Psoriasis Pathogenesis and Treatment

Abstract
1. Definition and Epidemiology
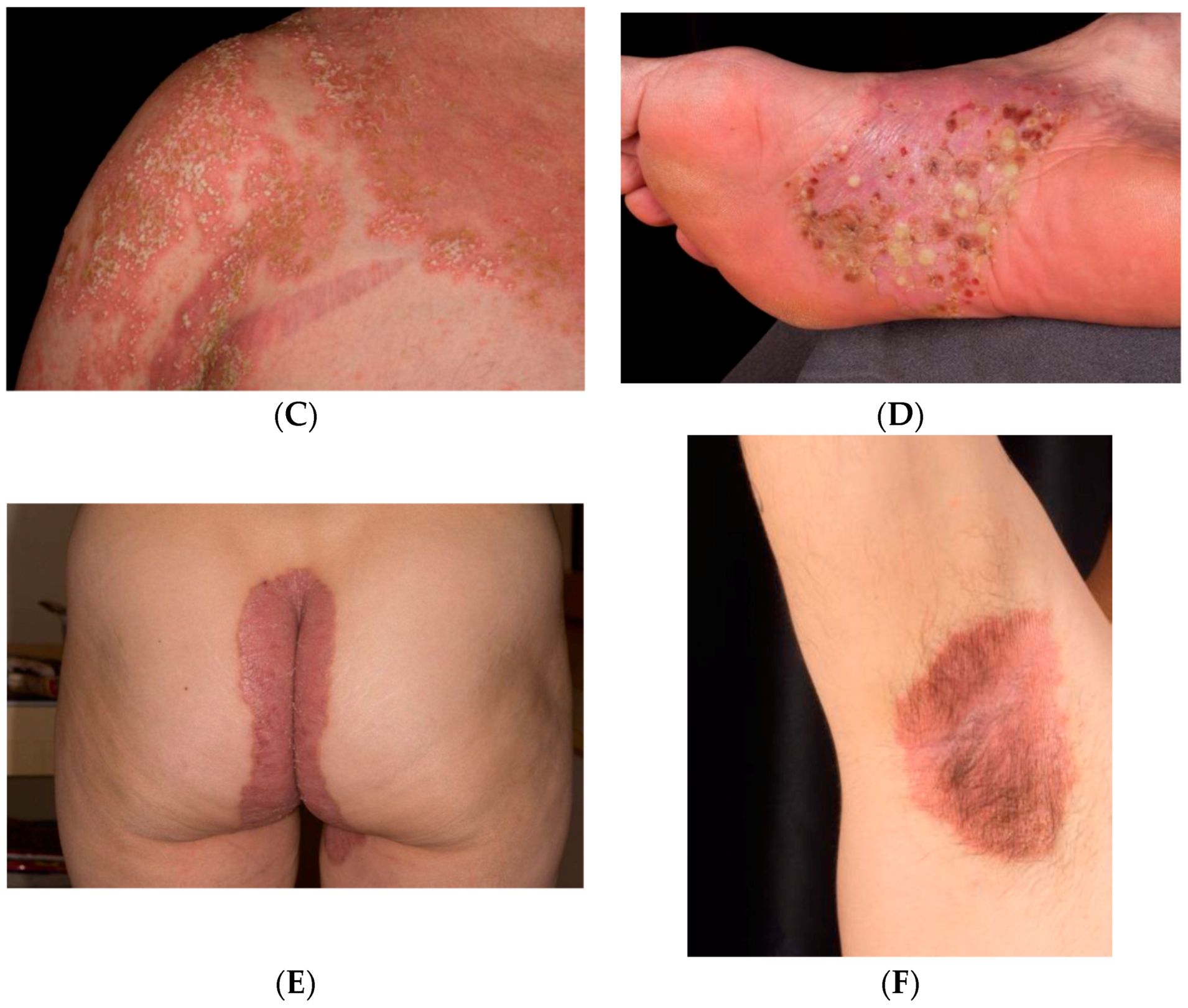
1.1. Clinical Classification
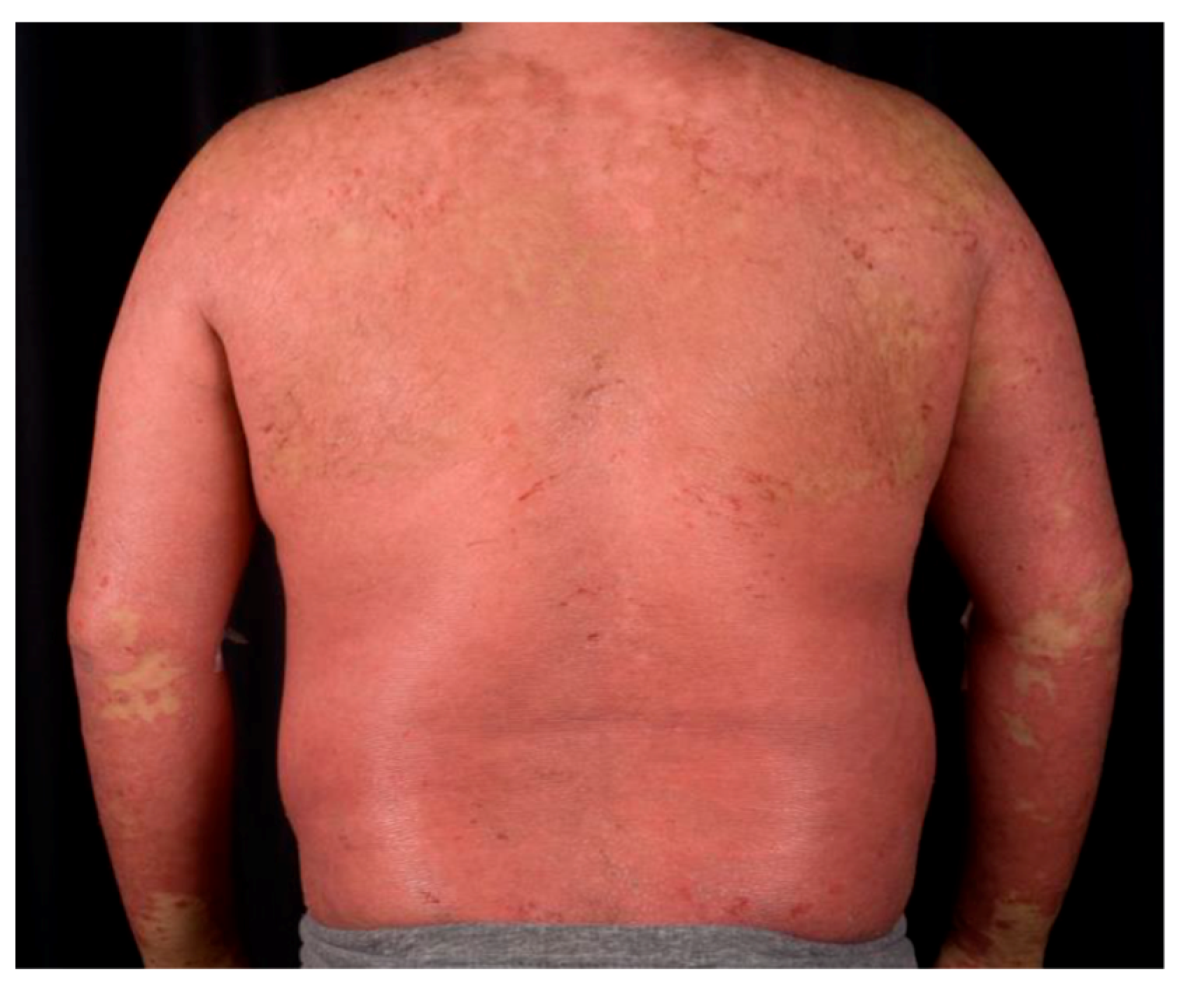
1.2. Psoriasis Vulgaris
1.3. Inverse Psoriasis
1.4. Guttate Psoriasis
1.5. Pustular psoriasis
1.6. Comorbidities in Psoriasis
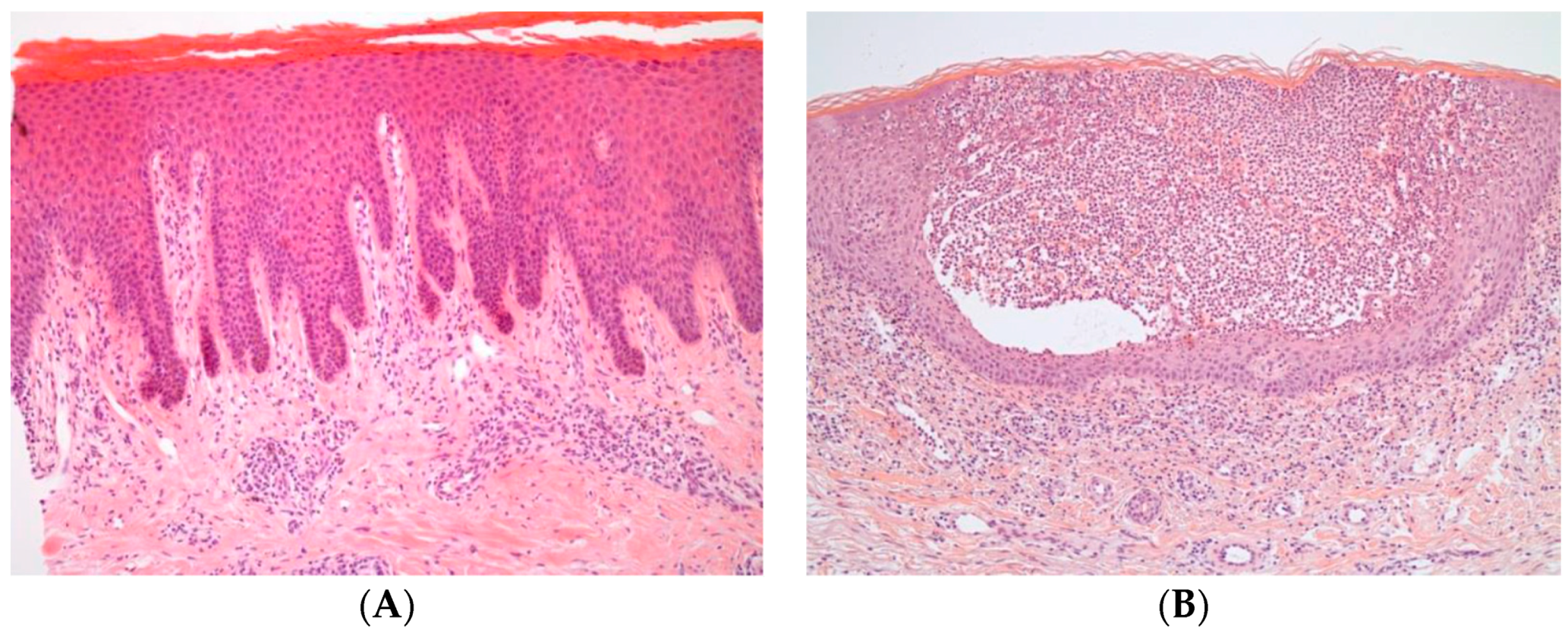
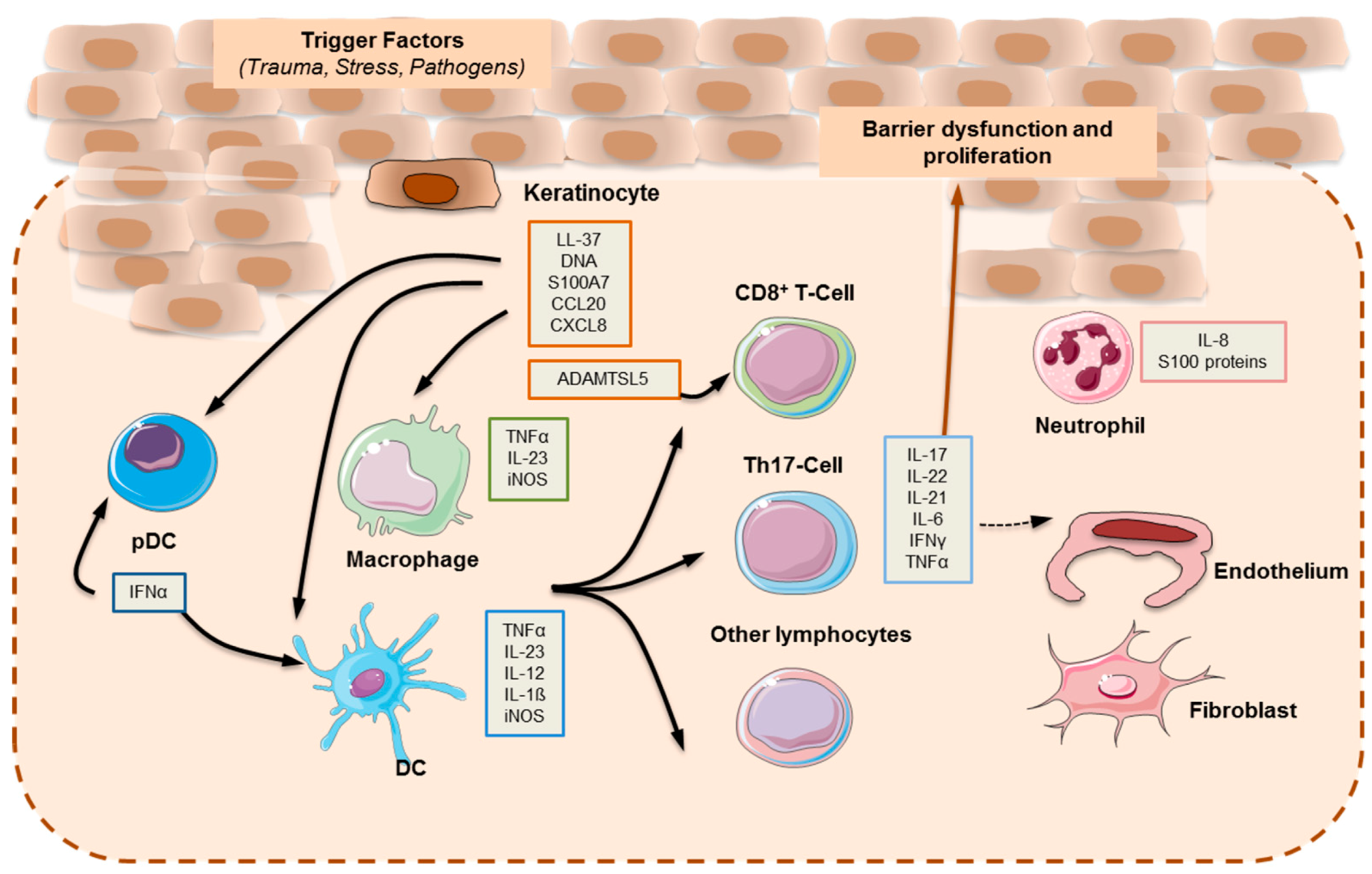
2. Pathogenesis
2.1. Main Cytokines and Cell Types in Plaque Psoriasis
2.2. Pathophysiology in Variants
2.3. Autoimmunity in Psoriasis
2.4. Genetics
2.4.1. Antigen Presentation
2.4.2. Genetic Variants Implicated in Aberrant Keratinocyte Proliferation and Differentiation
2.5. Epigenetics
2.6. Microbiome
3. Therapy
3.1. Small-Molecule Therapies
3.2. Biologics
3.2.1. TNF-α
3.2.2. IL23/Th17 axis
IL-23
IL-17
3.2.3. Biosimilars in Psoriasis
3.2.4. Drugs in the Research Pipeline
4. Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Christophers, E. Psoriasis—Epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef]
- Parisi, R.; Symmons, D.P.; Griffiths, C.E.; Ashcroft, D.M. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, S. Skin disease and socioeconomic conditions in rural Africa: Tanzania. Int. J. Dermatol. 1996, 35, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, T.D.; Schupp, C.W.; Armstrong, A.W. Psoriasis prevalence among adults in the united states. J. Am. Acad. Dermatol. 2014, 70, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, K.; Olsen, A.O.; Wilsgaard, T.; Furberg, A.S. Is the prevalence of psoriasis increasing? A 30-year follow-up of a population-based cohort. Br. J. Dermatol. 2013, 168, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.; Chimenti, S.; Luger, T.; Puig, L.; Reid, F.; Trueb, R.M. Scalp psoriasis: European consensus on grading and treatment algorithm. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.C.; Jwa, S.W.; Song, M.; Kim, M.B.; Kwon, K.S. Clinical course of guttate psoriasis: Long-term follow-up study. J. Dermatol. 2010, 37, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.A.; Chalmers, R.J.; Telfer, N.R. How great is the risk of further psoriasis following a single episode of acute guttate psoriasis? Arch. Dermatol. 1996, 132, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Koks, S.; Kingo, K.; Smith, C.; Barker, J.N.; Network, E. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Sommer, D.M.; Jenisch, S.; Suchan, M.; Christophers, E.; Weichenthal, M. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch. Dermatol. Res. 2006, 298, 321–328. [Google Scholar] [CrossRef]
- Gerdes, S.; Mrowietz, U.; Boehncke, W.H. Comorbidity in psoriasis. Hautarzt 2016, 67, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Herzog, C.; Rostock, A.; Ochsendorf, F.R.; Zollner, T.M.; Thaci, D.; Kaufmann, R.; Vogl, T.J.; Boehncke, W.H. Psoriasis: A possible risk factor for development of coronary artery calcification. Br. J. Dermatol. 2007, 156, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Dommasch, E.D.; Shin, D.B.; Azfar, R.S.; Kurd, S.K.; Wang, X.; Troxel, A.B. The risk of stroke in patients with psoriasis. J. Investig. Dermatol. 2009, 129, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Prodanovich, S.; Kirsner, R.S.; Kravetz, J.D.; Ma, F.; Martinez, L.; Federman, D.G. Association of psoriasis with coronary artery, cerebrovascular, and peripheral vascular diseases and mortality. Arch. Dermatol. 2009, 145, 700–703. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of myocardial infarction in patients with psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef]
- Ahlehoff, O.; Gislason, G.H.; Charlot, M.; Jorgensen, C.H.; Lindhardsen, J.; Olesen, J.B.; Abildstrom, S.Z.; Skov, L.; Torp-Pedersen, C.; Hansen, P.R. Psoriasis is associated with clinically significant cardiovascular risk: A danish nationwide cohort study. J. Intern. Med. 2011, 270, 147–157. [Google Scholar] [CrossRef]
- Kimball, A.B.; Guerin, A.; Latremouille-Viau, D.; Yu, A.P.; Gupta, S.; Bao, Y.; Mulani, P. Coronary heart disease and stroke risk in patients with psoriasis: Retrospective analysis. Am. J. Med. 2010, 123, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.S. Psoriasis is not a useful independent risk factor for cardiovascular disease. J. Investig. Dermatol. 2010, 130, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.S.; Huibregtse, A. Very severe psoriasis is associated with increased noncardiovascular mortality but not with increased cardiovascular risk. J. Investig. Dermatol. 2011, 131, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.J.; Harskamp, C.T.; Armstrong, A.W. Psoriasis and major adverse cardiovascular events: A systematic review and meta-analysis of observational studies. J. Am. Heart Assoc. 2013, 2, e000062. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, M.; Castelvecchio, S.; Ricci, C.; Pigatto, P.; Pellissero, G.; Cappato, R. Role of psoriasis as independent predictor of cardiovascular disease: A meta-regression analysis. Int. J. Cardiol. 2013, 168, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.J.; Weng, C.L.; Zhao, Y.T.; Liu, Q.H.; Yin, R.X. Psoriasis and risk of cardiovascular disease: A meta-analysis of cohort studies. Int. J. Cardiol. 2013, 168, 4992–4996. [Google Scholar] [CrossRef] [PubMed]
