


Assessing the Influence of HGT on the Evolution of Stress Responses in Microbial Communities from Shark Bay, Western Australia

Abstract
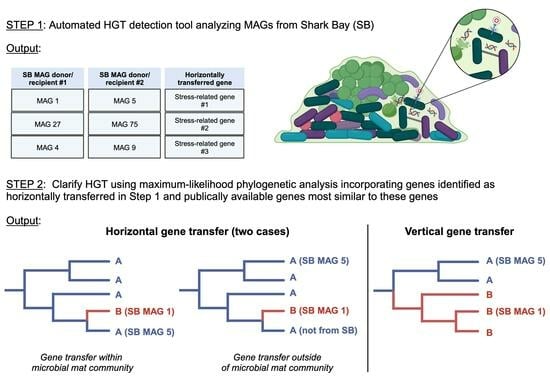
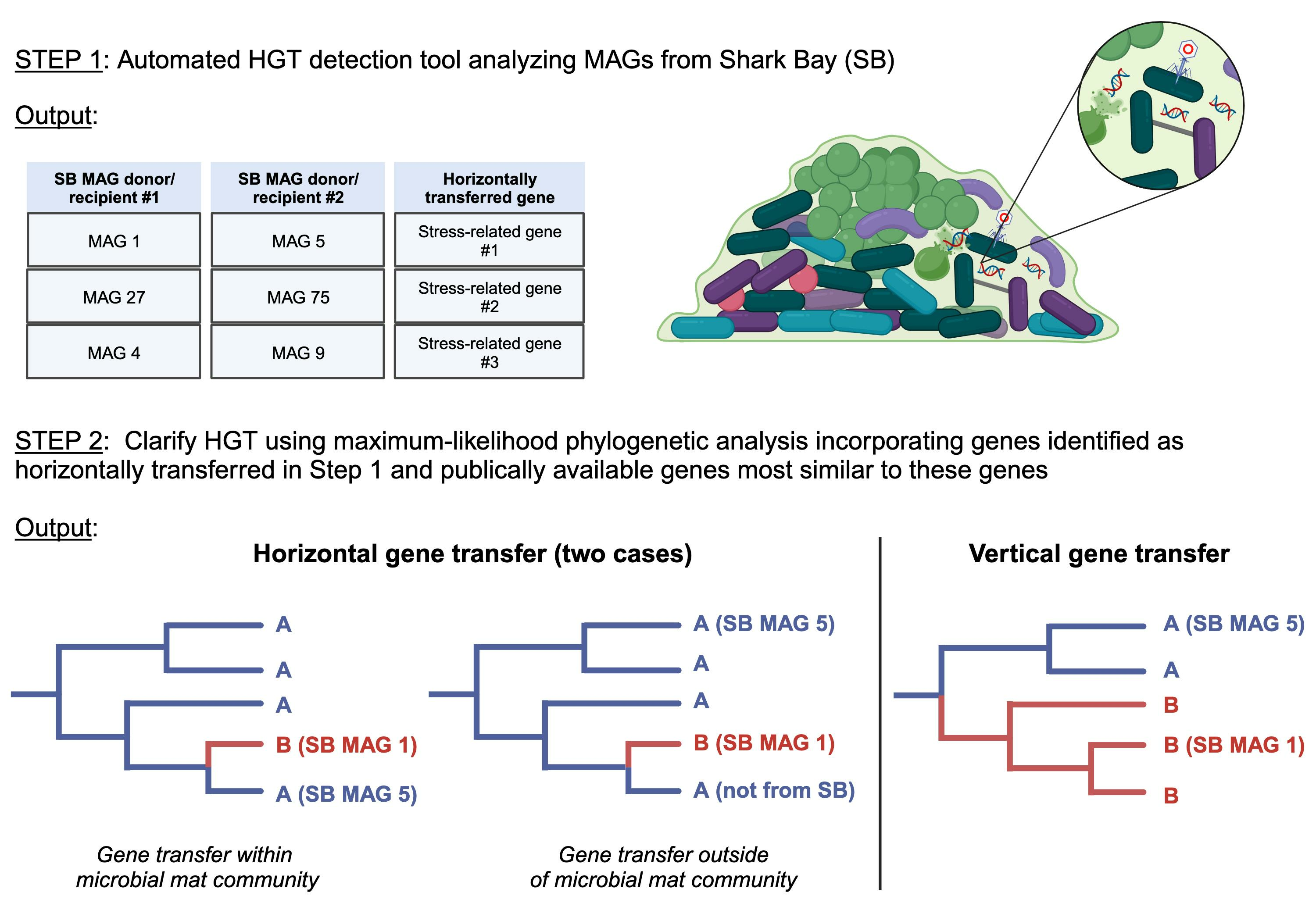
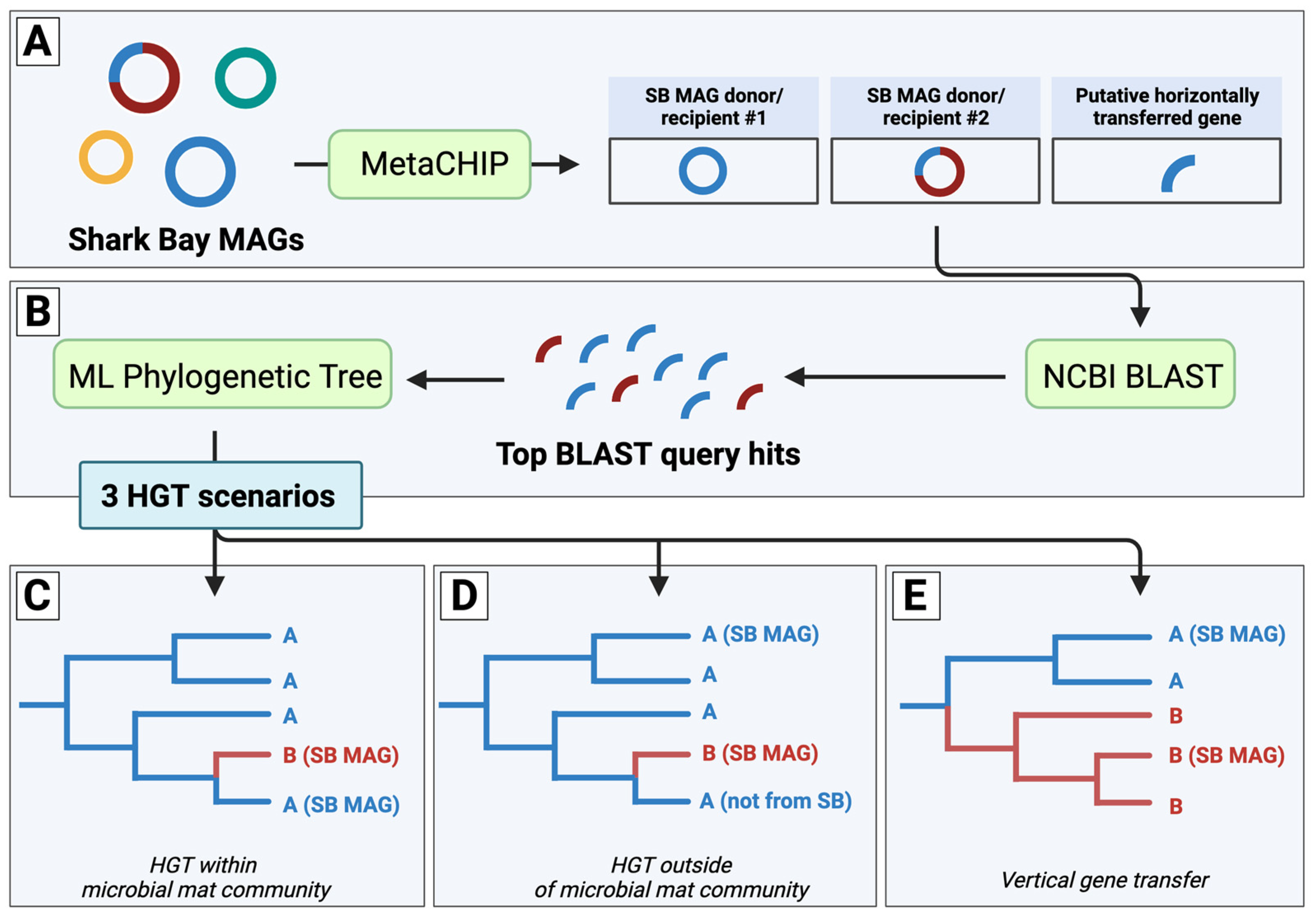
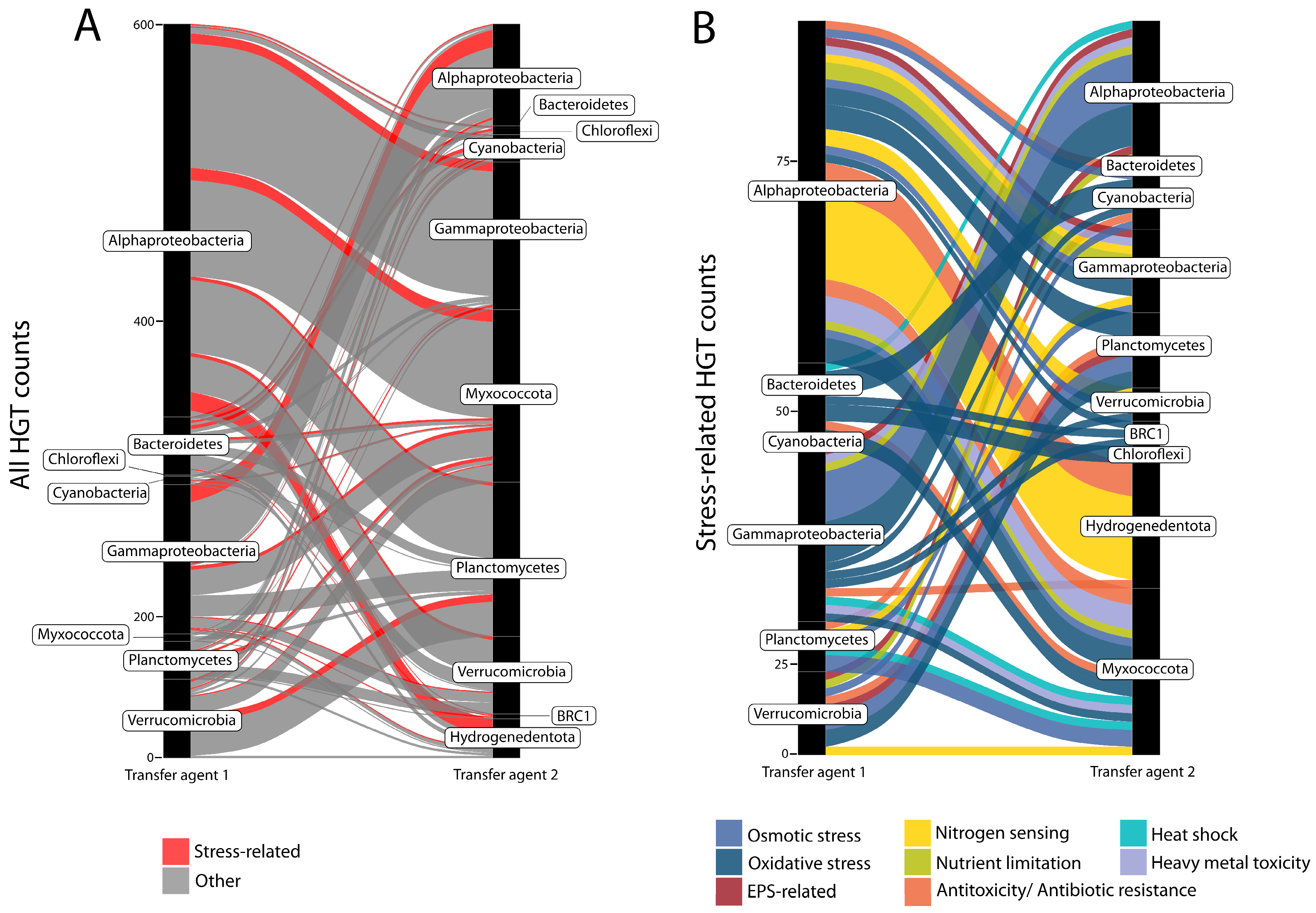
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Procurement
2.2. HGT Predictions
2.3. Phylogenomic and HGT Analyses
3. Results
3.1. Osmotic Stress
3.2. Oxidative Stress
3.3. Arsenic Toxicity
3.4. Relative Timing of HGT Events
4. Discussion
4.1. Adaptations to Osmotic Stress
4.2. Adaptations to Oxidative Stress
4.3. Adaptations to Arsenic Toxicity
4.4. Ecological Context of HGT Events
4.5. Relative Timing of Gene Transfers and Identification of Relationships among Microbes in Complex Communities
4.6. Augmenting ‘Stand-Alone’ HGT Detection Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Garcia-Pichel, F. Solar ultraviolet and the evolutionary history of cyanobacteria. Orig. Life Evol. Biosph. 1998, 28, 321–347. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pichel, F.; Sherry, N.D.; Castenholz, R.W. Evidence for an ultraviolet sunscreen role of the extracellular pigment scytonemin in the terrestrial cyanobacterium Chiorogloeopsis sp. Photochem. Photobiol. 1992, 56, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pichel, F.; Castenholz, R.W. Characterization and biological implications of scytonemin, a cyanobacterial sheath pigment. J. Phycol. 1991, 27, 395–409. [Google Scholar] [CrossRef]
- Goh, F.; Allen, M.A.; Leuko, S.; Kawaguchi, T.; Decho, A.W.; Burns, B.P.; Neilan, B.A. Determining the specific microbial populations and their spatial distribution within the stromatolite ecosystem of Shark Bay. ISME J. 2009, 3, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Skoog, E.J.; Moore, K.R.; Gong, J.; Ciccarese, D.; Momper, L.; Cutts, E.M.; Bosak, T. Metagenomic, (bio)chemical, and microscopic analyses reveal the potential for the cycling of sulfated EPS in Shark Bay pustular mats. ISME Commun. 2022, 2, 43. [Google Scholar] [CrossRef]
- Wong, H.L.; Smith, D.L.; Visscher, P.T.; Burns, B.P. Niche differentiation of bacterial communities at a millimeter scale in Shark Bay microbial mats. Sci. Rep. 2015, 5, 15607. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; White, R.A.; Visscher, P.T.; Charlesworth, J.C.; Vázquez-Campos, X.; Burns, B.P. Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes. ISME J. 2018, 12, 2619–2639. [Google Scholar] [CrossRef]
