1. Introduction
The chromatin of eukaryotes has different levels of organization. The nucleosome level is the lowest level, while on a larger scale chromatin forms topologically associating domains (TADs), separated from each other by boundaries [
1]. The boundary regions are usually marked by CTCF and cohesin—chromatin architecture factors. Another type of chromatin structure is the so-called A and B compartments, indicative of the spatial segregation of euchromatin and heterochromatin. Importantly, alterations in the chromatin architecture are linked with cancer and developmental diseases [
2,
3].
DNA double-strand breaks (DSB) change the chromatin architecture around them. In particular, microscopy showed that DSBs cause structural stabilization of chromatin [
4]. The study [
4] demonstrated that the proteins 53BP1 and RIF1 form a module that stabilizes the three-dimensional structure of chromatin at sites of DNA double-strand breaks. Their accumulation at specific regions of chromatin organizes neighboring structures into an ordered, circular arrangement, preventing aberrant DNA processing and maintaining the integrity of the genome and epigenetic information. Hi-C experiments revealed that a local DSB leads to the formation of a DNA damage repair foci around it, potentially formed by one-sided cohesin-mediated loop extrusion [
5]. Therefore, genotoxic anticancer drugs are expected to significantly alter the chromatin structure of cells at many loci or throughout the genome. However, how physiologically relevant concentrations of the DSBs-inducing anticancer drugs change the architecture of the genome is not studied. Therefore, we sought to investigate the consequence of doxorubicin treatment on the higher order chromatin structure, which induces DSBs in active promoters [
6].
Doxorubicin is a commonly used drug, DNA intercalator, and Topoisomerase 2 (Top2) inhibitor [
7]. Top2 inhibition by doxorubicin is well-characterized, as well as catalytic inhibition of Top2, and leads to cell cycle arrest [
8]. Mammalian cells have a “decatenation checkpoint” and spindle assembly checkpoint [
9]. Chemical inhibition of Top2 by topoisomerase poisons, catalytic inhibitors like ICRF-193, or the expression of a catalytically compromised Top2 leads to cell cycle arrest at the G2/M transition [
10]. Previously, physiological doses of doxorubicin and other anthracyclines have been shown to increase nucleosome turnover or eviction around active gene promoters [
7]. Interestingly, doxorubicin-induced DSBs also occur preferentially around promoters of active genes [
6]. Another Top2 inhibitor, etoposide, increases DSBs around promoters as well [
6]. A more recent study linked Top2-mediated DSBs to the chromatin architecture: they are enriched in chromatin loop anchors with high transcriptional activity [
11]. Based on these data we hypothesized that Top2-targeted chemotherapy affects the spatial organization of chromatin at active promoters, which could be detected by 3C methods.
Interestingly, human breast cancer cells with mutations, which cause doxorubicin resistance, have altered chromatin architecture in comparison with wild type cells [
12]. This long-term consequence of doxorubicin treatment indicates that genotoxic chemotherapy can have a mutation-mediated effect on chromatin structure. However, if doxorubicin treatment has a direct and immediate effect on chromatin spatial organization is not known.
Here, we show that physiological concentrations of doxorubicin significantly change the spatial chromatin structure of human RPE1 cells at many genomic loci. Doxorubicin treatment led to the reduction in Hi-C interactions at many regions of the genome. These regions covered clusters of active promoters. Additionally, we observed differential CTCF binding in active genomic regions after doxorubicin treatment. Doxorubicin-sensitive genomic regions had a preferential spatial organization: the treatment changed contacts between TADs. Moreover, both doxorubicin and ICRF193 (a catalytic inhibitor of Top2) similarly changed human chromatin compaction.
