The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease

Abstract
:1. Introduction
1.1. A Brief Introduction to Epigenetics
1.2. Environmental Epigenetics
1.3. Cancer: Current Status and Future Prospects
1.4. Article Overview
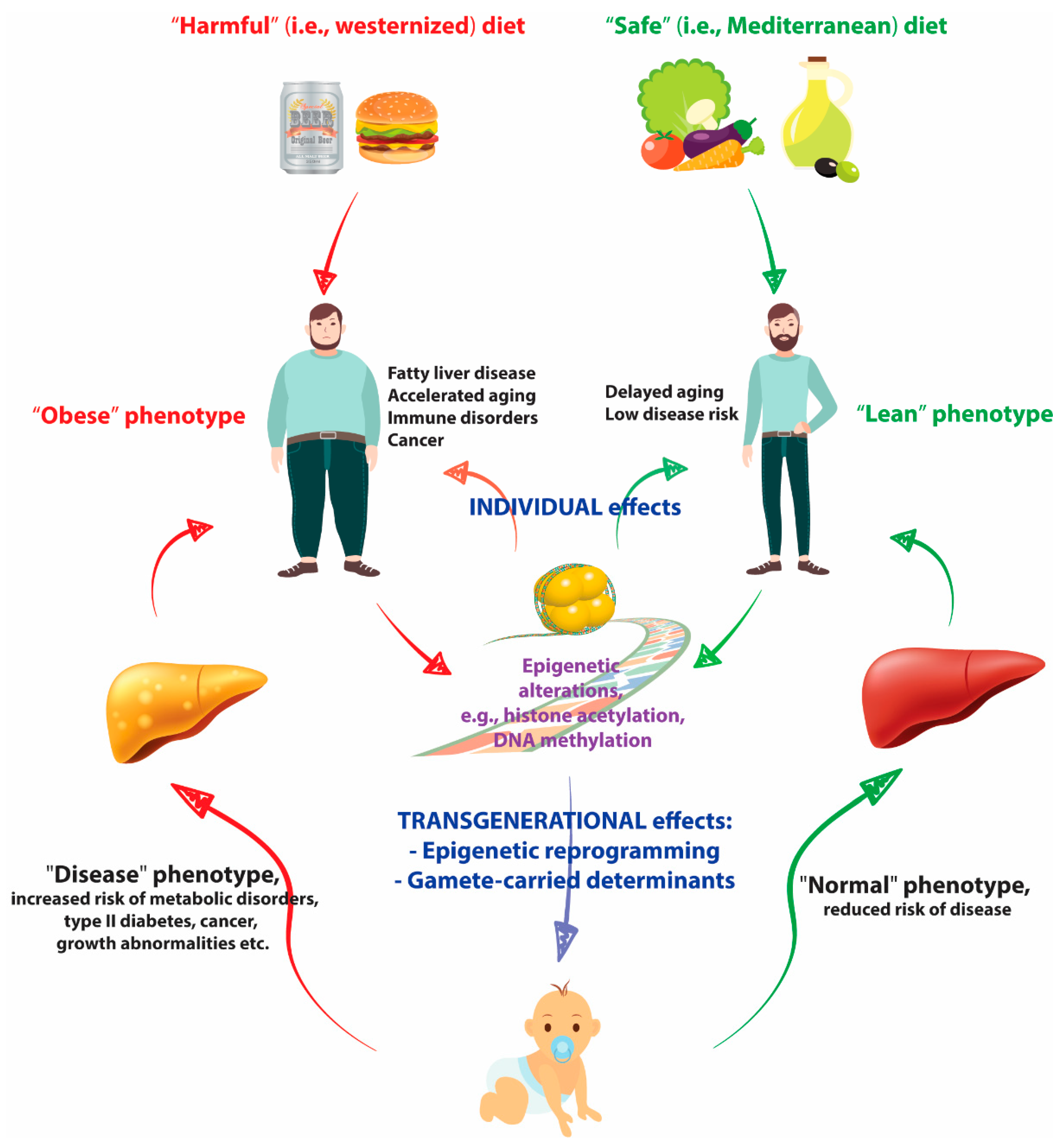
2. Nutritional Epigenetics
2.1. Nutritional Epigenetics in Health and Disease
2.2. Nutritional Epigenetics and Cancer
2.3. Nutritional Epigenetics: The Future
3. Chemical Environmental Epigenetics
3.1. Endocrine-Disrupting Chemicals (EDCs)
3.1.1. EDC Exposure Induces Abnormal Development, Behavior, and Disease
3.1.2. EDCs, Epigenetics, and Cancer
3.2. Other Toxin Exposures and Epigenetic Effects
4. Environmental Epigenetics and Cancer: Breast Cancer as a Specific Example
5. The Potential of Epigenetic Drugs
6. Conclusions
7. Take-Home Messages
- Nutrition—in particular maternal diet and dietary patterns—and chemical pollutants are two important environmental factors that impact human health.