- Moore, K.R.; Daye, M.; Gong, J.; Williford, K.; Konhauser, K.; Bosak, T. A review of microbial-environmental interactions recorded in Proterozoic carbonate-hosted chert. Geobiology 2023, 21, 3–27. [Google Scholar] [CrossRef]
- Hofmann, H.J. Precambrian microflora, Belcher Islands, Canada: Significance and systematics. J. Paleontol. 1976, 50, 1040–1073. [Google Scholar]
- Golubic, S.; Hofmann, H.J. Comparison of Holocene and mid-Precambrian Entophysalidaceae (Cyanophyta) in stromatolitic algal mats: Cell division and degradation. J. Paleontol. 1976, 50, 1074–1082. [Google Scholar]
- Brown, A.D. Microbial Water Stress. Bacteriol. Rev. 1976, 40, 803–846. [Google Scholar] [CrossRef] [PubMed]
- Galinski, E.A.; Trüper, H.G. Microbial behaviour in salt-stressed ecosystems. FEMS Microbiol. Rev. 1994, 15, 95–108. [Google Scholar] [CrossRef]
- Ventosa, A.; Nieto, J.J.; Oren, A. Biology of moderately halophilic aerobic bacteria. Microbiol. Mol. Biol. Rev. 1998, 62, 504–544. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Goh, F.; Jeon, Y.J.; Barrow, K.; Neilan, B.A.; Burns, B.P. Osmoadaptive strategies of the archaeon Halococcus hamelinensis isolated from a hypersaline stromatolite environment. Astrobiology 2011, 11, 529–536. [Google Scholar] [CrossRef]
- Ruvindy, R.; White, R.A.; Neilan, B.A.; Burns, B.P. Unravelling core microbial metabolisms in the hypersaline microbial mats of Shark Bay using high-throughput metagenomics. ISME J. 2016, 10, 183–196. [Google Scholar] [CrossRef]
- Campbell, M.A.; Grice, K.; Visscher, P.T.; Morris, T.; Wong, H.L.; White, R.A.; Burns, B.P.; Coolen, M.J.L. Functional gene expression in Shark Bay hypersaline microbial mats: Adaptive responses. Front. Microbiol. 2020, 11, 560336. [Google Scholar] [CrossRef]
- Datta, N.; Kontomichalou, P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 1965, 208, 239–241. [Google Scholar] [CrossRef]
- Abe, K.; Nomura, N.; Suzuki, S. Biofilms: Hot spots of horizontal gene transfer (HGT) in aquatic environments, with a focus on a new HGT mechanism. FEMS Microbiol. Ecol. 2021, 96, fiaa031. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Fulaz, S.; Vitale, S.; Quinn, L.; Casey, E. Nanoparticle–biofilm interactions: The role of the EPS matrix. Trends Microbiol. 2019, 27, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Hausner, M.; Wuertz, S. High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Appl. Environ. Microbiol. 1999, 65, 3710–3713. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, S.J.; Bailey, M.; Hansen, L.H.; Kroer, N.; Wuertz, S. Studying plasmid horizontal transfer in situ: A critical review. Nat. Rev. Microbiol. 2005, 3, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Arsène-Ploetze, F.; Koechler, S.; Marchal, M.; Coppée, J.Y.; Chandler, M.; Bonnefoy, V.; Brochier-Armanet, C.; Barakat, M.; Barbe, V.; Battaglia-Brunet, F.; et al. Structure, function, and evolution of the Thiomonas spp. genome. PLoS Genet. 2010, 6, e1000859. [Google Scholar] [CrossRef] [PubMed]
