Plasma Levels of Homocysteine and Cysteine Increased in Pediatric NAFLD and Strongly Correlated with Severity of Liver Damage

 ,
,  ,
,
Abstract
:
1. Introduction
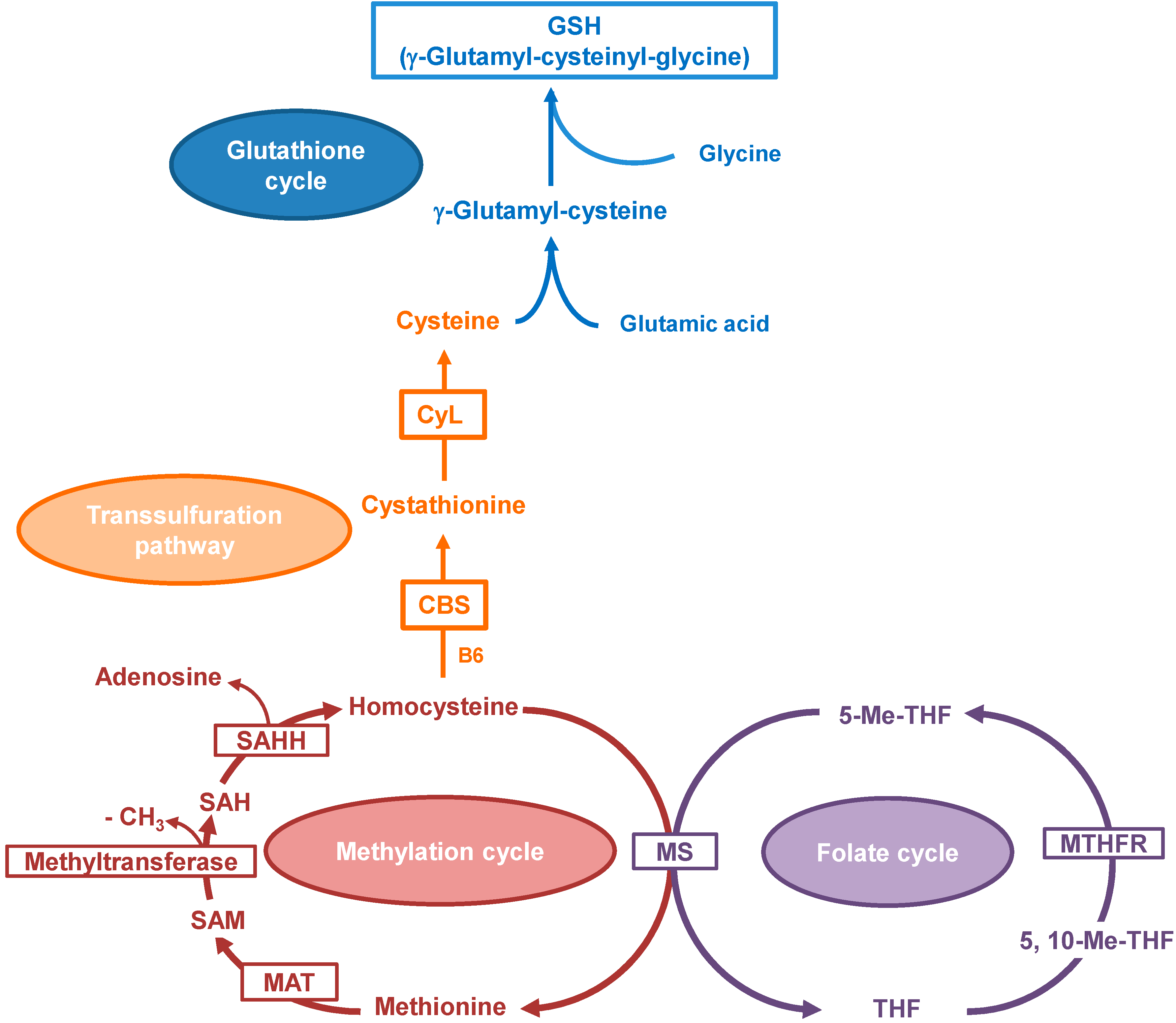
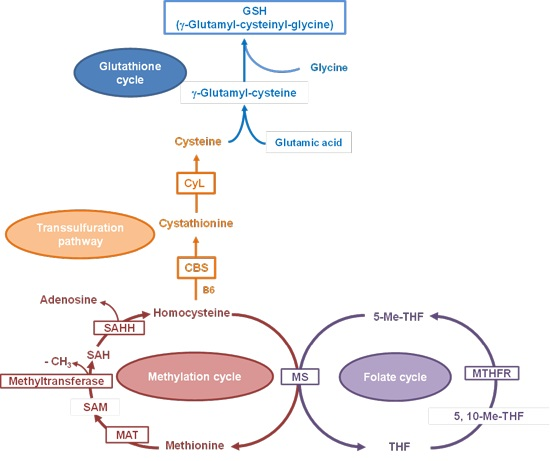
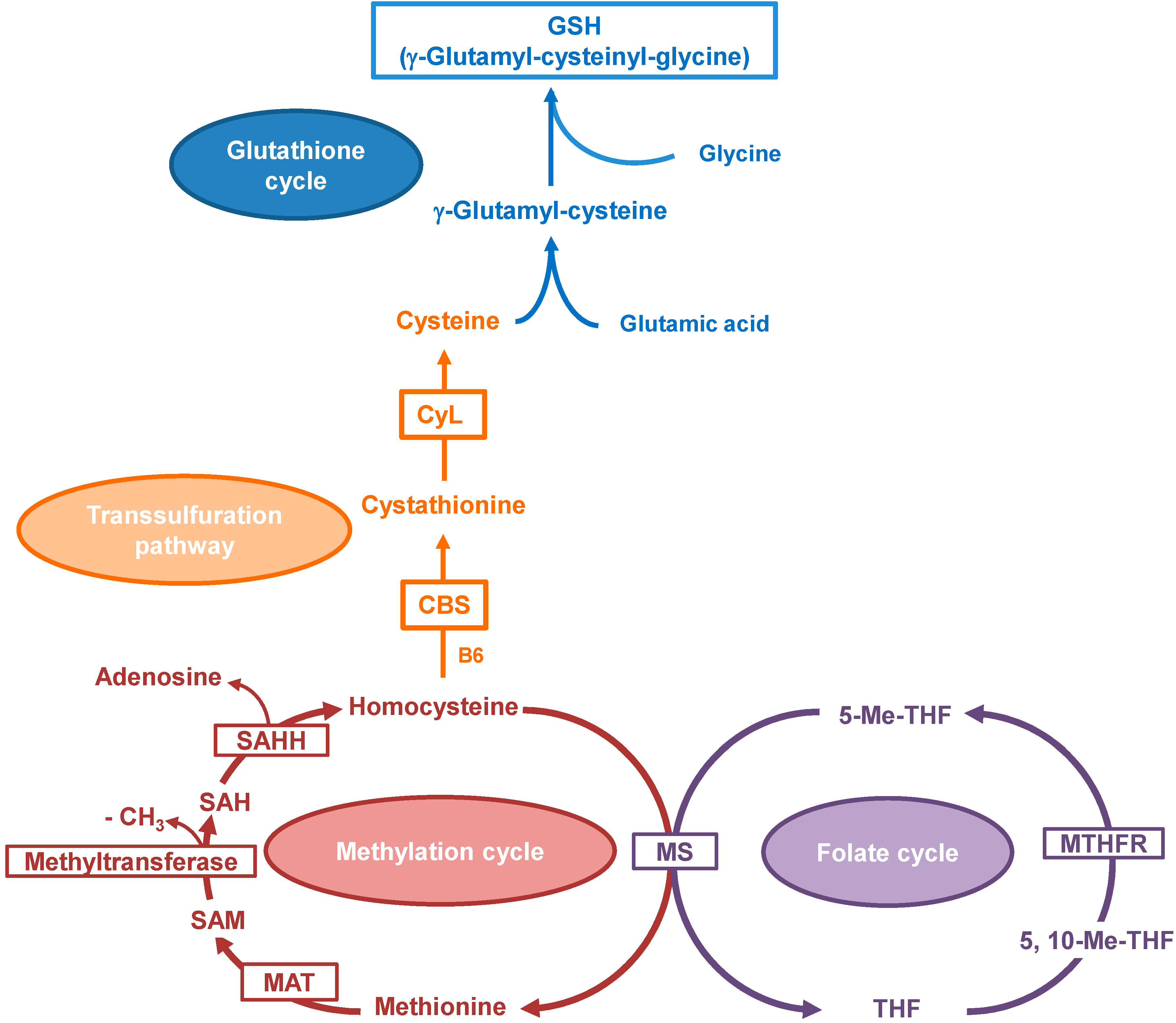
2. Results and Discussion
2.1. Results


{kind=link}
{kind=link}
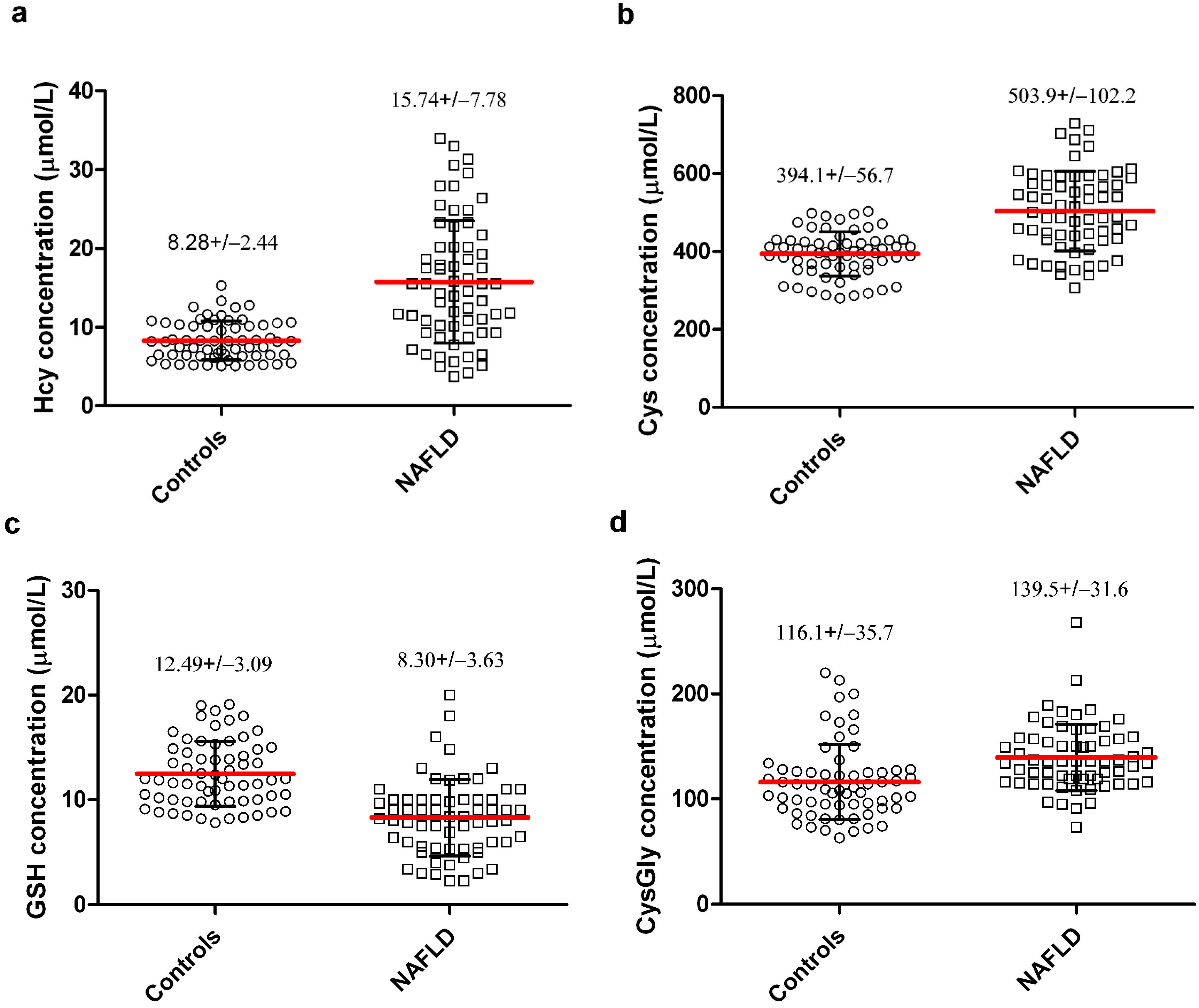{kind=link}
{kind=link}
| Parameters | Not-NASH (n = 34; 23 Male, 11 Female) | NASH (n = 30; 20 Male, 10 Female) | p * |
|---|---|---|---|
| Age (years) | 8.4 ± 2.4 | 9.9 ± 2.3 | 0.012 |
| Height (cm) | 131.2 ± 15.9 | 141.2 ± 18.6 | 0.023 |
| Weight (kg) | 42.3 ± 14.1 | 51.8 ± 16.9 | 0.017 |
| WC (cm) | 78.7 ± 10.2 | 82.1 ± 10.5 | 0.188 |
| BMI (kg/m2) | 23.9 ± 4.1 | 25.1 ± 3.7 | 0.263 |
| ALT (U/L) | 75.7 ± 53.6 | 83.1 ± 46.4 | 0.555 |
| AST (U/L) | 51.1 ± 19.7 | 53.2 ±24.8 | 0.712 |
| γGT (U/L) | 24.3 ±14.1 | 29.1 ± 20.1 | 0.283 |
| Triglycerides (mg/dL) | 100 ± 72 | 112 ± 79 | 0.557 |
| Cholesterol (mg/dL) | 149 ± 37 | 151 ± 33 | 0.821 |
| Glucose (mg/dL) | 80 ± 13 | 83 ± 14 | 0.437 |
| Insulin (mcU/mL) | 11.7 ± 8.4 | 13.8 ± 7.9 | 0.432 |
| HOMA | 2.4 ± 1.5 | 2.8 ± 1.4 | 0.546 |

| Histology | n | % |
|---|---|---|
| Steatosis | ||
| 1 | 13 | 20.3 |
| 2 | 39 | 60.9 |
| 3 | 12 | 18.8 |
| Inflammation | ||
| 0 | 6 | 9.4 |
| 1 | 39 | 60.9 |
| 2 | 18 | 28.1 |
| Ballooning | ||
| 0 | 28 | 43.7 |
| 1 | 24 | 37.5 |
| 2 | 12 | 18.8 |
| Fibrosis | ||
| 0 | 23 | 35.9 |
| 1 | 29 | 45.3 |
| 2 | 7 | 10.9 |
| 3 | 5 | 7.9 |
| Model | OR | CI | p |
|---|---|---|---|
| 1 (Hcy) | 1.141 | 1.040−1.251 | 0.005 |
| 2 (+ Cys) | 1.124 | 1.022−1.235 | 0.016 |
| 3 (+ Cys + GSH) | 1.126 | 1.024−1.240 | 0.016 |