- Horreau, C.; Pouplard, C.; Brenaut, E.; Barnetche, T.; Misery, L.; Cribier, B.; Jullien, D.; Aractingi, S.; Aubin, F.; Joly, P.; et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: A systematic literature review. J. Eur. Acad. Dermatol. Venereol. 2013, 27 (Suppl. 3), 12–29. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.M.; Ellervik, C.; Yazdanyar, S.; Jemec, G.B. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J. Am. Acad. Dermatol. 2013, 69, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, A.; Bartosinska, J.; Chodorowska, G.; Szepietowski, J.C.; Paluszkiewicz, P.; Schwartz, R.A. Cardiovascular aspects of psoriasis: An updated review. Int. J. Dermatol. 2013, 52, 153–162. [Google Scholar] [CrossRef]
- Samarasekera, E.J.; Neilson, J.M.; Warren, R.B.; Parnham, J.; Smith, C.H. Incidence of cardiovascular disease in individuals with psoriasis: A systematic review and meta-analysis. J. Investig. Dermatol. 2013, 133, 2340–2346. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, Y.H. Association of psoriasis with stroke and myocardial infarction: Meta-analysis of cohort studies. Br. J. Dermatol. 2012, 167, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, A.; Skov, L.; Joshi, A.A.; Mallbris, L.; Gislason, G.H.; Wu, J.J.; Rodante, J.; Lerman, J.B.; Ahlman, M.A.; Gelfand, J.M.; et al. The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J. Am. Acad. Dermatol. 2017, 77, 650–656.e3. [Google Scholar] [CrossRef]
- Mehta, N.N.; Yu, Y.; Saboury, B.; Foroughi, N.; Krishnamoorthy, P.; Raper, A.; Baer, A.; Antigua, J.; Van Voorhees, A.S.; Torigian, D.A.; et al. Systemic and vascular inflammation in patients with moderate to severe psoriasis as measured by [18f]-fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET/CT): A pilot study. Arch. Dermatol. 2011, 147, 1031–1039. [Google Scholar] [CrossRef]
- Joshi, A.A.; Lerman, J.B.; Aberra, T.M.; Afshar, M.; Teague, H.L.; Rodante, J.A.; Krishnamoorthy, P.; Ng, Q.; Aridi, T.Z.; Salahuddin, T.; et al. Glyca is a novel biomarker of inflammation and subclinical cardiovascular disease in psoriasis. Circ. Res. 2016, 119, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Ogdie, A.; Langan, S.; Love, T.; Haynes, K.; Shin, D.; Seminara, N.; Mehta, N.N.; Troxel, A.; Choi, H.; Gelfand, J.M. Prevalence and treatment patterns of psoriatic arthritis in the UK. Rheumatology 2013, 52, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, J.; Ren, L.M.; Wang, H.Y.; Liu, W.H.; Zhang, X.W.; Chen, S.; Mu, R.; He, J.; Zhao, Y.; et al. Epidemiology of eight common rheumatic diseases in china: A large-scale cross-sectional survey in Beijing. Rheumatology 2012, 51, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, J.N.; Paula, A.P.; Martins, G.A. Psoriatic arthritis in patients with psoriasis: Evaluation of clinical and epidemiological features in 133 patients followed at the university hospital of Brasilia. An. Bras. Dermatol. 2012, 87, 539–544. [Google Scholar] [CrossRef]
- Haroon, M.; Kirby, B.; FitzGerald, O. High prevalence of psoriatic arthritis in patients with severe psoriasis with suboptimal performance of screening questionnaires. Ann. Rheum. Dis. 2013, 72, 736–740. [Google Scholar] [CrossRef]
- Henes, J.C.; Ziupa, E.; Eisfelder, M.; Adamczyk, A.; Knaudt, B.; Jacobs, F.; Lux, J.; Schanz, S.; Fierlbeck, G.; Spira, D.; et al. High prevalence of psoriatic arthritis in dermatological patients with psoriasis: A cross-sectional study. Rheumatol. Int. 2014, 34, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Gladman, D.D.; Papp, K.A.; Khraishi, M.M.; Thaci, D.; Behrens, F.; Northington, R.; Fuiman, J.; Bananis, E.; Boggs, R.; et al. Prevalence of rheumatologist-diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. J. Am. Acad. Dermatol. 2013, 69, 729–735. [Google Scholar] [CrossRef]
- Reich, K.; Kruger, K.; Mossner, R.; Augustin, M. Epidemiology and clinical pattern of psoriatic arthritis in germany: A prospective interdisciplinary epidemiological study of 1511 patients with Plaque-type psoriasis. Br. J. Dermatol. 2009, 160, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.P.; Rouzaud, M.; Sevrain, M.; Barnetche, T.; Paul, C.; Richard, M.A.; Beylot-Barry, M.; Misery, L.; Joly, P.; Le Maitre, M.; et al. Prevalence of undiagnosed psoriatic arthritis among psoriasis patients: Systematic review and meta-analysis. J. Am. Acad. Dermatol. 2015, 73, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Stoll, M.L.; Zurakowski, D.; Nigrovic, L.E.; Nichols, D.P.; Sundel, R.P.; Nigrovic, P.A. Patients with juvenile psoriatic arthritis comprise two distinct populations. Arthritis Rheum. 2006, 54, 3564–3572. [Google Scholar] [CrossRef]
