
Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome

 , ,
, ,  ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case Report
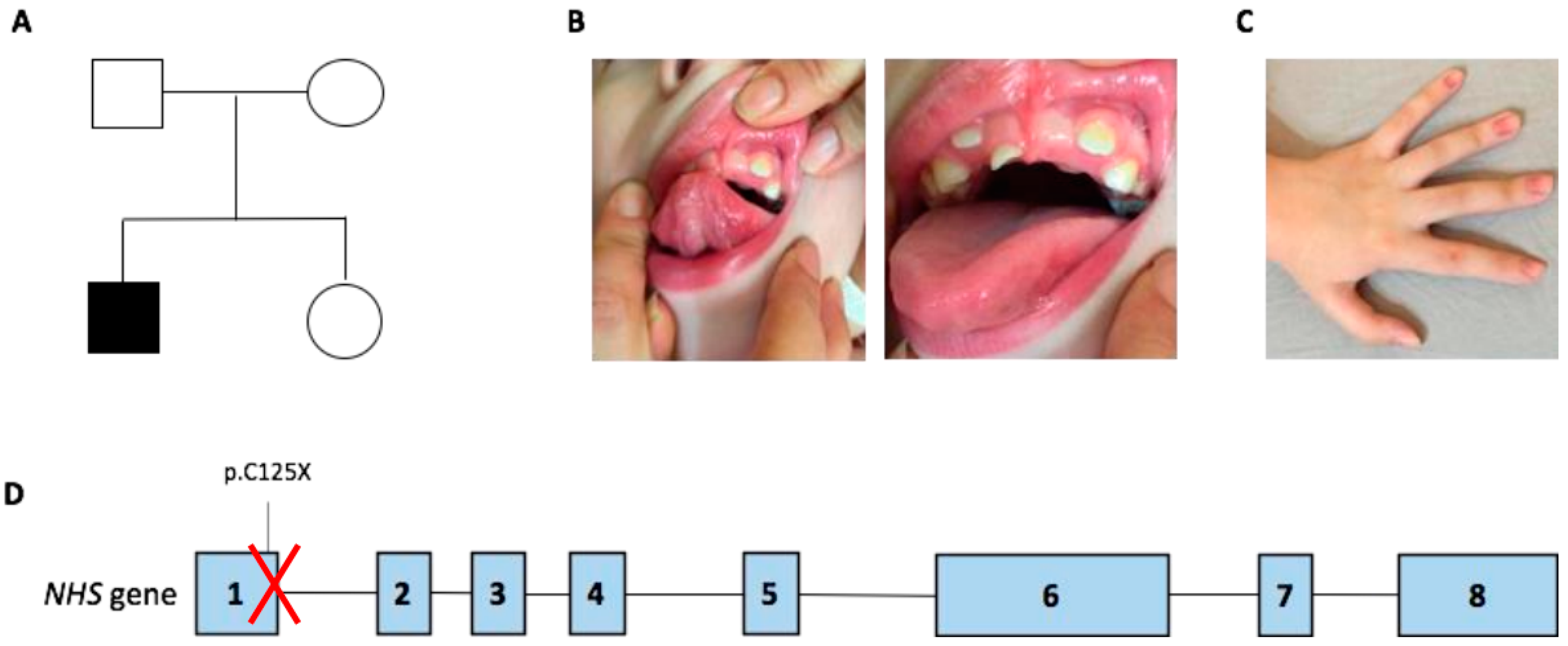
2.1. Patient Presentation
2.2. Whole Exome Sequencing Results
2.3. Expression and Protein-Protein Interaction Analyses
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salpietro, V.; Phadke, R.; Saggar, A.; Hargreaves, I.P.; Yates, R.; Fokoloros, C.; Mankad, K.; Hertecant, J.; Ruggieri, M.; McCormick, D.; et al. Zellweger syndrome and secondary mitochondrial myopathy. Eur. J. Pediatr. 2015, 174, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, A.; Schneider, R.B.; Augustine, E.F.; Ng, J.; Mankad, K.; Meyer, E.; McTague, A.; Ngoh, A.; Hemingway, C.; Robinson, R.; et al. Delineation of the movement disorders associated with FOXG1 mutations. Neurology 2016, 86, 1794–1800. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Malintan, N.T.; Llano-Rivas, I.; Spaeth, C.G.; Efthymiou, S.; Striano, P.; Vandrovcova, J.; Cutrupi, M.C.; Chimenz, R.; David, E.; et al. Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am. J. Hum. Genet. 2019, 104, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Pearson, N.; Charman, T.; Happé, F.; Bolton, P.F.; McEwen, F.S. Regression in autism spectrum disorder: Reconciling findings from retrospective and prospective research. Autism Res. 2018, 11, 1602–1620. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 12, 3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuray, C.; Maroofian, R.; Scala, M.; Sultan, T.; Pai, G.S.; Mojarrad, M.; Khashab, H.E.; de Holl, L.; Yue, W.; Alsaif, H.S.; et al. Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants. Brain 2020, 143, 2388–2397. [Google Scholar] [CrossRef]
- Manole, A.; Efthymiou, S.; O’Connor, E.; Mendes, M.I.; Jennings, M.; Maroofian, R.; Davagnanam, I.; Mankad, K.; Lopez, M.R.; Salpietro, V.; et al. De Novo and Bi-allelic Pathogenic Variants in NARS1 Cause Neurodevelopmental Delay Due to Toxic Gain-of-Function and Partial Loss-of-Function Effects. Am. J. Hum. Genet. 2020, 107, 311–324. [Google Scholar] [CrossRef]
- Salpietro, V.; Zollo, M.; Vandrovcova, J.; Ryten, M.; Botia, J.A.; Ferrucci, V.; Manole, A.; Efthymiou, S.; Al Mutairi, F.; Bertini, E.; et al. The phenotypic and molecular spectrum of PEHO syndrome and PEHO-like disorders. Brain 2017, 140, e49. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef] [Green Version]
- Dias, C.M.; Punetha, J.; Zheng, C.; Mazaheri, N.; Rad, A.; Efthymiou, S.; Petersen, A.; Dehghani, M.; Pehlivan, D.; Partlow, J.N.; et al. Homozygous Missense Variants in NTNG2, Encoding a Presynaptic Netrin-G2 Adhesion Protein, Lead to a Distinct Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 105, 1048–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piard, J.; Umanah, G.K.E.; Harms, F.L.; Abalde-Atristain, L.; Amram, D.; Chang, M.; Chen, R.; Alawi, M.; Salpietro, V.; Rees, M.I.; et al. A homozygous ATAD1 mutation impairs postsynaptic AMPA receptor trafficking and causes a lethal encephalopathy. Brain 2018, 141, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Patel, M.; Herzlich, A.A.; Sieving, P.C.; Chan, C.C. Ophthalmic pathology of Nance-Horan syndrome: Case report and review of the literature. Ophthalmic Genet. 2009, 30, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tug, E.; Dilek, N.F.; Javadiyan, S.; Burdon, K.P.; Percin, F.E. A Turkish family with Nance-Horan Syndrome due to a novel mutation. Gene 2013, 525, 141–145. [Google Scholar] [CrossRef]