| 4 (+ Cys + GSH + CysGly) | 1.126 | 1.024−1.240 | 0.016 |
2.2. Discussion
3. Experimental Section
3.1. Patients
3.2. Anthropometric Data
3.3. Blood Assays
3.4. Plasma Thiols Determinations
3.5. Liver Histology
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleiner, D.E.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathologic patterns and biopsy evaluation in clinical research. Semin. Liver Dis. 2012, 32, 3–13. [Google Scholar] [CrossRef]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Svegliati-Baroni, G.; Alisi, A.; Miele, L.; Valenti, L.; Vajro, P. A 360-degree overview of paediatric NAFLD: Recent insights. J. Hepatol. 2013, 58, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Finelli, C. What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J. Gastroenterol. 2013, 19, 3375–3384. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Feldstein, A.E.; Villani, A.; Raponi, M.; Nobili, V. Pediatric nonalcoholic fatty liver disease: A multidisciplinary approach. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Philippe, J.; Jornayvaz, F.R. Non-alcoholic fatty liver disease and insulin resistance: From bench to bedside. Diabetes Metab. 2013, 39, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Schattenberg, J.M. Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic approaches. J. Gastroenterol. Hepatol. 2013, 28, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol. 2010, 16, 4773–4783. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Pathways and regulation of homocysteine metabolism in mammals. Semin. Thromb. Hemost. 2000, 26, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of hepatic glutathione synthesis: Current concepts and controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [PubMed]