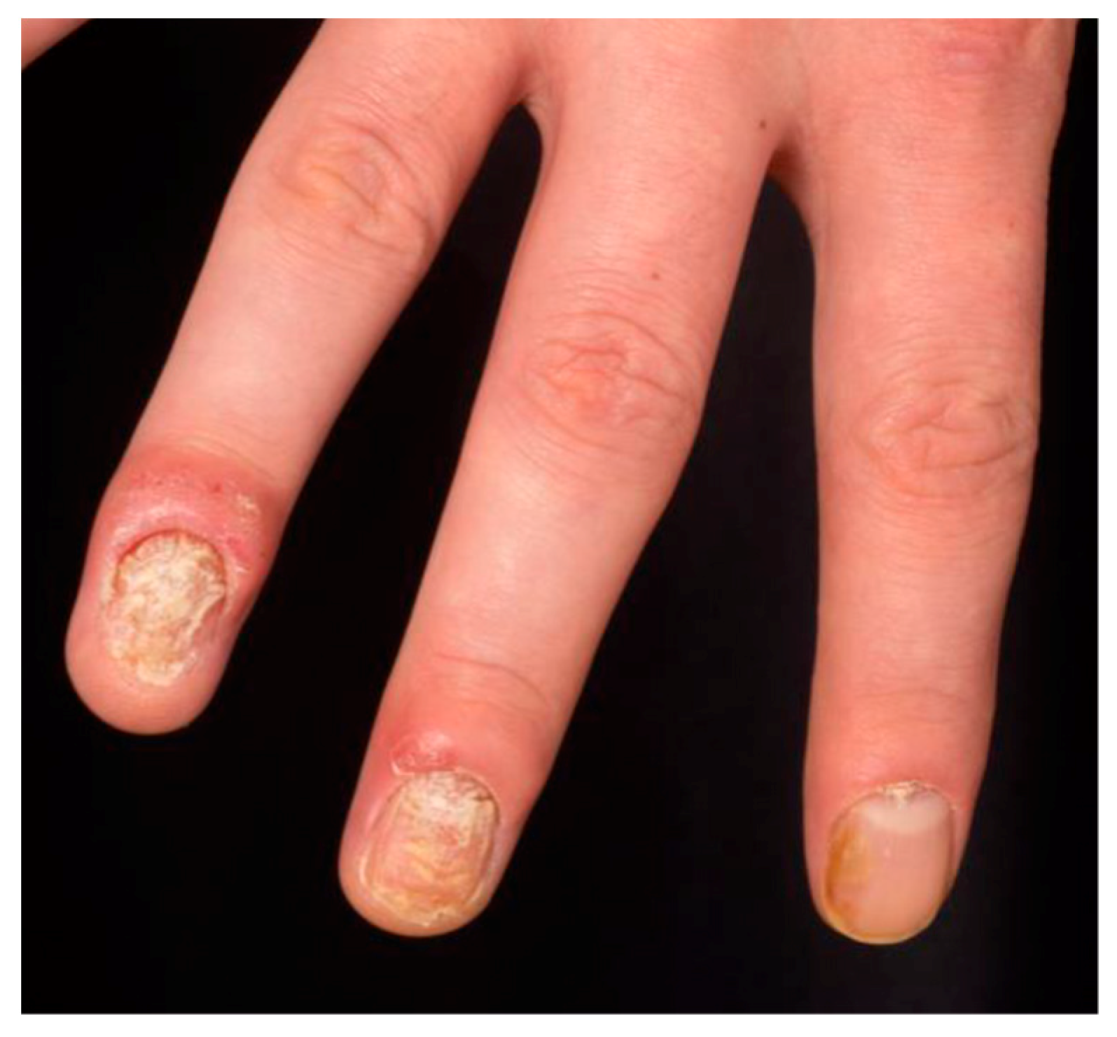
- Salomon, J.; Szepietowski, J.C.; Proniewicz, A. Psoriatic nails: A prospective clinical study. J. Cutan. Med. Surg. 2003, 7, 317–321. [Google Scholar] [CrossRef]
- Pasch, M.C. Nail psoriasis: A review of treatment options. Drugs 2016, 76, 675–705. [Google Scholar] [CrossRef] [PubMed]
- Langenbruch, A.; Radtke, M.A.; Krensel, M.; Jacobi, A.; Reich, K.; Augustin, M. Nail involvement as a predictor of concomitant psoriatic arthritis in patients with psoriasis. Br. J. Dermatol. 2014, 171, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Maejima, H.; Taniguchi, T.; Watarai, A.; Katsuoka, K. Evaluation of nail disease in psoriatic arthritis by using a modified nail psoriasis severity score index. Int. J. Dermatol. 2010, 49, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Ellinghaus, E.; Nair, R.P.; Stuart, P.E.; Esko, T.; Metspalu, A.; Debrus, S.; Raelson, J.V.; Tejasvi, T.; Belouchi, M.; et al. Combined analysis of genome-wide association studies for crohn disease and psoriasis identifies seven shared susceptibility loci. Am. J. Hum. Genet. 2012, 90, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Yeung, H.; Takeshita, J.; Mehta, N.N.; Kimmel, S.E.; Ogdie, A.; Margolis, D.J.; Shin, D.B.; Attor, R.; Troxel, A.B.; Gelfand, J.M. Psoriasis severity and the prevalence of major medical comorbidity: A population-based study. JAMA Dermatol. 2013, 149, 1173–1179. [Google Scholar] [CrossRef]
- Wan, J.; Wang, S.; Haynes, K.; Denburg, M.R.; Shin, D.B.; Gelfand, J.M. Risk of moderate to advanced kidney disease in patients with psoriasis: Population based cohort study. BMJ 2013, 347, f5961. [Google Scholar] [CrossRef] [PubMed]
- Rapp, S.R.; Feldman, S.R.; Exum, M.L.; Fleischer, A.B., Jr.; Reboussin, D.M. Psoriasis causes as much disability as other major medical diseases. J. Am. Acad. Dermatol. 1999, 41, 401–407. [Google Scholar] [CrossRef]
- Szepietowski, J.C.; Reich, A. Pruritus in psoriasis: An update. Eur. J. Pain 2016, 20, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.; Bai, J.W.; Pratt, M.; Sibbald, C.; Lynde, C.; Gulliver, W.P. The prevalence of anxiety in patients with psoriasis: A systematic review of observational studies and clinical trials. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Sampogna, F.; Tabolli, S.; Abeni, D. Living with psoriasis: Prevalence of shame, anger, worry, and problems in daily activities and social life. Acta Derm. Venereol. 2012, 92, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; Villanova, F.; Nestle, F.O. Psoriasis. Cold Spring Harb. Perspect. Med. 2014, 4, 6. [Google Scholar] [CrossRef]
- Harden, J.L.; Krueger, J.G.; Bowcock, A.M. The immunogenetics of psoriasis: A comprehensive review. J. Autoimmun. 2015, 64, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Gallo, R.L. Antimicrobial peptides in the pathogenesis of psoriasis. J. Dermatol. 2012, 39, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Yamasaki, K.; Muhleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide ll-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, J.; Meller, S.; Conrad, C.; Di Nardo, A.; Homey, B.; Lauerma, A.; Arai, N.; Gallo, R.L.; Digiovanni, J.; Gilliet, M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type i interferons. J. Exp. Med. 2010, 207, 2921–2930. [Google Scholar] [CrossRef]
- Santini, S.M.; Lapenta, C.; Donati, S.; Spadaro, F.; Belardelli, F.; Ferrantini, M. Interferon-α-conditioned human monocytes combine a TH1-orienting attitude with the induction of autologous TH17 responses: Role of IL-23 and IL-12. PLoS ONE 2011, 6, e17364. [Google Scholar] [CrossRef]
- Hansel, A.; Gunther, C.; Ingwersen, J.; Starke, J.; Schmitz, M.; Bachmann, M.; Meurer, M.; Rieber, E.P.; Schakel, K. Human slan (6-sulfo LacNAc) dendritic cells are inflammatory dermal dendritic cells in psoriasis and drive strong TH17/TH1 T-cell responses. J. Allergy Clin. Immunol. 2011, 127, 787–794. [Google Scholar] [CrossRef]
- Nestle, F.O.; Turka, L.A.; Nickoloff, B.J. Characterization of dermal dendritic cells in psoriasis. Autostimulation of t lymphocytes and induction of th1 type cytokines. J. Clin. Investig. 1994, 94, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Van der Fits, L.; Mourits, S.; Voerman, J.S.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Matsuzaki, G.; Umemura, M. Interleukin-17 family cytokines in protective immunity against infections: Role of hematopoietic cell-derived and non-hematopoietic cell-derived interleukin-17s. Microbiol. Immunol. 2018, 62, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef]