3. Results
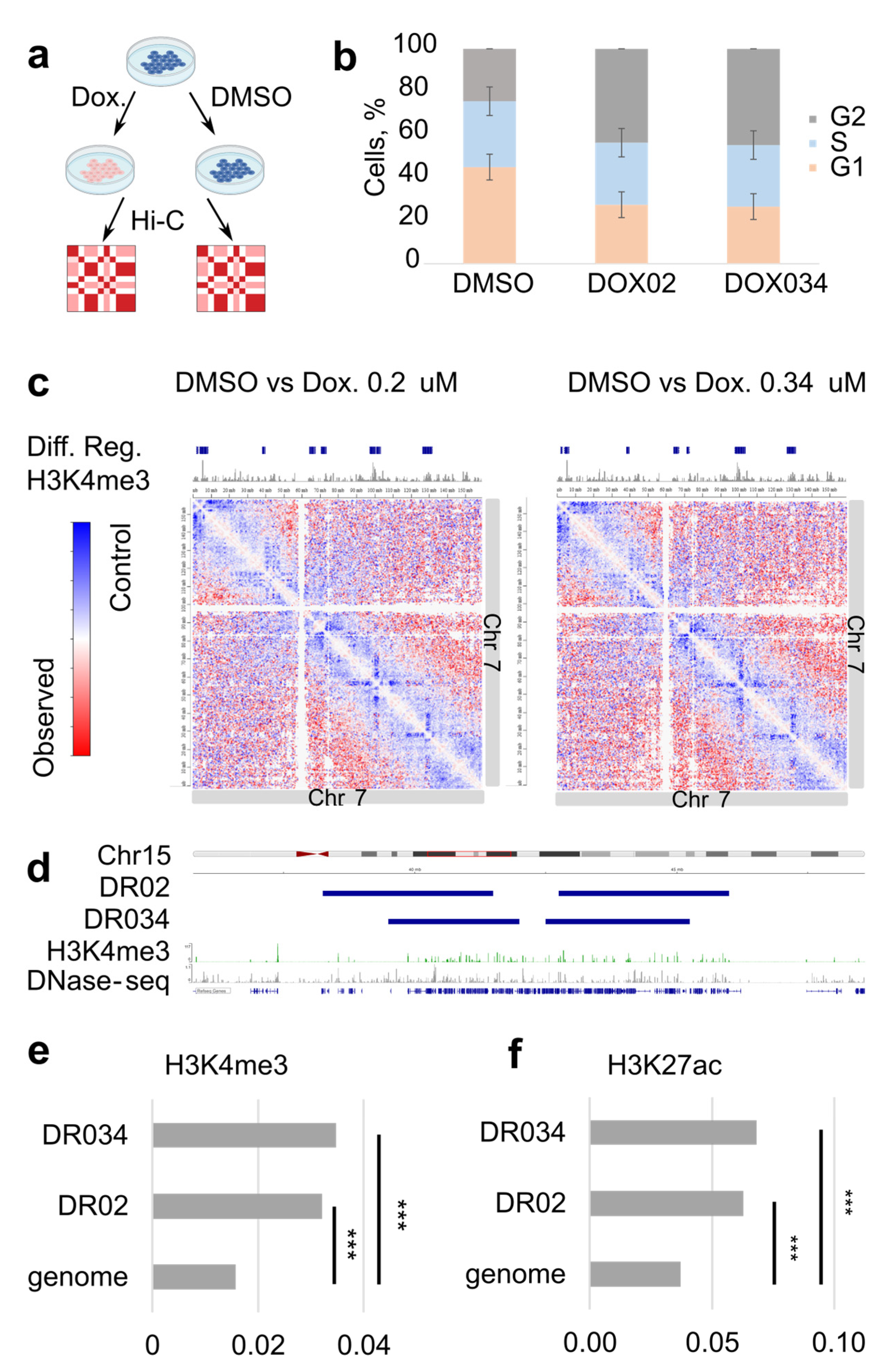
To gain insight into the chromatin architecture of the cells treated with doxorubicin, we treated human RPE1 cells with two physiologically relevant concentrations of the drug, 0.2 and 0.34 μM [
7,
13] (
Figure 1a). The treatment duration was set to 18 h, as this timing has been previously employed to investigate the effects of doxorubicin on chromatin [
7]; and after the 18-h treatment, doxorubicin was found to enhance nucleosome turnover around active promoters. We assessed the intranuclear localization of doxorubicin by detecting its autofluorescence (
Figure S1a), analyzed the cell cycle (
Figure 1b), and monitored cell viability. Furthermore, we conducted RNA-seq on both the treated and control cells to evaluate the RPE1 cell response to doxorubicin treatment under our experimental conditions. After 18 h, the presence of doxorubicin within the nucleus of RPE1 cells was clearly observable (
Figure S1a). This led to a significant increase in the proportion of cells in the G2/M stage of the cell cycle, indicating an arrest dependent on TOP2A (
Figure 1b) [
28]. Notably, we did not observe an increase in cell death within the culture at this time point, as cell viability remained around 95% for both the control and treated samples. Concurrently, we observed significant upregulation of stress-response genes and downregulation of cell cycle genes (
Table S1), suggesting that our treatment duration and concentrations were sufficient to induce the expected response in the cells.