- Al-Maskari, M.Y.; Waly, M.I.; Ali, A.; Al-Shuaibi, Y.S.; Ouhtit, A. Folate and vitamin B12 deficiency and hyperhomocysteinemia promote oxidative stress in adult type 2 diabetes. Nutrition 2012, 28, e23–e26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Austin, R.C. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. BioFactors 2009, 35, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, Y.; Ozkan, E.; Simşek, B. Plasma total homocysteine and cysteine levels as cardiovascular risk factors in coronary heart disease. Int. J. Cardiol. 2002, 82, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Noce, A.; di Giovamberardino, G.; de Stefano, A.; Callà, C.; Zenobi, R.; Dessì, M.; di Daniele, N. Homocysteine, cysteine, folate and vitamin B(12) status in type 2 diabetic patients with chronic kidney disease. J. Nephrol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Kiyohara, Y.; Kato, I.; Kitazono, T.; Tanizaki, Y.; Kubo, M.; Ueno, H.; Ibayashi, S.; Fujishima, M.; Iida, M. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: The Hisayama study. Stroke 2004, 35, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta 2012, 1822, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Lu, S.C. Homocysteine, the bad thiol. Hepatology 2005, 41, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Herrmann, W. Homocysteine and lipids: S-Adenosyl methionine as a key intermediate. FEBS Lett. 2009, 583, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Bravo, E.; Palleschi, S.; Aspichueta, P.; Buqué, X.; Rossi, B.; Cano, A.; Napolitano, M.; Ochoa, B.; Botham, K.M. High fat diet-induced non alcoholic fatty liver disease in rats is associated with