- These factors have a direct impact on the individual by contributing to the pathogenesis of many diseases, not least cancer.
- Furthermore, these factors probably span generations through epigenetic transmission, making them a major global public health problem not only for the individual but also future generations and society.
- Understanding the molecular mechanisms and signaling pathways involved in environmental epigenetics paves the way for both public health and targeted interventions to reduce their societal impact.
Funding
Conflicts of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 1999, 9, 40–48. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Huang, B.; Jiang, C.; Zhang, R. Epigenetics: The language of the cell? Epigenomics 2014, 6, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Weisbeck, A.; Jansen, R.J. Nutrients and the Pancreas: An Epigenetic Perspective. Nutrients 2017, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Hoile, S.P.; Grenfell, L.; Burdge, G.C. DNA methylation, ageing and the influence of early life nutrition. Proc. Nutr. Soc. 2014, 73, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussey, B.; Lindley, M.R.; Mastana, S. Epigenetics and epigenomics: The future of nutritional interventions. Future Sci. 2017, 3, FSO237. [Google Scholar] [CrossRef]
- Lo Re, O.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reamon-Buettner, S.M.; Mutschler, V.; Borlak, J. The next innovation cycle in toxicogenomics: Environmental epigenetics. Mutat. Res. 2008, 659, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Eurostat, E.C. Population Structure and Ageing; European Commission: Luxembourg, 2014. [Google Scholar]
- Malvezzi, M.; Bertuccio, P.; Rosso, T.; Rota, M.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2015: Does lung cancer have the highest death rate in EU women? Ann. Oncol. 2015, 26, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Chatenoud, L.; Bertuccio, P.; Bosetti, C.; Malvezzi, M.; Levi, F.; Negri, E.; La Vecchia, C. Trends in mortality from major cancers in the Americas: 1980–2010. Ann. Oncol. 2014, 25, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: Genetic and environmental aspects. Hum. Reprod. Update 2006, 12, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Strohsnitter, W.C.; Noller, K.L.; Hoover, R.N.; Robboy, S.J.; Palmer, J.R.; Titus-Ernstoff, L.; Kaufman, R.H.; Adam, E.; Herbst, A.L.; Hatch, E.E. Cancer risk in men exposed in utero to diethylstilbestrol. J. Natl. Cancer Inst. 2001, 93, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Argente, J.; Mehls, O.; Barrios, V. Growth and body composition in very young SGA children. Pediatr. Nephrol. 2010, 25, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Clark, P.M. Fetal undernutrition and disease in later life. Rev. Reprod. 1997, 2, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopik, V.S.; Maccani, M.A.; Francazio, S.; McGeary, J.E. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev. Psychopathol. 2012, 24, 1377–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, C.E.; Tarry-Adkins, J.L.; Ozanne, S.E. Transgenerational effects of maternal diet on metabolic and reproductive ageing. Mamm. Genome 2016, 27, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rando, O.J.; Simmons, R.A. I’m eating for two: Parental dietary effects on offspring metabolism. Cell 2015, 161, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Friso, S.; Choi, S.W. DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J. Nutr. Biochem. 2009, 20, 917–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.D.; Ross, S.A. Dietary components impact histone modifications and cancer risk. Nutr. Rev. 2007, 65, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Ions, L.J.; Alatawi, F.; Wakeling, L.A. The potential role of epigenetic responses to diet in ageing. Proc. Nutr. Soc. 2011, 70, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Alavian-Ghavanini, A.; Ruegg, J. Understanding Epigenetic Effects of Endocrine Disrupting Chemicals: From Mechanisms to Novel Test Methods. Basic Clin. Pharmacol. Toxicol. 