- Andres, J.; Arsène-Ploetze, F.; Barbe, V.; Brochier-Armanet, C.; Cleiss-Arnold, J.; Coppée, J.Y.; Dillies, M.A.; Geist, L.; Joublin, A.; Koechler, S.; et al. Life in an arsenic-containing gold mine: Genome and physiology of the autotrophic arsenite-oxidizing bacterium Rhizobium sp. NT-26. Genome Biol. Evol. 2013, 5, 934–953. [Google Scholar] [CrossRef] [PubMed]
- Hemme, C.L.; Green, S.J.; Rishishwar, L.; Prakash, O.; Pettenato, A.; Chakraborty, R.; Deutschbauer, A.M.; Van Nostrand, J.D.; Wu, L.; He, Z.; et al. Lateral gene transfer in a heavy metal-contaminated-groundwater microbial community. mBio 2016, 7, 02234-15. [Google Scholar] [CrossRef] [PubMed]
- Neira, G.; Vergara, E.; Cortez, D.; Holmes, D.S. A large-scale multiple genome comparison of acidophilic archaea (ph ≤ 5.0) extends our understanding of oxidative stress responses in polyextreme environments. Antioxidants 2022, 11, 59. [Google Scholar] [CrossRef]
- Probst, A.J.; Banfield, J.F. Homologous recombination and transposon propagation shape the population structure of an organism from the deep subsurface with minimal metabolism. Genome Biol. Evol. 2018, 10, 1115–1119. [Google Scholar] [CrossRef]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef]
- Tamames, J.; Moya, A. Estimating the extent of horizontal gene transfer in metagenomic sequences. BMC Genom. 2008, 9, 136. [Google Scholar] [CrossRef]
- Song, W.; Wemheuer, B.; Zhang, S.; Steensen, K.; Thomas, T. MetaCHIP: Community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches. Microbiome 2019, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, Y.; Li, S. LEMON: A method to construct the local strains at horizontal gene transfer sites in gut metagenomics. BMC Bioinform. 2019, 20, 702. [Google Scholar] [CrossRef] [PubMed]
- McInnes, R.S.; McCallum, G.E.; Lamberte, L.E.; van Schaik, W. Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 2020, 53, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhang, C.; Li, W.; Weng, S.; Song, W.; Li, J.; Wang, Y. Functional diversity of microbial communities in inactive seafloor sulfide deposits. FEMS Microbiol. Ecol. 2021, 97, fiab108. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Zhu, B.; Gao, G.F.; Guo, Y.; Hu, Y. Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 2021, 4, 1305. [Google Scholar] [CrossRef]