- Leung, D.Y.; Travers, J.B.; Giorno, R.; Norris, D.A.; Skinner, R.; Aelion, J.; Kazemi, L.V.; Kim, M.H.; Trumble, A.E.; Kotb, M.; et al. Evidence for a streptococcal superantigen-driven process in acute guttate psoriasis. J. Clin. Investig. 1995, 96, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Gudjonsson, J.E.; Sigmundsdottir, H.; Love, T.J.; Valdimarsson, H. Peripheral blood t cell responses to keratin peptides that share sequences with streptococcal m proteins are largely restricted to skin-homing CD8+ T cells. Clin. Exp. Immunol. 2004, 138, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Diluvio, L.; Vollmer, S.; Besgen, P.; Ellwart, J.W.; Chimenti, S.; Prinz, J.C. Identical TCR beta-chain rearrangements in streptococcal angina and skin lesions of patients with psoriasis vulgaris. J. Immunol. 2006, 176, 7104–7111. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef]
- Bissonnette, R.; Fuentes-Duculan, J.; Mashiko, S.; Li, X.; Bonifacio, K.M.; Cueto, I.; Suarez-Farinas, M.; Maari, C.; Bolduc, C.; Nigen, S.; et al. Palmoplantar pustular psoriasis (PPPP) is characterized by activation of the IL-17A pathway. J. Dermatol. Sci. 2017, 85, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Wilsmann-Theis, D.; Schnell, L.M.; Ralser-Isselstein, V.; Bieber, T.; Schon, M.P.; Huffmeier, U.; Mossner, R. Successful treatment with interleukin-17a antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J. Dermatol. 2018, 45, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Goldminz, A.M.; Au, S.C.; Kim, N.; Gottlieb, A.B.; Lizzul, P.F. Nf-kappab: An essential transcription factor in psoriasis. J. Dermatol. Sci. 2013, 69, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.A.; Nerviani, A.; Gallo Afflitto, G.; Pitzalis, C. Role of the IL-23/IL-17 axis in psoriasis and psoriatic arthritis: The clinical importance of its divergence in skin and joints. Int. J. Mol. Sci. 2018, 19, 530. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Bogdanos, D.P. Are psoriasis and psoriatic arthritis the same disease? The IL-23/IL-17 axis data. Autoimmun. Rev. 2017, 16, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Mensah, K.A.; Schwarz, E.M.; Ritchlin, C.T. Altered bone remodeling in psoriatic arthritis. Curr. Rheumatol. Rep. 2008, 10, 311–317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide ll37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Siewert, K.; Stohr, J.; Besgen, P.; Kim, S.M.; Ruhl, G.; Nickel, J.; Vollmer, S.; Thomas, P.; Krebs, S.; et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 2015, 212, 2203–2212. [Google Scholar] [CrossRef]
- Fuentes-Duculan, J.; Bonifacio, K.M.; Hawkes, J.E.; Kunjravia, N.; Cueto, I.; Li, X.; Gonzalez, J.; Garcet, S.; Krueger, J.G. Autoantigens ADAMTSL5 and LL37 are significantly upregulated in active psoriasis and localized with keratinocytes, dendritic cells and other leukocytes. Exp. Dermatol. 2017, 26, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Jarrett, R.; Subramaniam, S.; Salimi, M.; Gutowska-Owsiak, D.; Chen, Y.L.; Hardman, C.; Xue, L.; Cerundolo, V.; Ogg, G. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1A. J. Exp. Med. 2016, 213, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Yunusbaeva, M.; Valiev, R.; Bilalov, F.; Sultanova, Z.; Sharipova, L.; Yunusbayev, B. Psoriasis patients demonstrate HLA-Cw*06:02 allele dosage-dependent T cell proliferation when treated with hair follicle-derived keratin 17 protein. Sci. Rep. 2018, 8, 6098. [Google Scholar] [CrossRef]
- Farber, E.M.; Nall, M.L.; Watson, W. Natural history of psoriasis in 61 twin pairs. Arch. Dermatol. 1974, 109, 207–211. [Google Scholar] [CrossRef]
- Farber, E.M.; Nall, M.L. The natural history of psoriasis in 5600 patients. Dermatologica 1974, 148, 1–18. [Google Scholar] [CrossRef]
- Davidson, A.; Diamond, B. Autoimmune diseases. N. Engl. J. Med. 2001, 345, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Hayter, S.M.; Cook, M.C. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun. Rev. 2012, 11, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.M.; Krueger, J.G. Getting under the skin: The immunogenetics of psoriasis. Nat. Rev. Immunol. 2005, 5, 699–711. [Google Scholar] [CrossRef]
- Sagoo, G.S.; Cork, M.J.; Patel, R.; Tazi-Ahnini, R. Genome-wide studies of psoriasis susceptibility loci: A review. J. Dermatol. Sci. 2004, 35, 171–179. [Google Scholar] [CrossRef]
- Elder, J.T. Expanded genome-wide association study meta-analysis of psoriasis expands the catalog of common psoriasis-associated variants. J. Investig. Dermatol. Symp. Proc. 2018, 19, S77–S78. [Google Scholar] [CrossRef]
- Trembath, R.C.; Clough, R.L.; Rosbotham, J.L.; Jones, A.B.; Camp, R.D.; Frodsham, A.; Browne, J.; Barber, R.; Terwilliger, J.; Lathrop, G.M.; et al. Identification of a major susceptibility locus on chromosome 6p and evidence for further disease loci revealed by a two stage genome-wide search in psoriasis. Hum. Mol. Genet. 1997, 6, 813–820. [Google Scholar] [CrossRef]
- Nair, R.P.; Stuart, P.E.; Nistor, I.; Hiremagalore, R.; Chia, N.V.; Jenisch, S.; Weichenthal, M.; Abecasis, G.R.; Lim, H.W.; Christophers, E.; et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006, 78, 827–851. [Google Scholar] [CrossRef] [PubMed]