2018, 122, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Giovannucci, E.; Wolk, A. Folate intake, MTHFR polymorphisms, and risk of esophageal, gastric, and pancreatic cancer: A meta-analysis. Gastroenterology 2006, 131, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C. Histone Deacetylase Inhibition Restores Expression of Hypoxia-Inducible Protein NDRG1 in Pancreatic Cancer. Pancreas 2018, 47, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Semin. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duee, P.H.; Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Minelli, L.; Bertarelli, G.; Bacci, S. A western dietary pattern increases prostate cancer risk: A systematic review and meta-analysis. Nutrients 2016, 8, 626. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.; Hu, F.B.; Fuchs, C.; Giovannucci, E.; Hunter, D.J.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C. Major dietary patterns and the risk of colorectal cancer in women. Arch. Intern. Med. 2003, 163, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lytle, K.A.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid blocks progression of western diet-induced nonalcoholic steatohepatitis in obese Ldlr-/-mice. PLoS ONE 2017, 12, e0173376. [Google Scholar] [CrossRef] [PubMed]
- Jensen, V.S.; Hvid, H.; Damgaard, J.; Nygaard, H.; Ingvorsen, C.; Wulff, E.M.; Lykkesfeldt, J.; Fledelius, C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague–Dawley rats. Diabetol. Metab. Syndr. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyapal, S.; Kona, S.R.; Mullapudi, S.V.; Putcha, U.K.; Gurumurthy, P.; Ibrahim, A. Substitution of linoleic acid with α-linolenic acid or long chain n-3 polyunsaturated fatty acid prevents Western diet induced nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 10953. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, G.N.; Grigsby, K.B.; Kelty, T.J.; Zidon, T.M.; Childs, T.E.; Vieira-Potter, V.J.; Klinkebiel, D.L.; Matheny, M.; Scarpace, P.J.; Booth, F.W. Maternal Western diet age-specifically alters female offspring voluntary physical activity and dopamine-and leptin-related gene expression. FASEB J. 2017, 31, 5371–5383. [Google Scholar] [CrossRef] [PubMed]
- De La Rocha, C.; Pérez-Mojica, J.E.; Zenteno-De León, S.; Cervantes-Paz, B.; Tristán-Flores, F.E.; Rodríguez-Ríos, D.; Molina-Torres, J.; Ramírez-Chávez, E.; Alvarado-Caudillo, Y.; Carmona, F.J. Associations between whole peripheral blood fatty acids and DNA methylation in humans. Sci. Rep. 2016, 6, 25867. [Google Scholar] [CrossRef] [PubMed]
- Ramaiyan, B.; Talahalli, R.R. Dietary Unsaturated Fatty Acids Modulate Maternal Dyslipidemia-Induced DNA Methylation and Histone Acetylation in Placenta and Fetal Liver in Rats. Lipids 2018, 53, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, S.; Zhu, Y.; Zsombok, A.; Mauvais-Jarvis, F.; Zhao, J.; Lazartigues, E. Perinatal exposure to Western diet programs autonomic dysfunction in the male offspring. Cell. Mol. Neurobiol. 2018, 38, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J. Primary prevention of cardiovascular disease with a mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Shively, C.A.; Register, T.C.; Appt, S.E.; Clarkson, T.B.; Uberseder, B.; Clear, K.Y.; Wilson, A.S.; Chiba, A.; Tooze, J.A.; Cook, K.L. Consumption of Mediterranean versus Western Diet Leads to Distinct Mammary Gland Microbiome Populations. Cell Rep. 2018, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- House, J.S.; Mendez, M.; Maguire, R.L.; Gonzalez-Nahm, S.; Huang, Z.; Daniels, J.; Murphy, S.K.; Fuemmeler, B.F.; Wright, F.A.; Hoyo, C. Periconceptional Maternal Mediterranean Diet Is Associated with Favorable Offspring Behaviors and Altered CpG Methylation of Imprinted Genes. Front. Cell Dev. Biol. 2018, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, D.K. Environment and Life Style Related Epigenetics: A clinical Preview. J. Clin. Epigenet. 2016, 2, 3. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodemus-Johnson, J.; Sinnott, R.A. Fruit and Juice Epigenetic Signatures Are Associated with Independent Immunoregulatory Pathways. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Possible Developmental Early Effects of Endocrine Disrupters on Child Health; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Boudalia, S.; Berges, R.; Chabanet, C.; Folia, M.; Decocq, L.; Pasquis, B.; Abdennebi-Najar, L.; Canivenc-Lavier, M.C. A multi-generational study on low-dose BPA exposure in Wistar rats: Effects on maternal behavior, flavor intake and development. Neurotoxicol. Teratol. 2014, 41, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, L.S.; Fenton, S.E. Cancer and developmental exposure to endocrine disruptors. Environ. Health Perspect. 2003, 111, 389–394. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, J.A.; Newbold, R.R.; Bullock, B.C. Long-term effects on the female mouse genital tract associated with prenatal exposure to diethylstilbestrol. Cancer Res. 1980, 40, 3988–3999. [Google Scholar] [PubMed]
- Palmer, J.R.; Wise, L.A.; Hatch, E.E.; Troisi, R.; Titus-Ernstoff, L.; Strohsnitter, W.; Kaufman, R.; Herbst, A.L.; Noller, K.L.; Hyer, M.; et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.A.; Potischman, N.A.; Brinton, L.A.; Brogan, D.; Coates, R.J.; Gammon, M.D.; Malone, K.E.; Schoenberg, J.B. Prenatal and perinatal risk factors for breast cancer in young women. Epidemiology 1997, 8, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; Cho, E.; Onojafe, I.; Raygada, M.; Clarke, R. Maternal exposure to genistein during pregnancy increases carcinogen-induced mammary tumorigenesis in female rat offspring. Oncol. Rep. 1999, 6, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.A.; Grinspon, R.P. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.; Saunders, P.T.; Fisken, M.; Scott, H.M.; Hutchison, G.R.; Smith, L.B.; Sharpe, R.M. Identification in rats of a programming window for reproductive tract masculinization, disruption of which leads to hypospadias and cryptorchidism. J. Clin. Investig. 2008, 118, 1479–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, A.M.; Sonnenschein, C. Environmental causes of cancer: Endocrine disruptors as carcinogens. Nat. Rev. Endocrinol. 2010, 6, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Newbold, R.R.; Hanson, R.B.; Jefferson, W.N.; Bullock, B.C.; Haseman, J.; McLachlan, J.A. Proliferative lesions and reproductive tract tumors in male descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis 2000, 21, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newbold, R.R.; Hanson, R.B.; Jefferson, W.N.; Bullock, B.C.; Haseman, J.; McLachlan, J.A. Increased tumors but uncompromised fertility in the female descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis 1998, 19, 1655–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, R.M.; Skakkebaek, N.E. Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract? Lancet 1993, 341, 1392–1395. [Google Scholar] [CrossRef]
- Sweeney, M.F.; Hasan, N.; Soto, A.M.; Sonnenschein, C. Environmental endocrine disruptors: Effects on the human male reproductive system. Rev. Endocr. Metab. Disord. 2015, 16, 341–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, L.S. The mechanism of dioxin toxicity: Relationship to risk assessment. Environ. Health Perspect. 1994, 102 (Suppl. 9), 157–167. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Polychlorinated dibenzo-para-dioxins and polychlorinated dibenzofurans. IARC Monogr. Eval. Carcinog. Risks Hum. 1997, 69, 228–238. [Google Scholar]
- Paustenbach, D.J. The US EPA Science Advisory Board evaluation (2001) of the EPA dioxin reassessment. Regul. Toxicol. Pharmacol. 2002, 36, 211–219. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. IARC working group on the evaluation of carcinogenic risks to humans. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 45–119. [Google Scholar]