- Song, W.; Wemheuer, B.; Steinberg, P.D.; Marzinelli, E.M.; Thomas, T. Contribution of horizontal gene transfer to the functionality of microbial biofilm on a macroalgae. ISME J. 2021, 15, 807–817. [Google Scholar] [CrossRef]
- Hazzouri, K.M.; Sudalaimuthuasari, N.; Saeed, E.E.; Kundu, B.; Al-Maskari, R.S.; Nelson, D.; AlShehhi, A.A.; Aldhuhoori, M.A.; Almutawa, D.S.; Alshehhi, F.R.; et al. Salt flat microbial diversity and dynamics across salinity gradient. Sci. Rep. 2022, 12, 11293. [Google Scholar] [CrossRef]
- Kuppa Baskaran, D.K.; Umale, S.; Zhou, Z.; Raman, K.; Anantharaman, K. Metagenome-based metabolic modelling predicts unique microbial interactions in deep-sea hydrothermal plume microbiomes. ISME Commun. 2023, 3, 42. [Google Scholar] [CrossRef]
- Wang, H.; Min, C.; Xia, F.; Xia, Y.; Tang, M.; Li, J.; Hu, Y.; Zou, M. Metagenomic analysis reveals the short-term influences on conjugation of blaNDM-1 and microbiome in hospital wastewater by silver nanoparticles at environmental-related concentration. Environ. Res. 2023, 228, 115866. [Google Scholar] [CrossRef]
- Douglas, G.M.; Langille, M.G.I. Current and promising approaches to identify horizontal gene transfer events in metagenomes. Genome Biol. Evol. 2019, 11, 2750–2766. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.; Diaconis, P. On the histogram as a density estimator: L2 Theory. Probab. Theory Relat. Fields 1981, 57, 453–476. [Google Scholar] [CrossRef]
- Kunte, H.J. Osmoregulation in Bacteria: Compatible solute accumulation and osmosensing. Environ. Chem. 2006, 3, 94–99. [Google Scholar] [CrossRef]
- Chen, S.Y.; Lai, M.C.; Lai, S.J.; Lee, Y.C. Characterization of osmolyte betaine synthesizing sarcosine dimethylglycine N-methyltransferase from Methanohalophilus portucalensis. Arch. Microbiol. 2009, 191, 735–743. [Google Scholar] [CrossRef]
- Nannapaneni, P.; Hertwig, F.; Depke, M.; Hecker, M.; Mäder, U.; Völker, U.; Steil, L.; van Hijum, S.A.F.T. Defining the structure of the general stress regulon of Bacillus subtilis using targeted microarray analysis and random forest classification. Microbiology 2012, 158, 696–707. [Google Scholar] [CrossRef]
- Hoffmann, T.; von Blohn, C.; Stanek, A.; Moses, S.; Barzantny, H.; Bremer, E. Synthesis, release, and recapture of compatible solute proline by osmotically stressed Bacillus subtilis cells. Appl. Environ. Microbiol. 2012, 78, 5753–5762. [Google Scholar] [CrossRef]
- Spiegelhalter, F.; Bremer, E. Osmoregulation of the opuE proline transport gene from Bacillus subtilis: Contributions of the sigma A- and sigma B-dependent stress-responsive promoters. Mol. Microbiol. 1998, 29, 285–296. [Google Scholar] [CrossRef]
