Arany, I.; Megyesi, J.K.; Kaneto, H.; Price, P.M.; Safirstein, R.L. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2004, 287, F543–F549. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Tsuji, T.; Yasuda, H.; Sun, Y.; Fujigaki, Y.; Hishida, A. The molecular mechanisms of the attenuation of cisplatin-induced acute renal failure by N-acetylcysteine in rats. Nephrol. Dial. Transplant. 2008, 23, 2198–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Akcay, A.; Nguyen, Q.; Edelstein, C.L. Mediators of inflammation in acute kidney injury. Mediat. Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3beta: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, 930710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Havasi, A.; Gall, J.; Bonegio, R.; Li, Z.; Mao, H.; Schwartz, J.H.; Borkan, S.C. GSK3beta promotes apoptosis after renal ischemic injury. J. Am. Soc. Nephrol. 2010, 21, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef] [Green Version]
- Jamadar, A.; Rao, R. Glycogen Synthase Kinase-3 Signaling in Acute Kidney Injury. Nephron 2020, 144, 609–612. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, W.C.; Wang, C.Y.; Tsai, C.C.; Chen, C.L.; Chang, Y.T.; Kai, J.I.; Lin, C.F. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br. J. Pharm. 2009, 157, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Ge, Y.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Howard, C.; Tao, S.; Yang, H.C.; Fogo, A.B.; Woodgett, J.R.; Harris, R.C.; Rao, R. Specific deletion of glycogen synthase kinase-3beta in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int. 2012, 82, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, L.; Skaltsounis, A.L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodova, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Siskind, L.J. Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiol. Ren. Physiol. 2017, 313, F835–F841. [Google Scholar] [CrossRef] [Green Version]
- Perse, M.; Veceric-Haler, Z. Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics and Challenges. Biomed Res. Int. 2018, 2018, 1462802. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Wang, J.; Xin, X.; Ke, Z.; Luo, J. Phosphorylation of glycogen synthase kinase-3 beta at serine 9 confers cisplatin resistance in ovarian cancer cells. Int. J. Oncol. 2007, 31, 657–662. [Google Scholar]
- Li, Z.; Tan, F.; Thiele, C.J. Inactivation of glycogen synthase kinase-3beta contributes to brain-derived neutrophic factor/TrkB-induced resistance to chemotherapy in neuroblastoma cells. Mol. Cancer 2007, 6, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Gohr, K.; Hamacher, A.; Engelke, L.H.; Kassack, M.U. Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38. BMC Cancer 2017, 17, 711. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3—At the crossroads of cell death and survival. J. Cell Sci. 2014, 127, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perazella, M.A.; Moeckel, G.W. Nephrotoxicity from chemotherapeutic agents: Clinical manifestations, pathobiology, and prevention/therapy. Semin. Nephrol. 2010, 30, 570–581. [Google Scholar] [CrossRef]
- Sears, S.; Siskind, L. Potential Therapeutic Targets for Cisplatin-Induced Kidney Injury: Lessons from Other Models of AKI and Fibrosis. J. Am. Soc. Nephrol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2007, 293, F1282–F1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francescato, H.D.; Costa, R.S.; Junior, F.B.; Coimbra, T.M. Effect of JNK inhibition on cisplatin-induced renal damage. Nephrol. Dial. Transplant. 2007, 22, 2138–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francescato, H.D.; Costa, R.S.; Silva, C.G.; Coimbra, T.M. Treatment with a p38 MAPK inhibitor attenuates cisplatin nephrotoxicity starting after the beginning of renal damage. Life Sci. 2009, 84, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Guo, F.; Xia, Z.; Liang, Y.; Lei, S.; Tan, Z.; Ma, L.; Fu, P. Activation of GPR120 by TUG891 ameliorated cisplatin-induced acute kidney injury via repressing ER stress and apoptosis. Biomed. Pharm. 2020, 126, 110056. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Chien, C.C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol 2006, 17, 765–774. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Iyoda, M.; Matsumoto, K.; Shindo-Hirai, Y.; Kuno, Y.; Yamamoto, Y.; Suzuki, T.; Saito, T.; Iseri, K.; Shibata, T. Epidermal growth factor receptor inhibition with erlotinib partially prevents cisplatin-induced nephrotoxicity in rats. PLoS ONE 2014, 9, e111728. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabla, N.; Dong, Z. Curtailing side effects in chemotherapy: A tale of PKCdelta in cisplatin treatment. Oncotarget 2012, 3, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, G.; Fredriksson, L.; Herpers, B.; Meerman, J.; van de Water, B.; de Graauw, M. TNF-alpha-mediated NF-kappaB survival signaling impairment by cisplatin enhances JNK activation allowing synergistic apoptosis of renal proximal tubular cells. Biochem. Pharm. 2013, 85, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharm. Exp. 2007, 322, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef]
- Kelly, K.J.; Meehan, S.M.; Colvin, R.B.; Williams, W.W.; Bonventre, J.V. Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int. 1999, 56, 922–931. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Giambelluca, M.S.; Bertheau-Mailhot, G.; Laflamme, C.; Rollet-Labelle, E.; Servant, M.J.; Pouliot, M. TNF-alpha expression in neutrophils and its regulation by glycogen synthase kinase-3: A potentiating role for lithium. FASEB J. 2014, 28, 3679–3690. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Role of glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB activation and apoptosis in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G204–G211. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xu, S.; Cai, Q.; Zhou, W.; Jiao, X.; Bao, M.; Yu, X. Wnt/beta-catenin agonist BIO alleviates cisplatin-induced nephrotoxicity without compromising its efficacy of anti-proliferation in ovarian cancer. Life Sci. 2020, 263, 118672. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsing, C.-H.; Tsai, C.-C.; Chen, C.-L.; Lin, Y.-H.; Tseng, P.-C.; Satria, R.D.; Lin, C.-F. Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury. Biomedicines 2021, 9, 887. https://doi.org/10.3390/biomedicines9080887
Hsing C-H, Tsai C-C, Chen C-L, Lin Y-H, Tseng P-C, Satria RD, Lin C-F. Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury. Biomedicines. 2021; 9(8):887. https://doi.org/10.3390/biomedicines9080887
Chicago/Turabian StyleHsing, Chung-Hsi, Cheng-Chieh Tsai, Chia-Ling Chen, Yu-Hui Lin, Po-Chun Tseng, Rahmat Dani Satria, and Chiou-Feng Lin. 2021. "Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury" Biomedicines 9, no. 8: 887. https://doi.org/10.3390/biomedicines9080887
APA StyleHsing, C.-H., Tsai, C.-C., Chen, C.-L., Lin, Y.-H., Tseng, P.-C., Satria, R. D., & Lin, C.-F. (2021). Pharmacologically Inhibiting Glycogen Synthase Kinase-3β Ameliorates Renal Inflammation and Nephrotoxicity in an Animal Model of Cisplatin-Induced Acute Kidney Injury. Biomedicines, 9(8), 887. https://doi.org/10.3390/biomedicines9080887