Next, we generated and compared Hi-C maps of the treated and control cells (
Figure S1b–e). The Hi-C maps of both control and doxorubicin-treated cells had typical structural features, such as TADs, boundaries, and compartments (
Figure S1b–d). First, we analyzed whether there are whole-genome changes between Hi-C maps of the treated and control samples. We performed TAD-calling followed by pileup analysis of TADs and did not observe differences between the samples (
Figure S1c). The aggregated analysis of compartments (
Figure S1d) showed that in the doxorubicin-treated samples, the A and B compartments are present and are as well defined, as in control cells (
Figure S1d). However, we observed a reproducible whole-genome difference in the distance decay of the Hi-C contacts (
Figure S1e). Doxorubicin-treated cells demonstrated an increase in chromatin interactions at a distance of 1 Mb and a decrease at the distance of 10 Mb compared to the control (
Figure S1e). Overall, these data suggest that the genome-wide chromatin structure is mostly intact after doxorubicin treatment, but the chromatin compaction changes.
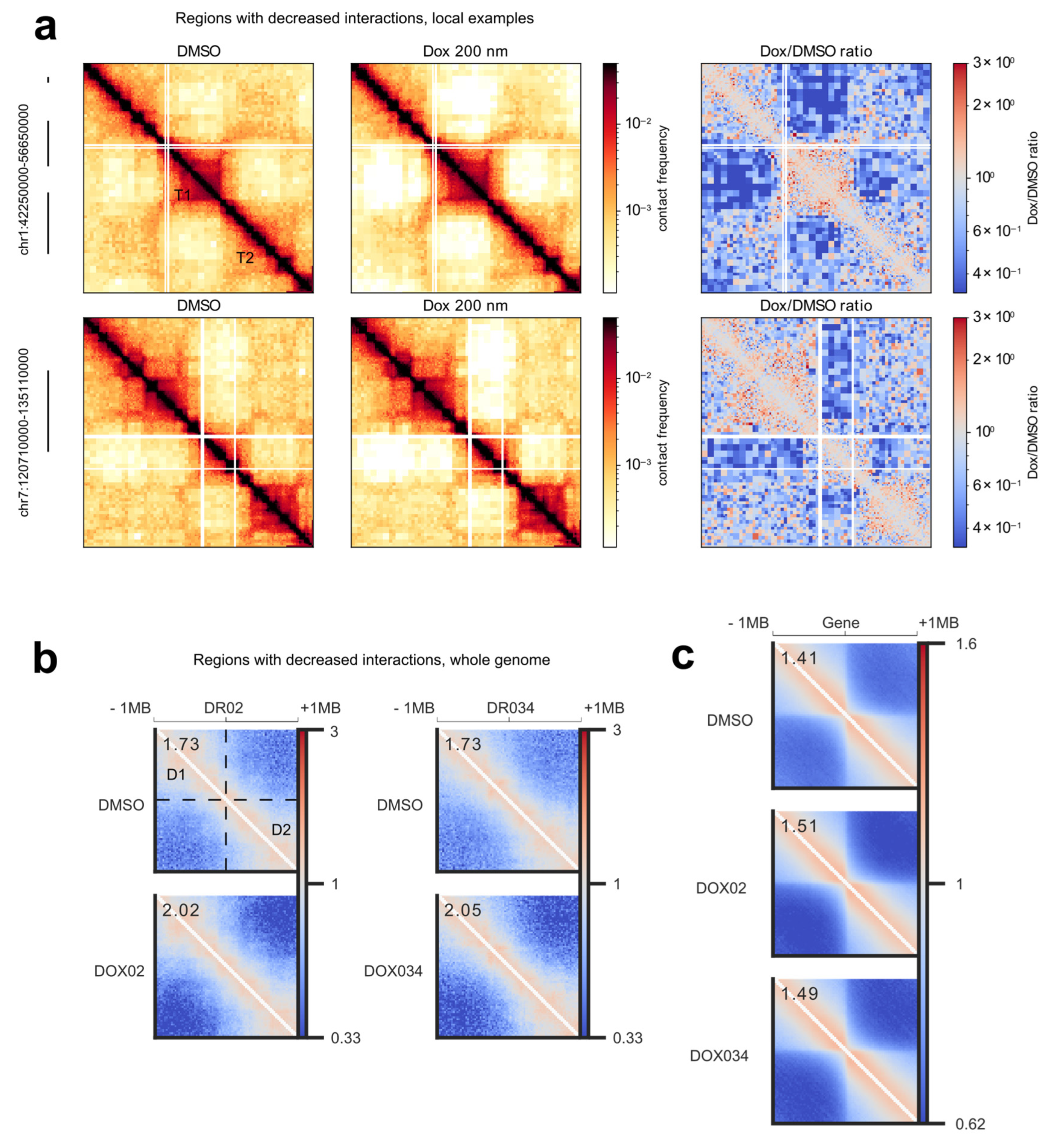
Next, we searched for local changes on the Hi-C maps by pairwise comparison of the treated and control chromosomes (
Figure 1c). To perform the comparison, we divided Hi-C maps of the treated samples by control map, resulting in differential maps for all chromosomes. On the differential map, we observed a clear reduction in the local Hi-C contacts upon treatment at many loci close to the main diagonal (
Figure 1c). This observation indicates that anticancer treatment can affect the structure of the chromatin locally.