- Mallon, E.; Bunce, M.; Savoie, H.; Rowe, A.; Newson, R.; Gotch, F.; Bunker, C.B. HLA-C and guttate psoriasis. Br. J. Dermatol. 2000, 143, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Karason, A.; Antonsdottir, A.; Runarsdottir, E.H.; Hauksson, V.B.; Upmanyu, R.; Gulcher, J.; Stefansson, K.; Valdimarsson, H. Psoriasis patients who are homozygous for the Hla-Cw*0602 allele have a 2.5-fold increased risk of developing psoriasis compared with Cw6 heterozygotes. Br. J. Dermatol. 2003, 148, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.H.; Ameen, H.; Veal, C.; Evans, J.; Ramrakha-Jones, V.S.; Marsland, A.M.; Burden, A.D.; Griffiths, C.E.; Trembath, R.C.; Barker, J.N. The major psoriasis susceptibility locus psors1 is not a risk factor for late-onset psoriasis. J. Investig. Dermatol. 2005, 124, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Berki, D.M.; Liu, L.; Choon, S.E.; David Burden, A.; Griffiths, C.E.M.; Navarini, A.A.; Tan, E.S.; Irvine, A.D.; Ranki, A.; Ogo, T.; et al. Activating card14 mutations are associated with generalized pustular psoriasis but rarely account for familial recurrence in psoriasis vulgaris. J. Investig. Dermatol. 2015, 135, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Yang, C.F.; Fann, C.S.; Chen, C.L.; Tsai, T.F.; Chien, Y.H.; Chiang, S.C.; Chen, C.H.; Hung, S.I.; Wu, J.Y.; et al. Mapping of psoriasis to 17q terminus. J. Med. Genet. 2005, 42, 152–158. [Google Scholar] [CrossRef]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Tomfohrde, J.; Silverman, A.; Barnes, R.; Fernandez-Vina, M.A.; Young, M.; Lory, D.; Morris, L.; Wuepper, K.D.; Stastny, P.; Menter, A.; et al. Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science 1994, 264, 1141–1145. [Google Scholar] [CrossRef]
- Capon, F.; Novelli, G.; Semprini, S.; Clementi, M.; Nudo, M.; Vultaggio, P.; Mazzanti, C.; Gobello, T.; Botta, A.; Fabrizi, G.; et al. Searching for psoriasis susceptibility genes in italy: Genome scan and evidence for a new locus on chromosome 1. J. Investig. Dermatol. 1999, 112, 32–35. [Google Scholar] [CrossRef]
- De Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C.; et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef]
- Oh, I.Y.; de Guzman Strong, C. The molecular revolution in cutaneous biology: EDC and locus control. J. Investig. Dermatol. 2017, 137, e101–e104. [Google Scholar] [CrossRef]
- Riveira-Munoz, E.; He, S.M.; Escaramis, G.; Stuart, P.E.; Huffmeier, U.; Lee, C.; Kirby, B.; Oka, A.; Giardina, E.; Liao, W.; et al. Meta-analysis confirms the LCE3C_LCE3B deletion as a risk factor for psoriasis in several ethnic groups and finds interaction with HLA-Cw6. J. Investig. Dermatol. 2011, 131, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.T. Genome-wide association scan yields new insights into the immunopathogenesis of psoriasis. Genes Immun. 2009, 10, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Knight, J.; Tejasvi, T.; Kang, H.M.; Allen, M.H.; Lambert, S.; et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 2015, 6, 7001. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Low, H.Q.; Wang, L.; Li, Y.; Ellinghaus, E.; Han, J.; Estivill, X.; Sun, L.; Zuo, X.; Shen, C.; et al. Genome-wide meta-analysis identifies multiple novel associations and ethnic heterogeneity of psoriasis susceptibility. Nat. Commun. 2015, 6, 6916. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Andres, R.M.; Hald, A.; Johansen, C.; Kragballe, K.; Iversen, L. Studies of jak/stat3 expression and signalling in psoriasis identifies STAT3-SER727 phosphorylation as a modulator of transcriptional activity. Exp. Dermatol. 2013, 22, 323–328. [Google Scholar] [CrossRef]
- Di Meglio, P.; Di Cesare, A.; Laggner, U.; Chu, C.C.; Napolitano, L.; Villanova, F.; Tosi, I.; Capon, F.; Trembath, R.C.; Peris, K.; et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced TH17 effector response in humans. PLoS ONE 2011, 6, e17160. [Google Scholar] [CrossRef]
- Kopp, T.; Riedl, E.; Bangert, C.; Bowman, E.P.; Greisenegger, E.; Horowitz, A.; Kittler, H.; Blumenschein, W.M.; McClanahan, T.K.; Marbury, T.; et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature 2015, 521, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Eken, A.; Singh, A.K.; Oukka, M. Interleukin 23 in crohn’s disease. Inflamm. Bowel. Dis. 2014, 20, 587–595. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Selectivity and therapeutic inhibition of kinases: To be or not to be? Nat. Immunol. 2009, 10, 356–360. [Google Scholar] [CrossRef]