- Nothlings, U.; Wilkens, L.R.; Murphy, S.P.; Hankin, J.H.; Henderson, B.E.; Kolonel, L.N. Meat and fat intake as risk factors for pancreatic cancer: The multiethnic cohort study. J. Natl. Cancer Inst. 2005, 97, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, C.E.; Gerde, P.; Hanberg, A.; Jernstrom, B.; Johansson, C.; Kyrklund, T.; Rannug, A.; Tornqvist, M.; Victorin, K.; Westerholm, R. Cancer risk assessment, indicators, and guidelines for polycyclic aromatic hydrocarbons in the ambient air. Environ. Health Perspect. 2002, 110 (Suppl. 3), 451–488. [Google Scholar] [PubMed]
- Baird, W.M.; Hooven, L.A.; Mahadevan, B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ. Mol. Mutagen. 2005, 45, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, S.; Wang, H.; Tao, S.; Kiyama, R. Biological impact of environmental polycyclic aromatic hydrocarbons (ePAHs) as endocrine disruptors. Environ. Pollut. 2016, 213, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Sram, R.J.; Binkova, B.; Dejmek, J.; Bobak, M. Ambient air pollution and pregnancy outcomes: A review of the literature. Environ. Health Perspect. 2005, 113, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Suades-Gonzalez, E.; Gascon, M.; Guxens, M.; Sunyer, J. Air Pollution and Neuropsychological Development: A Review of the Latest Evidence. Endocrinology 2015, 156, 3473–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winans, B.; Humble, M.C.; Lawrence, B.P. Environmental toxicants and the developing immune system: A missing link in the global battle against infectious disease? Reprod. Toxicol. 2011, 31, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wormley, D.D.; Ramesh, A.; Hood, D.B. Environmental contaminant-mixture effects on CNS development, plasticity, and behavior. Toxicol. Appl. Pharmacol. 2004, 197, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Doherty, S.P.; Grabowski, J.; Hoffman, C.; Ng, S.P.; Zelikoff, J.T. Early life insult from cigarette smoke may be predictive of chronic diseases later in life. Biomarkers 2009, 14 (Suppl. 1), 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ducci, F.; Goldman, D. Genetic approaches to addiction: Genes and alcohol. Addiction 2008, 103, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Farris, S.P.; Wolen, A.R.; Miles, M.F. Using expression genetics to study the neurobiology of ethanol and alcoholism. Int. Rev. Neurobiol. 2010, 91, 95–128. [Google Scholar] [PubMed]
- Delclos, K.B.; Bucci, T.J.; Lomax, L.G.; Latendresse, J.R.; Warbritton, A.; Weis, C.C.; Newbold, R.R. Effects of dietary genistein exposure during development on male and female CD (Sprague-Dawley) rats. Reprod. Toxicol. 2001, 15, 647–663. [Google Scholar] [CrossRef]
- Latendresse, J.R.; Bucci, T.J.; Olson, G.; Mellick, P.; Weis, C.C.; Thorn, B.; Newbold, R.R.; Delclos, K.B. Genistein and ethinyl estradiol dietary exposure in multigenerational and chronic studies induce similar proliferative lesions in mammary gland of male Sprague-Dawley rats. Reprod. Toxicol. 2009, 28, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Brisken, C.; Schaeberle, C.; Sonnenschein, C. Does cancer start in the womb? Altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J. Mammary Gland Biol. Neoplas. 2013, 18, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Schaeberle, C.M.; Rubin, B.S.; Sonnenschein, C.; Soto, A.M. The male mammary gland: A target for the xenoestrogen bisphenol A. Reprod. Toxicol. 2013, 37, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhimolea, E.; Wadia, P.R.; Murray, T.J.; Settles, M.L.; Treitman, J.D.; Sonnenschein, C.; Shioda, T.; Soto, A.M. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS ONE 2014, 9, e99800. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yi, B.R.; Go, R.E.; Hwang, K.A.; Nam, K.H.; Choi, K.C. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen receptor-dependent pathway. Environ. Toxicol. Pharmacol. 2014, 37, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Hamm, J.T.; Birnbaum, L.S.; Youngblood, G.L. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 2002, 67, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.H.; Russo, J. Developmental stage of the rat mammary gland as determinant of its susceptibility to 7,12-dimethylbenz[a]anthracene. J. Natl. Cancer Inst. 1978, 61, 1439–1449. [Google Scholar] [PubMed]