We hypothesized that decreased interactions can be the result of the lower coverage of the Hi-C map in these genomic regions. To verify this, we calculated and compared the coverage of the control and doxorubicin-treated map (
Figure S2). We did not observe differences in coverage for the regions with decreased interaction, neither for normalized nor for raw maps (
Figure S2). These data suggest that the observed differences are not connected with sequencing issues but rather with the biological effect of doxorubicin.
We next assessed whether regions of decreased interactions colocalize with active promoters, since doxorubicin-induced DSBs and histone exchange accumulate there. We mapped differential Hi-C regions and compared these to the H3K4me3 ChIP-seq which marks active promoters (
Figure 1c,d). Regions of decreased interactions usually covered genomic regions with clusters of many active promoters (
Figure 1d). Hypergeometric testing demonstrated that the H3K4me3 peaks are significantly enriched in the regions of decreased interactions in comparison with the entire genome (
p-value < 10
−16) (
Figure 1e). Peaks of H3K27ac demonstrated significant enrichment in these regions too (
p-value < 10
−16) (
Figure 1f). Based on these observations, we suggest that the changes in the local chromatin architecture harbor active regulatory regions of the genome.
Next, we conducted a more detailed analysis of the structure of the altered regions. Firstly, through visual examination of the regions exhibiting decreased interactions, we observed that these interactions frequently occurred between TADs (
Figure 2a). Secondly, we aimed to determine if the regions with decreased Hi-C interactions displayed similarities in chromatin structure, specifically, whether their positioning at the borders of chromatin domains was a genome-wide trend. To investigate the structural similarity of these areas, we performed a pileup analysis of the chromatin architecture within the altered regions (
Figure 2b). We accumulated Hi-C contacts from all identified sites with decreased interactions, along with 1 MB flanking regions, in a single plot. If the chromatin structure of the regions were random, the pileup analysis would reveal no discernible pattern, as the structural domains would be averaged out. We would only observe an increase in interactions closer to the diagonal. However, as anticipated from our analysis of local regions (
Figure 2a), the pileup analysis demonstrated a consistent structural pattern on average within the altered regions (
Figure 2b). Specifically, we observed two TADs separated from each other, and doxorubicin treatment reduced interactions between these two domains (
Figure 2b). Additionally, we identified smaller domains along the diagonal, likely corresponding to sub-TADs.
A comparison of the piled chromatin structures between the control and treatment conditions clearly revealed an overall increase in insulation between the two TADs in the treatment group. This allowed for a quantitative comparison of the insulation increase induced by the two concentrations of doxorubicin (
Figure 2b). Notably, both physiologically relevant drug concentrations resulted in the exact same average increase in insulation.