- Zhang, F.; Meng, G.; Strober, W. Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 2008, 9, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Craiglow, B.G.; Boyden, L.M.; Hu, R.; Virtanen, M.; Su, J.; Rodriguez, G.; McCarthy, C.; Luna, P.; Larralde, M.; Humphrey, S.; et al. CARD14-associated papulosquamous eruption: A spectrum including features of psoriasis and pityriasis rubra pilaris. J. Am. Acad. Dermatol. 2018, 79, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-κB/RELA in normal and psoriatic epidermis and downregulation of NF-κB in response to treatment with etanercept. J. Investig. Dermatol. 2005, 124, 1275–1283. [Google Scholar] [CrossRef]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Stuart, P.E.; Nair, R.P.; Ellinghaus, E.; Ding, J.; Tejasvi, T.; Gudjonsson, J.E.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C.; et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Huffmeier, U.; Uebe, S.; Ekici, A.B.; Bowes, J.; Giardina, E.; Korendowych, E.; Juneblad, K.; Apel, M.; McManus, R.; Ho, P.; et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010, 42, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K. The genetic background of generalized pustular psoriasis: Il36rn mutations and card14 gain-of-function variants. J. Dermatol. Sci. 2014, 74, 187–192. [Google Scholar] [CrossRef]
- Tian, S.; Krueger, J.G.; Li, K.; Jabbari, A.; Brodmerkel, C.; Lowes, M.A.; Suarez-Farinas, M. Meta-analysis derived (mad) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS ONE 2012, 7, e44274. [Google Scholar] [CrossRef] [PubMed]
- Ainali, C.; Valeyev, N.; Perera, G.; Williams, A.; Gudjonsson, J.E.; Ouzounis, C.A.; Nestle, F.O.; Tsoka, S. Transcriptome classification reveals molecular subtypes in psoriasis. BMC Genom. 2012, 13, 472. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, A.; Suarez-Farinas, M.; Fuentes-Duculan, J.; Cueto, I.; Li, K.; Tian, S.; Brodmerkel, C.; Krueger, J.G. Increased expression of interleukin-17 pathway genes in nonlesional skin of moderate-to-severe psoriasis vulgaris. Br. J. Dermatol. 2016, 174, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Stuart, P.E.; Sarkar, M.K.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Cellular dissection of psoriasis for transcriptome analyses and the post-GWAS era. BMC Med. Genom. 2014, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Grjibovski, A.M.; Olsen, A.O.; Magnus, P.; Harris, J.R. Psoriasis in norwegian twins: Contribution of genetic and environmental effects. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.; Wapinski, O.L.; Yang, Y.W.; Bureau, J.F.; Gopinath, S.; Monack, D.M.; Chang, H.Y.; Brahic, M.; Kirkegaard, K. The nest long NCRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef]
- Gupta, R.; Ahn, R.; Lai, K.; Mullins, E.; Debbaneh, M.; Dimon, M.; Arron, S.; Liao, W. Landscape of long noncoding RNAS in psoriatic and healthy skin. J. Investig. Dermatol. 2016, 136, 603–609. [Google Scholar] [CrossRef]
- Sonkoly, E.; Bata-Csorgo, Z.; Pivarcsi, A.; Polyanka, H.; Kenderessy-Szabo, A.; Molnar, G.; Szentpali, K.; Bari, L.; Megyeri, K.; Mandi, Y.; et al. Identification and characterization of a novel, psoriasis susceptibility-related noncoding RNA gene, PRINS. J. Biol. Chem. 2005, 280, 24159–24167. [Google Scholar] [CrossRef]
- Szegedi, K.; Sonkoly, E.; Nagy, N.; Nemeth, I.B.; Bata-Csorgo, Z.; Kemeny, L.; Dobozy, A.; Szell, M. The anti-apoptotic protein G1P3 is overexpressed in psoriasis and regulated by the non-coding RNA, PRINS. Exp. Dermatol. 2010, 19, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Iyer, M.K.; Stuart, P.E.; Swindell, W.R.; Gudjonsson, J.E.; Tejasvi, T.; Sarkar, M.K.; Li, B.; Ding, J.; Voorhees, J.J.; et al. Analysis of long non-coding RNAS highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. 2015, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.C.; Wang, K.C. Long noncoding RNA: Significance and potential in skin biology. Cold Spring Harb. Perspect. Med. 2014, 4, a015404. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Nguyen, G.H.; Fujita, M.; Florell, S.R.; Callis Duffin, K.; Krueger, G.G.; O’Connell, R.M. Micrornas in psoriasis. J. Investig. Dermatol. 2016, 136, 365–371. [Google Scholar] [CrossRef]
- Lovendorf, M.B.; Zibert, J.R.; Gyldenlove, M.; Ropke, M.A.; Skov, L. MicroRNA-223 and MIR-143 are important systemic biomarkers for disease activity in psoriasis. J. Dermatol. Sci. 2014, 75, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.Y.; Han, L.; Weiland, M.; Lu, C.J.; McKinnon, K.; Zhou, L.; Lim, H.W.; Elder, J.T.; Mi, Q.S. Emerging biomarkers in psoriatic arthritis. IUBMB Life 2015, 67, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Meisgen, F.; Butler, L.M.; Han, G.; Wang, X.J.; Soderberg-Naucler, C.; Stahle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. J. Immunol. 2013, 190, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Guinea-Viniegra, J.; Jiménez, M.; Schonthaler, H.B.; Navarro, R.; Delgado, Y.; José Concha-Garzón, M.; Tschachler, E.; Obad, S.; Daudén, E.; Wagner, E.F. Targeting MIR-21 to treat psoriasis. Sci. Transl. Med. 2014, 6, 225re221. [Google Scholar] [CrossRef]
- Joyce, C.E.; Zhou, X.; Xia, J.; Ryan, C.; Thrash, B.; Menter, A.; Zhang, W.; Bowcock, A.M. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum. Mol. Genet. 2011, 20, 4025–4040. [Google Scholar] [CrossRef]