hyperhomocysteinemia caused by down regulation of the transsulphuration pathway. Lipids Health Dis. 2011, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Aissa, A.F.; Tryndyak, V.; de Conti, A.; Melnyk, S.; Gomes, T.D.; Bianchi, M.L.; James, S.J.; Beland, F.A.; Antunes, L.M.; Pogribny, I.P. Effect of methionine-deficient and methionine-supplemented diets on the hepatic one-carbon and lipid metabolism in mice. Mol. Nutr. Food Res. 2014, 58, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Gulsen, M.; Yesilova, Z.; Bagci, S.; Uygun, A.; Ozcan, A.; Ercin, C.N.; Erdil, A.; Sanisoglu, S.Y.; Cakir, E.; Ates, Y.; et al. Elevated plasma homocysteine concentrations as a predictor of steatohepatitis in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2005, 20, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Patsiaoura, K.; Katsiki, E.; Zafeiriadou, E.; Deretzi, G.; Zavos, C.; Gavalas, E.; Katsinelos, P.; Mane, V.; et al. Serum homocysteine levels in patients with nonalcoholic fatty liver disease. Ann. Hepatol. 2012, 11, 68–76. [Google Scholar] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Pastore, A.; Gaeta, L.M.; Tozzi, G.; Comparcola, D.; Sartorelli, M.R.; Marcellini, M.; Bertini, E.; Piemonte, F. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin. Chim. Acta 2005, 355, 105–111. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, S.C.; Muniz, M.T.; Siqueira, M.D.; Siqueira, E.R.; Gomes, A.V.; Silva, K.A.; Bezerra, L.C.; D’Almeida, V.; de Oliveira, C.P.; Pereira, L.M. Plasmatic higher levels of homocysteine in non-alcoholic fatty liver disease (NAFLD). Nutr. J. 2013, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, C.P.; Stefano, J.T.; Cavaleiro, A.M.; Fortes, M.A.; Vieira, S.M.; Lima, V.M.; Santos, T.E.; Santos, V.N.; de