- Von Blohn, C.; Kempf, B.; Kappes, R.M.; Bremer, E. Osmostress response in Bacillus subtilis: Characterization of a proline uptake system (OpuE) regulated by high osmolarity and the alternative transcription factor sigma B. Mol. Microbiol. 1997, 25, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M. Diverse and common features of trehalases and their contributions to microbial trehalose metabolism. Appl. Microbiol. Biotechnol. 2020, 104, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Revsbech, N.P.; Jorgensen, B.B.; Blackburn, T.H.; Cohen, Y. Microelectrode studies of the photosynthesis and O2, H2S, and pH profiles of a microbial mat. Limnol. Oceanogr. 1983, 28, 1062–1074. [Google Scholar] [CrossRef]
- Latifi, A.; Ruiz, M.; Zhang, C.C. Oxidative stress in cyanobacteria. FEMS Microbiol. Rev. 2009, 33, 258–278. [Google Scholar] [CrossRef] [PubMed]
- Dryden, M.S.; Cooke, J.; Salib, R.J.; Holding, R.E.; Biggs, T.; Salamat, A.A.; Allan, R.N.; Newby, R.S.; Halstead, F.; Oppenheim, B.; et al. Reactive oxygen: A novel antimicrobial mechanism for targeting biofilm-associated infection. J. Glob. Antimicrob. Resist. 2017, 8, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Drlica, K. Reactive oxygen species and the bacterial response to lethal stress. Curr. Opin. Microbiol. 2014, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef]
- Meibom, K.L.; Dubail, I.; Dupuis, M.; Barel, M.; Lenco, J.; Stulik, J.; Golovliov, I.; Sjöstedt, A.; Charbit, A. The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol. Microbiol. 2008, 67, 1384–1401. [Google Scholar] [CrossRef]
- Krajewska, J.; Modrak-Wójcik, A.; Arent, Z.J.; Wiȩckowski, D.; Zolkiewski, M.; Bzowska, A.; Kȩdzierska-Mieszkowska, S. Characterization of the molecular chaperone ClpB from the pathogenic spirochaete Leptospira interrogans. PLoS ONE 2017, 12, e0181118. [Google Scholar] [CrossRef]
- Tripathi, P.; Singh, L.K.; Kumari, S.; Hakiem, O.R.; Batra, J.K. ClpB is an essential stress regulator of Mycobacterium tuberculosis and endows survival advantage to dormant bacilli. Int. J. Med. Microbiol. 2020, 310, 151402. [Google Scholar] [CrossRef]
- Alam, A.; Bröms, J.E.; Kumar, R.; Sjöstedt, A. The role of ClpB in bacterial stress responses and virulence. Front. Mol. Biosci. 2021, 8, 668910. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Tamás, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef]