- Zibert, J.R.; Lovendorf, M.B.; Litman, T.; Olsen, J.; Kaczkowski, B.; Skov, L. Micrornas and potential target interactions in psoriasis. J. Dermatol. Sci. 2010, 58, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zeng, J.; Yuan, J.; Deng, X.; Huang, Y.; Chen, L.; Zhang, P.; Feng, H.; Liu, Z.; Wang, Z.; et al. MicroRNA-210 overexpression promotes psoriasis-like inflammation by inducing TH1 and TH17 cell differentiation. J. Clin. Investig. 2018, 128, 2551–2568. [Google Scholar] [CrossRef]
- Lovendorf, M.B.; Mitsui, H.; Zibert, J.R.; Ropke, M.A.; Hafner, M.; Dyring-Andersen, B.; Bonefeld, C.M.; Krueger, J.G.; Skov, L. Laser capture microdissection followed by next-generation sequencing identifies disease-related micrornas in psoriatic skin that reflect systemic microRNA changes in psoriasis. Exp. Dermatol. 2015, 24, 187–193. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, S.; Arias-Santiago, S.; Orgaz-Molina, J.; Magro-Checa, C.; Valenzuela, I.; Navarro, P.; Naranjo-Sintes, R.; Sancho, J.; Zubiaur, M. Abnormal levels of expression of plasma microRNA-33 in patients with psoriasis. Actas. Dermosifiliogr. 2014, 105, 497–503. [Google Scholar] [CrossRef]
- Chatzikyriakidou, A.; Voulgari, P.V.; Georgiou, I.; Drosos, A.A. The role of microrna-146a (miR-146a) and its target IL-1R-associated kinase (IRAK1) in psoriatic arthritis susceptibility. Scand. J. Immunol. 2010, 71, 382–385. [Google Scholar] [CrossRef]
- Zhang, W.; Yi, X.; Guo, S.; Shi, Q.; Wei, C.; Li, X.; Gao, L.; Wang, G.; Gao, T.; Wang, L.; et al. A single-nucleotide polymorphism of mir-146a and psoriasis: An association and functional study. J. Cell. Mol. Med. 2014, 18, 2225–2234. [Google Scholar] [CrossRef]
- Xu, L.; Leng, H.; Shi, X.; Ji, J.; Fu, J.; Leng, H. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed. Pharmacother. 2017, 90, 524–530. [Google Scholar] [CrossRef]
- Primo, M.N.; Bak, R.O.; Schibler, B.; Mikkelsen, J.G. Regulation of pro-inflammatory cytokines TNFα and IL24 by microRNA-203 in primary keratinocytes. Cytokine 2012, 60, 741–748. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.T.; Liang, G.P.; Zhang, P.; Deng, X.J.; Tang, Q.; Zhai, H.Y.; Chang, C.C.; Su, Y.W.; Lu, Q.J. Up-regulation of microRNA-210 induces immune dysfunction via targeting FOXP3 in CD4+ T cells of psoriasis vulgaris. Clin. Immunol. 2014, 150, 22–30. [Google Scholar] [CrossRef]
- Tsuru, Y.; Jinnin, M.; Ichihara, A.; Fujisawa, A.; Moriya, C.; Sakai, K.; Fukushima, S.; Ihn, H. MiR-424 levels in hair shaft are increased in psoriatic patients. J. Dermatol. 2014, 41, 382–385. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Krueger, G. A role for epigenetics in psoriasis: Methylated cytosine-guanine sites differentiate lesional from nonlesional skin and from normal skin. J. Investig. Dermatol. 2012, 132, 506–508. [Google Scholar] [CrossRef]
- Roberson, E.D.; Liu, Y.; Ryan, C.; Joyce, C.E.; Duan, S.; Cao, L.; Martin, A.; Liao, W.; Menter, A.; Bowcock, A.M. A subset of methylated CPG sites differentiate psoriatic from normal skin. J. Investig. Dermatol. 2012, 132, 583–592. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Fahlen, A.; Engstrand, L.; Baker, B.S.; Powles, A.; Fry, L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch. Dermatol. Res. 2012, 304, 15–22. [Google Scholar] [CrossRef]
- Miyoshi, J.; Chang, E.B. The gut microbiota and inflammatory bowel diseases. Transl. Res. 2017, 179, 38–48. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.H.; Strober, B.E.; Pei, Z.; Blaser, M.J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 2008, 3, e2719. [Google Scholar] [CrossRef]
- Alekseyenko, A.V.; Perez-Perez, G.I.; De Souza, A.; Strober, B.; Gao, Z.; Bihan, M.; Li, K.; Methe, B.A.; Blaser, M.J. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 2013, 1, 31. [Google Scholar] [CrossRef]
- Fry, L.; Baker, B.S. Triggering psoriasis: The role of infections and medications. Clin. Dermatol. 2007, 25, 606–615. [Google Scholar] [CrossRef]
- Takemoto, A.; Cho, O.; Morohoshi, Y.; Sugita, T.; Muto, M. Molecular characterization of the skin fungal microbiome in patients with psoriasis. J. Dermatol. 2015, 42, 166–170. [Google Scholar] [CrossRef]
- Statnikov, A.; Alekseyenko, A.V.; Li, Z.; Henaff, M.; Perez-Perez, G.I.; Blaser, M.J.; Aliferis, C.F. Microbiomic signatures of psoriasis: Feasibility and methodology comparison. Sci. Rep. 2013, 3, 2620. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.H.; Pei, Z.; Blaser, M.J. Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl. Acad. Sci. USA 2007, 104, 2927–2932. [Google Scholar] [CrossRef]
- Mrowietz, U.; Kragballe, K.; Reich, K.; Spuls, P.; Griffiths, C.E.; Nast, A.; Franke, J.; Antoniou, C.; Arenberger, P.; Balieva, F.; et al. Definition of treatment goals for moderate to severe psoriasis: A European consensus. Arch. Dermatol. Res. 2011, 303, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hone, S.W.; Donnelly, M.J.; Powell, F.; Blayney, A.W. Clearance of recalcitrant psoriasis after tonsillectomy. Clin. Otolaryngol. Allied Sci. 1996, 21, 546–547. [Google Scholar] [CrossRef] [PubMed]