- Li, A.; Li, B.; Wu, L.; Yang, L.; Chen, N.; Ma, Z. Identification of a novel NHS mutation in a Chinese family with Nance-Horan syndrome. Curr. Eye Res. 2015, 40, 434–438. [Google Scholar] [CrossRef]
- Tian, Q.; Li, Y.; Kousar, R.; Guo, H.; Peng, F.; Zheng, Y.; Yang, X.; Long, Z.; Tian, R.; Xia, K.; et al. A novel NHS mutation causes Nance-Horan Syndrome in a Chinese family. BMC Med. Genet. 2017, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Toutain, A.; Ayrault, A.D.; Moraine, C. Mental retardation in Nance-Horan syndrome: Clinical and neuropsychological assessment in four families. Am. J. Med. Genet. 1997, 71, 305–314. [Google Scholar] [CrossRef]
- Liao, H.M.; Niu, D.M.; Chen, Y.J.; Fang, J.S.; Chen, S.J.; Chen, C.H. Identification of a microdeletion at Xp22.13 in a Taiwanese family presenting with Nance-Horan syndrome. J. Hum. Genet. 2011, 56, 8–11. [Google Scholar] [CrossRef] [Green Version]
- Coccia, M.; Brooks, S.P.; Webb, T.R.; Christodoulou, K.; Wozniak, I.O.; Murday, V.; Balicki, M.; Yee, H.A.; Wangensteen, T.; Riise, R.; et al. X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum. Mol. Genet. 2009, 18, 2643–2655. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Aldahmesh, M.A.; Mohamed, J.Y.; Alkuraya, F.S. Phenotype-genotype correlation in potential female carriers of X-linked developmental cataract (Nance-Horan syndrome). Ophthalmic Genet. 2012, 33, 89–95. [Google Scholar] [CrossRef]
- Sharma, S.; Burdon, K.P.; Dave, A.; Jamieson, R.V.; Yaron, Y.; Billson, F.; Van Maldergem, L.; Lorenz, B.; Gécz, J.; Craig, J.E. Novel causative mutations in patients with Nance-Horan syndrome and altered localization of the mutant NHS-A protein isoform. Mol. Vis. 2008, 14, 1856–1864. [Google Scholar] [PubMed]
- Burdon, K.P.; McKay, J.D.; Sale, M.M.; Russell-Eggitt, I.M.; Mackey, D.A.; Wirth, M.G.; Elder, J.E.; Nicoll, A.; Clarke, M.P.; FitzGerald, L.M.; et al. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 2003, 73, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Accogli, A.; Traverso, M.; Madia, F.; Bellini, T.; Vari, M.S.; Pinto, F.; Capra, V. A novel Xp22.13 microdeletion in Nance-Horan syndrome. Birth Defects Res. 2017, 109, 866–868. [Google Scholar] [CrossRef]
- Shoshany, N.; Avni, I.; Morad, Y.; Weiner, C.; Einan-Lifshitz, A.; Pras, E. NHS Gene Mutations in Ashkenazi Jewish Families with Nance-Horan Syndrome. Curr. Eye Res. 2017, 42, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Jansen, A.; Bauters, M.; Froyen, G.; Fryns, J.P. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am. J. Med. Genet. A 2007, 143, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Mathys, R.; Deconinck, H.; Keymolen, K.; Jansen, A.; Van Esch, H. Severe visual impairment and retinal changes in a boy with a deletion of the gene for Nance-Horan syndrome. Bull. Soc. Belge Ophtalmol. 2007, 305, 49–53. [Google Scholar]