Azevedo Salgado, A.L.; Parise, E.R.; et al. Association of polymorphisms of glutamate-cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2010, 25, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.; Kugiyama, K.; Sugiyama, S.; Nakamura, S.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Dominy, J.E., Jr.; Lee, J.I.; Coloso, R.M. Mammalian cysteine metabolism: New insights into regulation of cysteine metabolism. J. Nutr. 2006, 136, 1652S–1659S. [Google Scholar] [PubMed]
- Oz, H.S.; Chen, T.S.; Neuman, M. Methionine deficiency and hepatic injury in a dietary steatohepatitis model. Dig. Dis. Sci. 2008, 53, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Dahlhoff, C.; Worsch, S.; Sailer, M.; Hummel, B.A.; Fiamoncini, J.; Uebel, K.; Obeid, R.; Scherling, C.; Geisel, J.; Bader, B.L.; et al. Methyl-donor supplementation in obese mice prevents the progression of NAFLD, activates AMPK and decreases acyl-carnitine levels. Mol. Metab. 2014, 3, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Garcia-Trevijano, E.R.; Rodriguez, J.A.; Carretero, M.V.; Bustos, M.; Fernández, E.; Eguinoa, E.; Mato, J.M.; Avila, M.A. Induction of TIMP-1 expression in rat hepatic stellate cells and hepatocytes: A new role for homocysteine in liver fibrosis. Biochim. Biophys. Acta 1999, 1455, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Matté, C.; Stefanello, F.M.; Mackedanz, V.; Pederzolli, C.D.; Lamers, M.L.; Dutra-Filho, C.S.; Dos Santos, M.F.; Wyse, A.T. Homocysteine induces oxidative stress, inflammatory infiltration, fibrosis and reduces glycogen/glycoprotein content in liver of rats. Int. J. Dev. Neurosci. 