Next, we addressed the relationship between the structure of the regions with decreased interactions and the clusters of active promoters. In one of the pioneering studies of the human genome architecture, it was shown that house-keeping genes are often located on the TAD’s boundaries [
29]. Furthermore, a more recent investigation demonstrated that individual genes act as insulators, with active genes exhibiting stronger insulation compared to inactive genes [
30]. Given our observation that regions with decreased interactions surround clusters of active promoters, we hypothesized that transcriptional activity contributes to the structure of doxorubicin-sensitive regions. To verify this hypothesis, we initially focused on the chromatin architecture surrounding all active genes in the RPE1 cell line (
Figure 2c, top). Our analysis confirmed a previously observed phenomenon in a distinct experimental system: transcribed genes tended to be located preferentially at the boundaries of TADs (
Figure 2c, top). Subsequently, we examined whether treatment with doxorubicin altered the insulation properties of these boundaries (
Figure 2c, middle and bottom). We showed that doxorubicin reproducibly increased insulation around active genes (
Figure 2c). Consequently, we conclude that doxorubicin impacts the chromatin structure surrounding active genes in general. When these active genes form clusters, the resultant changes accumulate locally, and we observe these genomic locations as regions with decreased interactions. Considering that normally transcribed genes themselves possess insulation properties and tend to be situated at TAD boundaries, we discern a repetitive structural pattern within the regions exhibiting decreased interactions.
Next, we tested whether the changes in the Hi-C contacts are independent of cell cycle arrest and are rather explained by the DSBs and histone exchange. We treated cells with ICRF193—a catalytic inhibitor of Top2 [
31], which leads to Top2-dependent cell cycle arrest, similar to doxorubicin. After 18 h of treatment, two different concentrations of ICRF193 (5 μM and 0.5 μM) led to the same significant increase in the cells in the G2/M stages of the cell cycle and almost complete depletion of the cells in the G1 stage of the cell cycle (
Figure 3a). These data indicate that ICRF193 efficiently leads to TOP2-dependent cell cycle arrest in our experimental system.
We performed Hi-C on ICRF193-treated cells and compared them with the control. Importantly, decreased interactions did not appear in the ICRF193-treated sample, indicating that they are independent of Top2 inhibition and cell cycle arrest (
Figure 3b).
Interestingly, treatment with ICRF193 led to the same change in the distance decay of Hi-C contacts as doxorubicin (
Figure 3c and
Figure S1e). The result appeared reproducible with two different concentrations of ICRF193 (
Figure 3c). That means that the change in general chromatin compaction is a result of Top2-inhibition or Top2-dependent cell cycle arrest (
Figure 3c). Our data indicate that both catalytic inhibition of Top2 by ICRF193 and indirect inhibition of Top2 by doxorubicin lead to the change of the distance decay of Hi-C contacts. Therefore, we speculate that Top2 inhibitors in general change the chromatin compaction state of cycling cells.
Next, to test if the regions with decreased interactions are enriched for differentially expressed genes, we performed RNA-seq for the control and doxorubicin-treated cells (
Figure 3d–f). We observed no enrichment of differentially expressed genes at the regions with decreased interactions (
Figure 3f). Gene ontology analysis showed up-regulation of apoptotic genes and downregulation of the genes responsible for cell cycle progression in treated samples (
Table S1). These results indicate that the decreased genome interactions around active regulatory regions are independent of differential gene expression in these regions. Meanwhile, the differential expression could be explained by the stress response in the cells treated with doxorubicin.
Furthermore, we investigate what other chromatin changes are occurring upon genotoxic drug treatment. We performed ChIP-seq for CTCF and RAD21 under the same experimental conditions and compared their binding between treated samples and control cells (
Figure 4a,b,
Table S2). We detected differential binding of CTCF in the doxorubicin-treated cells but not in ICRF193-treated cells (
Table S2). Of the differential CTCF peaks, 89% corresponded to increased binding in treatment (
Figure 4b,c,
Table S2). Meanwhile, for RAD21 we did not detect differential peaks with the chosen threshold (FDR < 0.05) in any of the samples (
Table S2). Next, we checked whether the differential CTCF peaks are overrepresented in active regulatory regions marked by H3K27 acetylation. We performed a hypergeometric test and showed significant enrichment of differential CTCF in H3K27ac regions compared to the whole genome (
Figure 4d). This means that in doxorubicin-treated cells, some CTCF binding sites inside active regulatory regions become accessible/more accessible to the factor.
Next, we evaluated if increased CTCF binding can contribute to the chromatin structure changes after doxorubicin treatment. We aligned the coordinates of the differential CTCF peaks with differential Hi-C regions (
Figure 4c,e). We observed that sometimes regions with decreased interactions contain a differential CTCF peak (
Figure 4e), including de-novo appeared peaks (
Figure 4c). However, many regions with decreased Hi-C contacts did not overlap with the differential CTCF sites (
Figure 4e). Therefore, we speculated that increased CTCF binding is not necessary for the appearance of decreased Hi-C contacts but can facilitate it, if present.