- Niccolini, F.; Mencacci, N.E.; Yousaf, T.; Rabiner, E.A.; Salpietro, V.; Pagano, G.; Balint, B.; Efthymiou, S.; Houlden, H.; Gunn, R.N.; et al. PDE10A and ADCY5 mutations linked to molecular and microstructural basal ganglia pathology. Mov. Disord. 2018, 33, 1961–1965. [Google Scholar] [CrossRef]
- Ghosh, S.G.; Becker, K.; Huang, H.; Dixon-Salazar, T.; Chai, G.; Salpietro, V.; Al-Gazali, L.; Waisfisz, Q.; Wang, H.; Vaux, K.K.; et al. Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. Am. J. Hum. Genet. 2018, 103, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Perez-Dueñas, B.; Nakashima, K.; San Antonio-Arce, V.; Manole, A.; Efthymiou, S.; Vandrovcova, J.; Bettencourt, C.; Mencacci, N.E.; Klein, C.; et al. A homozygous loss-of-function mutation in PDE2A associated to early-onset hereditary chorea. Mov. Disord. 2018, 33, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zollo, M.; Ahmed, M.; Ferrucci, V.; Salpietro, V.; Asadzadeh, F.; Carotenuto, M.; Maroofian, R.; Al-Amri, A.; Singh, R.; Scognamiglio, I.; et al. PRUNE is crucial for normal brain development and mutated in microcephaly with neurodevelopmental impairment. Brain 2017, 140, 940–952. [Google Scholar] [CrossRef]
- Coleman, J.; Jouannot, O.; Ramakrishnan, S.K.; Zanetti, M.N.; Wang, J.; Salpietro, V.; Houlden, H.; Rothman, J.E.; Krishnakumar, S.S. PRRT2 Regulates Synaptic Fusion by Directly Modulating SNARE Complex Assembly. Cell Rep. 2018, 22, 820–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.U.; Khalid, H.; Salpietro, V.; Weber, K.T. Idiopathic intracranial hypertension associated with either primary or secondary aldosteronism. Am. J. Med. Sci. 2013, 346, 194–198. [Google Scholar] [CrossRef]
- Salpietro, V.; Lin, W.; Vedove, A.D.; Storbeck, M.; Liu, Y.; Efthymiou, S.; Manole, A.; Wiethoff, S.; Ye, Q.; Saggar, A.; et al. Homozygous mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann. Neurol. 2017, 81, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manole, A.; Jaunmuktane, Z.; Hargreaves, I.; Ludtmann, M.H.R.; Salpietro, V.; Bello, O.D.; Pope, S.; Pandraud, A.; Horga, A.; Scalco, R.S.; et al. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain 2017, 140, 2820–2837. [Google Scholar] [CrossRef] [Green Version]
- Pavlidou, E.; Salpietro, V.; Phadke, R.; Hargreaves, I.P.; Batten, L.; McElreavy, K.; Pitt, M.; Mankad, K.; Wilson, C.; Cutrupi, M.C.; et al. Pontocerebellar hypoplasia type 2D and optic nerve atrophy further expand the spectrum associated with selenoprotein biosynthesis deficiency. Eur. J. Paediatr. Neurol. 2016, 20, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Efthymiou, S.; Manole, A.; Maurya, B.; Wiethoff, S.; Ashokkumar, B.; Cutrupi, M.C.; Dipasquale, V.; Manti, S.; Botia, J.A.; et al. A loss-of-function homozygous mutation in DDX59 implicates a conserved DEAD-box RNA helicase in nervous system development and function. Hum. Mutat. 2018, 39, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Efthymiou, S.; Salpietro, V.; Malintan, N.; Poncelet, M.; Kriouile, Y.; Fortuna, S.; De Zorzi, R.; Payne, K.; Henderson, L.B.; Cortese, A.; et al. Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain 2019, 142, 2948–2964. [Google Scholar] [CrossRef] [PubMed]