- Koechler, S.; Farasin, J.; Cleiss-Arnold, J.; Arsène-Ploetze, F. Toxic metal resistance in biofilms: Diversity of microbial responses and their evolution. Res. Microbiol. 2015, 166, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Babilonia, J.; Conesa, A.; Casaburi, G.; Pereira, C.; Louyakis, A.S.; Reid, R.P.; Foster, J.S. Comparative metagenomics provides insight into the ecosystem functioning of the Shark Bay stromatolites, Western Australia. Front. Microbiol. 2018, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Takezaki, N.; Rzhetsky, A.; Nei, M. Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. 1995, 12, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Escobar, J.; Beltrán, Y.; Bergman, B.; Díez, B.; Ininbergs, K.; Souza, V.; Falcón, L.I. Phylogenetic and molecular clock inferences of cyanobacterial strains within Rivulariaceae from distant environments. FEMS Microbiol. Lett. 2011, 316, 90–99. [Google Scholar] [CrossRef]
- Fournier, G.P.; Moore, K.R.; Rangel, L.T.; Payette, J.G.; Momper, L.; Bosak, T. The Archean origin of oxygenic photosynthesis and extant cyanobacterial lineages. Proc. R. Soc. B Biol. Sci. 2021, 288, 20210675. [Google Scholar] [CrossRef]
- Moore, K.R.; Pajusalu, M.; Gong, J.; Sojo, V.; Matreux, T.; Braun, D.; Bosak, T. Biologically mediated silicification of marine cyanobacteria and implications for the Proterozoic fossil record. Geology 2020, 48, 862–866. [Google Scholar] [CrossRef]
- Moore, K.R.; Gong, J.; Pajusalu, M.; Skoog, E.J.; Xu, M.; Soto Feliz, T.; Sojo, V.; Matreux, T.; Baldes, M.J.; Braun, D.; et al. A new model for silicification of cyanobacteria in Proterozoic tidal flats. Geobiology 2021, 19, 438–449. [Google Scholar] [CrossRef]
- Li, S.J.; Hua, Z.S.; Huang, L.N.; Li, J.; Shi, S.H.; Chen, L.X.; Kuang, J.L.; Liu, J.; Hu, M.; Shu, W.S. Microbial communities evolve faster in extreme environments. Sci. Rep. 2014, 4, 6205. [Google Scholar] [CrossRef] [PubMed]
- Boos, W.; Ehmann, U.; Bremer, E.; Middendorf, A.; Postma, P. Trehalase of Escherichia Coli. Mapping and cloning of its structural gene and identification of the enzyme as a periplasmic protein induced under high osmolarity growth conditions. J. Biol. Chem. 1987, 262, 13212–13218. [Google Scholar] [CrossRef] [PubMed]
- Tamre, E.; Fournier, G.P. Inferred ancestry of scytonemin biosynthesis proteins in cyanobacteria indicates a response to Paleoproterozoic oxygenation. Geobiology 2022, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Bernroitner, M.; Zamocky, M.; Furtmüller, P.G.; Peschek, G.A.; Obinger, C. Occurrence, phylogeny, structure, and function of catalases and peroxidases in cyanobacteria. J. Exp. Bot. 2009, 60, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Burow, L.C.; Woebken, D.; Marshall, I.P.G.; Singer, S.W.; Pett-Ridge, J.; Prufert-Bebout, L.; Spormann, A.M.; Bebout, B.M.; Weber, P.K.; Hoehler, T.M. Identification of Desulfobacterales as primary hydrogenotrophs in a complex microbial mat community. Geobiology 2014, 12, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ben Hania, W.; Joseph, M.; Schumann, P.; Bunk, B.; Fiebig, A.; Spröer, C.; Klenk, H.P.; Fardeau, M.L.; Spring, S. Complete genome sequence and description of Salinispira pacifica gen. nov., sp. nov., a novel spirochaete isolated form a hypersaline microbial mat. Stand. Genom. Sci. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Z.; Craig Everroad, R.; Karaoz, U.; Detweiler, A.M.; Pett-Ridge, J.; Weber, P.K.; Prufert-Bebout, L.; Bebout, B.M. Metagenomics reveals niche partitioning within the phototrophic zone of a microbial mat. PLoS ONE 2018, 13, e0202792. [Google Scholar] [CrossRef]
- Campbell, M.A.; Coolen, M.J.L.; Visscher, P.T.; Morris, T.; Grice, K. Structure and function of shark bay microbial communities following tropical cyclone Olwyn: A metatranscriptomic and organic geochemical perspective. Geobiology 2021, 19, 642–664. [Google Scholar] [CrossRef]
- Spring, S.; Rohde, M.; Bunk, B.; Spröer, C.; Will, S.E.; Neumann-Schaal, M. New insights into the energy metabolism and taxonomy of Deferribacteres revealed by the characterization of a new isolate from a hypersaline microbial mat. Environ. Microbiol. 2022, 24, 2543–2575. [Google Scholar] [CrossRef]
- Lumppio, H.L.; Shenvi, N.V.; Summers, A.O.; Voordouw, G.; Kurtz, J. Rubrerythrin and rubredoxin oxidoreductase in Desulfovibrio vulgaris: A novel oxidative stress protection system. J. Bacteriol. 2001, 183, 101–108. [Google Scholar] [CrossRef]
- Sztukowska, M.; Bugno, M.; Potempa, J.; Travis, J.; Kurtz, D.M. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol. Microbiol. 2002, 44, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Brioukhanov, A.; Pieulle, L.; Dolla, A. Antioxidative defense systems of anaerobic sulfate-reducing microorganisms. In Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology; Formatex Research Center: Badajoz, Spain, 2010; pp. 148–159. [Google Scholar]
- Bergman, I.A.; Kolesov, G.M. Arsenic, antimony, and bismuth as indicators of the genesis of ore material in Early Precambrian ferrous quartzite formations. Geochem. Int. 2012, 50, 816–831. [Google Scholar] [CrossRef]
- Sforna, M.C.; Philippot, P.; Somogyi, A.; Van Zuilen, M.A.; Medjoubi, K.; Schoepp-Cothenet, B.; Nitschke, W.; Visscher, P.T. Evidence for arsenic metabolism and cycling by microorganisms 2.7 Billion Years Ago. Nat. Geosci. 2014, 7, 811–815. [Google Scholar] [CrossRef]