Finally, we evaluated this assumption and checked if increased CTCF binding can contribute to the changes in chromatin structure. We performed a pileup analysis of the chromatin architecture, surrounding differential CTCF peaks. To do that we took coordinates of all the CTCF peaks overrepresented in treatment, added 1 Mb of flanking regions from both sides, and averaged Hi-C contacts for all such regions on one plot (
Figure 4f). Interestingly, the pileup analysis of the control Hi-C showed that new CTCF peaks appear at the pre-existing strong boundaries (
Figure 4f, top). Doxorubicin treatment led to a slight but reproducible increase in the insulation in these boundaries (
Figure 4f, bottom). Therefore, we conclude that increased CTCF binding contributes to the changes in chromatin structure after doxorubicin treatment.
Finally, our objective was to investigate the mechanistic factors contributing to the emergence of regions with decreased interactions. We focused on two key observations: (i) regions exhibiting decreased interactions were found surrounding active promoters marked by H3K27ac (
Figure 1d–f), and (ii) doxorubicin induces DNA double-strand breaks (DSBs) in the vicinity of active promoters [
6]. Previous studies have demonstrated that DSBs occurring at transcribed genes are repaired at a faster rate, suggesting a process known as transcription-coupled DNA double-strand break repair [
32]. It is noteworthy that both RNA polymerase II and DSBs can influence the distribution of cohesin complexes in their proximity [
5,
30,
33]. Therefore, we aimed to investigate whether the distribution of cohesin within regions displaying decreased interactions would undergo changes (
Figure 5a and
Figure S3).
To address this, we plotted the RAD21 signal around the H3K27ac peaks within the regions exhibiting decreased interactions, and interestingly, we observed a broader distribution of the RAD21 signal around these sites (
Figure 5a). This result was consistent across the differential regions identified from the differential Hi-C maps for both concentrations of doxorubicin. Notably, a higher concentration of the drug resulted in a slightly more pronounced redistribution of RAD21 at these sites (
Figure 5a and
Figure S3). Consequently, in our experimental system, doxorubicin treatment did not induce the appearance of significantly differential RAD21 peaks, but it did lead to the redistribution of the RAD21 signal around active and acetylated sites.
4. Discussion
Here, we investigated the effects of the physiological concentrations of the anticancer genotoxic drug doxorubicin on the chromatin structure. We showed that treatment of human cells with doxorubicin leads to the appearance of local areas of reduced Hi-C contacts around active promoters.
An increasing body of evidence supports the role of cohesin in maintaining genome integrity [
5,
34,
35]. For instance, cohesin is necessary to suppress transcription at DNA DSBs during interphase [
34]. Cohesin accumulates at sites of damaged DNA [
36]. Moreover, cohesin is involved in sister chromatid cohesion during homologous recombination at DSBs in S/G2 phase cells [
37]. Mechanistically, cohesin accumulates on both sides of a DSB, regardless of the repair pathway used, resulting in one-sided loop extrusion that extends towards the surrounding regions on both sides of the break [
5]. Consistent with these findings, we observed a broader distribution of cohesin around active regulatory elements marked by H3K27ac following doxorubicin treatment. Instead of detecting differential peaks for cohesin, we observed an expanded range for the ChIP-seq signal around these sites. This outcome was expected, as we did not induce local breaks but rather studied randomly positioned breaks, which tend to occur preferentially around active promoters. We propose that the accumulation and redistribution of cohesin contribute to increased insulation within these regions after doxorubicin treatment.
Based on the presented data and existing literature, we propose a model for the formation of regions with decreased interactions following doxorubicin treatment (
Figure 5b). The barrier function of RNA polymerase contributes to the accumulation of cohesin at active promoters [
30]. Doxorubicin treatment induces DNA double-strand breaks (DSBs), which disrupt transcription and result in the accumulation and redistribution of cohesin (
Figure 5a) through at least two pathways: (i) RNA polymerase dependent [
30] and (ii) DSBs dependent [
5]. This leads to increased insulation surrounding active genes in general (
Figure 2c). In cases where active genes cluster together, the increase in insulation is accumulated around each active promoter, leading to distinct changes in insulation that are visually apparent on Hi-C maps at the local level (
Figure 1c,
Figure 2a, and
Figure 5b).