- Den Hond, E.; Roels, H.A.; Hoppenbrouwers, K.; Nawrot, T.; Thijs, L.; Vandermeulen, C.; Winneke, G.; Vanderschueren, D.; Staessen, J.A. Sexual maturation in relation to polychlorinated aromatic hydrocarbons: Sharpe and Skakkebaek’s hypothesis revisited. Environ. Health Perspect. 2002, 110, 771–776. [Google Scholar] [CrossRef] [PubMed]
- El Sheikh Saad, H.; Meduri, G.; Phrakonkham, P.; Berges, R.; Vacher, S.; Djallali, M.; Auger, J.; Canivenc-Lavier, M.C.; Perrot-Applanat, M. Abnormal peripubertal development of the rat mammary gland following exposure in utero and during lactation to a mixture of genistein and the food contaminant vinclozolin. Reprod. Toxicol. 2011, 32, 15–25. [Google Scholar] [CrossRef] [PubMed]
- El Sheikh Saad, H.; Toullec, A.; Vacher, S.; Pocard, M.; Bieche, I.; Perrot-Applanat, M. In utero and lactational exposure to vinclozolin and genistein induces genomic changes in the rat mammary gland. J. Endocrinol. 2013, 216, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bosagna, C.; Covert, T.R.; Haque, M.M.; Settles, M.; Nilsson, E.E.; Anway, M.D.; Skinner, M.K. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod. Toxicol. 2012, 34, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Bosagna, C.; Settles, M.; Lucker, B.; Skinner, M.K. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Li, W.; Zhao, W.; Wang, R. Targeting epigenetic regulations in cancer. Acta Biochim. Biophys. Sin. 2016, 48, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, C.M.; Schulz, W.A.; Skowron, M.A.; Hoffmann, M.J.; Niegisch, G. Tumor immunotherapy—The potential of epigenetic drugs to overcome resistance. Transl. Cancer Res. 2018, 7, 1151–1160. [Google Scholar] [CrossRef]
- Tiffon, C.E.; Adams, J.E.; van der Fits, L.; Wen, S.; Townsend, P.A.; Ganesan, A.; Hodges, E.; Vermeer, M.H.; Packham, G. The histone deacetylase inhibitors vorinostat and romidepsin downmodulate IL-10 expression in cutaneous T-cell lymphoma cells. Br. J. Pharmacol. 2011, 162, 1590–1602. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Nutrient | Food Origin | Epigenetic Role |
|---|---|---|
| Methionine | Sesame seeds, brazil nuts, fish, peppers, spinach | SAM synthesis |
| Folic Acid | Leafy vegetables, sunflower seeds, baker’s yeast, liver | Methionine synthesis |
| Vitamin B12 | Meat, liver, shellfish, milk | Methionine synthesis |
| Vitamin B6 | Meats, whole grain products, vegetables, nuts | Methionine synthesis |
| SAM-e (SAM) | Popular dietary supplement pill; unstable in food | Enzymes transfer methyl groups from SAM directly to the DNA |
| Choline | Egg yolks, liver, soy, cooked beef, chicken, veal and turkey | Methyl donor to SAM |
| Betaine | Wheat, spinach, shellfish, and sugar beets | Break down the toxic byproducts of SAM synthesis |
| Resveratrol | Red wine | Removes acetyl groups from histones, improving health (shown in lab mice) |
| Genistein | Soy, soy products | Increased methylation, cancer prevention, unknown mechanism |
| Sulforaphane | Broccoli | Increased histone acetylation turning on anti-cancer genes |
| Butyrate | A compound produced in the intestine when dietary fiber is fermented | Increased histone acetylation turning on ‘protective’ genes, increased lifespan (shown in the lab in flies) |
| Diallyl sulphide (DADS) | Garlic | Increased histone acetylation turning on anti-cancer genes |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. https://doi.org/10.3390/ijms19113425
Tiffon C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. International Journal of Molecular Sciences. 2018; 19(11):3425. https://doi.org/10.3390/ijms19113425
Chicago/Turabian StyleTiffon, Céline. 2018. "The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease" International Journal of Molecular Sciences 19, no. 11: 3425. https://doi.org/10.3390/ijms19113425
APA StyleTiffon, C. (2018). The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. International Journal of Molecular Sciences, 19(11), 3425. https://doi.org/10.3390/ijms19113425