- Stokke, R.; Dahle, H.; Roalkvam, I.; Wissuwa, J.; Daae, F.L.; Tooming-Klunderud, A.; Thorseth, I.H.; Pedersen, R.B.; Steen, I.H. Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ. Microbiol. 2015, 17, 4063–4077. [Google Scholar] [CrossRef] [PubMed]
- Borsodi, A.K.; Anda, D.; Makk, J.; Krett, G.; Dobosy, P.; Büki, G.; Erőss, A.; Mádl-Szőnyi, J. Biofilm forming bacteria and archaea in thermal karst springs of Gellért Hill discharge area (Hungary). J. Basic Microbiol. 2018, 58, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.; Burne, R.A.; Wu, H.; Koo, H. Oral biofilms: Pathogens, matrix, and polymicrobial interactions in microenvironments. Trends Microbiol. 2018, 26, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Savio, D.; Stadler, P.; Reischer, G.H.; Demeter, K.; Linke, R.B.; Blaschke, A.P.; Mach, R.L.; Kirschner, A.K.T.; Stadler, H.; Farnleitner, A.H. Spring water of an alpine karst aquifer is dominated by a taxonomically stable but discharge-responsive bacterial community. Front. Microbiol. 2019, 10, 28. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef]
- Nazir, R.; Zaffar, M.R.; Amin, I. Bacterial Biofilms: The Remarkable Heterogeneous Biological Communities and Nitrogen Fixing Microorganisms in Lakes; Elsevier Inc.: Oxford, UK, 2019; ISBN 9780128174951. [Google Scholar]
- Kochetkova, T.V.; Zayulina, K.S.; Zhigarkov, V.S.; Minaev, N.V.; Chichkov, B.N.; Novikov, A.A.; Toshchakov, S.V.; Elcheninov, A.G.; Kublanov, I.V. Tepidiforma bonchosmolovskayae gen. nov., sp. nov., a moderately thermophilic Chloroflexi bacterium from a Chukotka hot spring (Arctic, Russia), representing a novel class, Tepidiformia, which includes the previously uncultivated lineage OLB14. Int. J. Syst. Evol. Microbiol. 2020, 70, 1192–1202. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoog, E.J.; Fournier, G.P.; Bosak, T. Assessing the Influence of HGT on the Evolution of Stress Responses in Microbial Communities from Shark Bay, Western Australia. Genes 2023, 14, 2168. https://doi.org/10.3390/genes14122168
Skoog EJ, Fournier GP, Bosak T. Assessing the Influence of HGT on the Evolution of Stress Responses in Microbial Communities from Shark Bay, Western Australia. Genes. 2023; 14(12):2168. https://doi.org/10.3390/genes14122168
Chicago/Turabian StyleSkoog, Emilie J., Gregory P. Fournier, and Tanja Bosak. 2023. "Assessing the Influence of HGT on the Evolution of Stress Responses in Microbial Communities from Shark Bay, Western Australia" Genes 14, no. 12: 2168. https://doi.org/10.3390/genes14122168