- Horan, M.; Billson, F. X-linked cataract and Hutchinsonian teeth. J. Paediatr. Child Health 1974, 10, 98–102. [Google Scholar] [CrossRef]
- Nance, W.E.; Warburg, M.; Bixler, D.; Helveston, E.M. Congenital X-linked cataract, dental anomalies and brachymetacarpalia. Birth Defects Orig. Artic. Ser. 1974, 10, 285–291. [Google Scholar]
- Stambolian, D.; Lewis, R.A.; Buetow, K.; Bond, A.; Nussbaum, R. Nance-Horan syndrome: Localization within the region Xp21.1-Xp22.3 by linkage analysis. Am. J. Hum. Genet. 1990, 47, 13–19. [Google Scholar]
- Eran, P.; Almogit, A.; David, Z.; Wolf, H.R.; Hana, G.; Yaniv, B.; Elon, P.; Isaac, A. The D144E substitution in the VSX1 gene: A non-pathogenic variant or a disease causing mutation? Ophthalmic Genet. 2008, 29, 53–59. [Google Scholar] [CrossRef]
- Musleh, M.; Hall, G.; Lloyd, I.C.; Gillespie, R.L.; Waller, S.; Douzgou, S.; Clayton-Smith, J.; Kehdi, E.; Black, G.C.; Ashworth, J. Diagnosing the cause of bilateral paediatric cataracts: Comparison of standard testing with a next-generation sequencing approach. Eye 2016, 30, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Wirth, M.G.; Russell-Eggitt, I.M.; Craig, J.E.; Elder, J.E.; Mackey, D.A. Aetiology of congenital and paediatric cataract in an Australian population. Br. J. Ophthalmol. 2002, 86, 782–786. [Google Scholar] [CrossRef]
- Trumler, A.A. Evaluation of pediatric cataracts and systemic disorders. Curr. Opin. Ophthalmol. 2011, 22, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014, 121, 2124–2137. [Google Scholar] [CrossRef] [PubMed]
- Gjørup, H.; Haubek, D.; Jacobsen, P.; Ostergaard, J.R. Nance-Horan syndrome-the oral perspective on a rare disease. Am. J. Med. Genet. A 2017, 173, 88–98. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casto, C.; Dipasquale, V.; Ceravolo, I.; Gambadauro, A.; Aliberto, E.; Galletta, K.; Granata, F.; Ceravolo, G.; Falzia, E.; Riva, A.; et al. Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome. Brain Sci. 2021, 11, 1150. https://doi.org/10.3390/brainsci11091150
Casto C, Dipasquale V, Ceravolo I, Gambadauro A, Aliberto E, Galletta K, Granata F, Ceravolo G, Falzia E, Riva A, et al. Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome. Brain Sciences. 2021; 11(9):1150. https://doi.org/10.3390/brainsci11091150
Chicago/Turabian StyleCasto, Celeste, Valeria Dipasquale, Ida Ceravolo, Antonella Gambadauro, Emanuela Aliberto, Karol Galletta, Francesca Granata, Giorgia Ceravolo, Emanuela Falzia, Antonella Riva, and et al. 2021. "Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome" Brain Sciences 11, no. 9: 1150. https://doi.org/10.3390/brainsci11091150
APA StyleCasto, C., Dipasquale, V., Ceravolo, I., Gambadauro, A., Aliberto, E., Galletta, K., Granata, F., Ceravolo, G., Falzia, E., Riva, A., Piccolo, G., Cutrupi, M. C., Striano, P., Accogli, A., Zara, F., Di Rosa, G., Gitto, E., Calì, E., Efthymiou, S., ... Chimenz, R. (2021). Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome. Brain Sciences, 11(9), 1150. https://doi.org/10.3390/brainsci11091150