Notably, when local DSBs were studied using Hi-C, the broken regions had increased genomic interactions in comparison with the control [
5], while we see the decrease in Hi-C contacts, but on a larger scale (
Figure 2a,b). The difference most likely comes from the fact that upon doxorubicin treatment many DSBs accumulate around many active promoters, and they are being repaired. Therefore, under our conditions, it is impossible to dissect a change in chromatin structure caused by a single break. We rather detect a general trend, resulting from the accumulation of DNA damage and increased histone turnover, reflecting the situation in the cells of the patients.
Our data indicate differential binding of CTCF in active regulatory regions of the genome after doxorubicin treatment. Interestingly, CTCF directly binds DNA and is a nucleosome-dependent factor [
38], while RAD21 is part of the cohesin complex, which is expected to be nucleosome independent. Based on this, we speculate that the increase in CTCF binding in doxorubicin-treated samples could be the result of nucleosome destabilization around active regulatory regions of the genome [
7]. Both CTCF and RAD21 form ChIP-seq peaks on TADs boundaries. Their activity is tightly linked in the loop extrusion model, according to which an ATP-dependent motor cohesin gradually enlarges chromatin loops until it reaches boundaries marked by CTCF [
39]. We suggest, as a consequence of nucleosome destabilization, CTCF can bind in the vicinity of active regulatory regions. That would facilitate chromatin rearrangement and compaction, making boundaries stronger (
Figure 4f).
An alternative explanation for the observed differential CTCF binding could be attributed to the role of DNA methylation. DNA methylation has been shown to interfere with CTCF binding [
40]. Experimental studies involving targeted de novo methylation of a CTCF loop anchor site using dCas9-Dnmt3a have demonstrated blocked CTCF binding and disrupted DNA looping, resulting in altered gene expression [
40]. Furthermore, DNA methylation has been found to co-occur with H3K27ac in bivalent regulatory regions [
41]. Thus, it is plausible that doxorubicin treatment may impair DNA methylation, leading to increased CTCF binding. Further investigations are needed to elucidate the precise mechanisms underlying the relationship between doxorubicin, DNA methylation and/or nucleosome stability, and CTCF binding, which will enhance our understanding of the impact of doxorubicin on chromatin dynamics and gene regulation.
Interestingly, our analysis reveals that the chromatin architecture of regions with decreased interactions is not random. Frequently, doxorubicin treatment leads to a reduction in genomic contacts between two large domains containing subdomains (
Figure 2a,b). We attribute this observation to the preferential positioning of active genes on the boundaries of TADs in RPE1 cells, as well as in other previously studied cell lines [
29,
30]. This finding suggests that specific chromatin structures may be more susceptible to the effects of doxorubicin and possibly other genotoxic drugs. It is known that loop boundaries represent more fragile genomic regions [
42]. Expanding on this knowledge, our results demonstrate that boundaries with clusters of active genes undergo the most significant spatial reorganization following doxorubicin treatment.
Previously, the Top2-inhibiting drug CBL0137 was shown to alter the chromatin architecture [
43]. In contrast to the effects shown here, CBL0137 weakened the boundaries, increasing contacts between different TADs [
43]. In particular, CBL0137 is a DNA intercalator as well as doxorubicin and all other curaxins and anthracyclines [
44]. These data suggest that treatments with DNA intercalators tend to cause changes in the 3D chromatin structure, affecting genomic contacts and interactions of certain proteins with DNA. And the specificity of the change depends on the type of the intercalator.
Anthracyclines, such as doxorubicin, are widely utilized and effective anticancer drugs in the treatment of various solid tumors and hematologic malignancies [
45]. The changes in genome architecture demonstrated in this study could potentially represent a common side effect of anticancer drug treatments. It is worth noting that cohesin subunits are frequently mutated in cancer [
5]. For instance, there are known cancer-associated mutations in the SA2 subunit, which impair its ability to repress transcription at DSBs while still supporting sister chromatid cohesion [
5]. The presence of mutated cohesin, along with a favorable chromatin structure for its accumulation, may create a positive feedback loop that promotes the accumulation of mutations in these hotspots. Therefore, in the future, it would be significant to understand how the changes in chromatin architecture contribute to the therapeutic effects and side effects of chemotherapy.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}