2009, 27, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.H.; Prathapasinghe, G.A.; Siow, Y.L.; O, K. Hyperhomocysteinemia induces liver injury in rat: Protective effect of folic acid supplementation. Biochim. Biophys. Acta 2006, 1762, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastore, A.; Alisi, A.; Di Giovamberardino, G.; Crudele, A.; Ceccarelli, S.; Panera, N.; Dionisi-Vici, C.; Nobili, V. Plasma Levels of Homocysteine and Cysteine Increased in Pediatric NAFLD and Strongly Correlated with Severity of Liver Damage. Int. J. Mol. Sci. 2014, 15, 21202-21214. https://doi.org/10.3390/ijms151121202
Pastore A, Alisi A, Di Giovamberardino G, Crudele A, Ceccarelli S, Panera N, Dionisi-Vici C, Nobili V. Plasma Levels of Homocysteine and Cysteine Increased in Pediatric NAFLD and Strongly Correlated with Severity of Liver Damage. International Journal of Molecular Sciences. 2014; 15(11):21202-21214. https://doi.org/10.3390/ijms151121202
Chicago/Turabian StylePastore, Anna, Anna Alisi, Gianna Di Giovamberardino, Annalisa Crudele, Sara Ceccarelli, Nadia Panera, Carlo Dionisi-Vici, and Valerio Nobili. 2014. "Plasma Levels of Homocysteine and Cysteine Increased in Pediatric NAFLD and Strongly Correlated with Severity of Liver Damage" International Journal of Molecular Sciences 15, no. 11: 21202-21214. https://doi.org/10.3390/ijms151121202
APA StylePastore, A., Alisi, A., Di Giovamberardino, G., Crudele, A., Ceccarelli, S., Panera, N., Dionisi-Vici, C., & Nobili, V. (2014). Plasma Levels of Homocysteine and Cysteine Increased in Pediatric NAFLD and Strongly Correlated with Severity of Liver Damage. International Journal of Molecular Sciences, 15(11), 21202-21214. https://doi.org/10.3390/ijms151121202