- McMillin, B.D.; Maddern, B.R.; Graham, W.R. A role for tonsillectomy in the treatment of psoriasis? Ear Nose Throat. J. 1999, 78, 155–158. [Google Scholar] [CrossRef]
- Rachakonda, T.D.; Dhillon, J.S.; Florek, A.G.; Armstrong, A.W. Effect of tonsillectomy on psoriasis: A systematic review. J. Am. Acad. Dermatol. 2015, 72, 261–275. [Google Scholar] [CrossRef]
- Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Sigurgeirsson, B.; Olafsson, J.H.; Petersen, H.; Sigurdsson, M.I.; Gudjonsson, J.E.; Johnston, A.; Valdimarsson, H. HLA-Cw6 homozygosity in plaque psoriasis is associated with streptococcal throat infections and pronounced improvement after tonsillectomy: A prospective case series. J. Am. Acad. Dermatol. 2016, 75, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Sigurgeirsson, B.; Olafsson, J.H.; Sigurdsson, M.I.; Petersen, H.; Arnadottir, S.; Gudjonsson, J.E.; Johnston, A.; Valdimarsson, H. Improvement of psoriasis after tonsillectomy is associated with a decrease in the frequency of circulating T cells that recognize streptococcal determinants and homologous skin determinants. J. Immunol. 2012, 188, 5160–5165. [Google Scholar] [CrossRef]
- Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Sigurgeirsson, B.; Olafsson, J.H.; Sigurdsson, M.I.; Petersen, H.; Gudjonsson, J.E.; Johnston, A.; Valdimarsson, H. Patient-reported outcomes and clinical response in patients with moderate-to-severe plaque psoriasis treated with tonsillectomy: A randomized controlled trial. Acta Derm. Venereol. 2017, 97, 340–345. [Google Scholar] [CrossRef]
- Revicki, D.; Willian, M.K.; Saurat, J.H.; Papp, K.A.; Ortonne, J.P.; Sexton, C.; Camez, A. Impact of adalimumab treatment on health-related quality of life and other patient-reported outcomes: Results from a 16-week randomized controlled trial in patients with moderate to severe plaque psoriasis. Br. J. Dermatol. 2008, 158, 549–557. [Google Scholar] [CrossRef]
- Saurat, J.H.; Stingl, G.; Dubertret, L.; Papp, K.; Langley, R.G.; Ortonne, J.P.; Unnebrink, K.; Kaul, M.; Camez, A.; Investigators, C.S. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. Methotrexate vs. Placebo in patients with psoriasis (champion). Br. J. Dermatol. 2008, 158, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, T.; Salah, L.A.; Gillstedt, M.; Wennberg, A.M.; Osmancevic, A. Methotrexate management in psoriasis: Are we following the guidelines? Acta Derm. Venereol. 2018, 98, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.C.; Helliwell, P.S. Methotrexate efficacy in the tight control in psoriatic arthritis study. J. Rheumatol. 2016, 43, 356–361. [Google Scholar] [CrossRef]
- West, J.; Ogston, S.; Berg, J.; Palmer, C.; Fleming, C.; Kumar, V.; Foerster, J. Hla-cw6-positive patients with psoriasis show improved response to methotrexate treatment. Clin. Exp. Dermatol. 2017, 42, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.C.; Griffiths, C.E.; Berth-Jones, J.; Papp, K.A.; Vanaclocha, F.; Dauden, E.; Beard, A.; Puvanarajan, L.; Paul, C. Intermittent short courses of cyclosporine microemulsion for the long-term management of psoriasis: A 2-year cohort study. J. Am. Acad. Dermatol. 2001, 44, 643–651. [Google Scholar] [CrossRef]
- Brand, N.; Petkovich, M.; Krust, A.; Chambon, P.; de The, H.; Marchio, A.; Tiollais, P.; Dejean, A. Identification of a second human retinoic acid receptor. Nature 1988, 332, 850–853. [Google Scholar] [CrossRef]
- Harper, R.A. Specificity in the synergism between retinoic acid and EGF on the growth of adult human skin fibroblasts. Exp. Cell Res. 1988, 178, 254–263. [Google Scholar] [CrossRef]
- Lee, J.H.; Youn, J.I.; Kim, T.Y.; Choi, J.H.; Park, C.J.; Choe, Y.B.; Song, H.J.; Kim, N.I.; Kim, K.J.; Lee, J.H.; et al. A multicenter, randomized, open-label pilot trial assessing the efficacy and safety of etanercept 50 mg twice weekly followed by etanercept 25 mg twice weekly, the combination of etanercept 25 mg twice weekly and acitretin, and acitretin alone in patients with moderate to severe psoriasis. BMC Dermatol. 2016, 16, 11. [Google Scholar]
- Gesser, B.; Johansen, C.; Rasmussen, M.K.; Funding, A.T.; Otkjaer, K.; Kjellerup, R.B.; Kragballe, K.; Iversen, L. Dimethylfumarate specifically inhibits the mitogen and stress-activated kinases 1 and 2 (MSK1/2): Possible role for its anti-psoriatic effect. J. Investig. Dermatol. 2007, 127, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.C.; Listopad, J.J.; Rentzsch, C.U.; Igney, F.H.; von Bonin, A.; Hennekes, H.H.; Asadullah, K.; Docke, W.D. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J. Investig. Dermatol. 2007, 127, 835–845. [Google Scholar] [CrossRef]
- Gillard, G.O.; Collette, B.; Anderson, J.; Chao, J.; Scannevin, R.H.; Huss, D.J.; Fontenot, J.D. Dmf, but not other fumarates, inhibits NF-κB activity in vitro in an NRF2-independent manner. J. Neuroimmunol. 2015, 283, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Oehrl, S.; Olaru, F.; Kunze, A.; Maas, M.; Pezer, S.; Schmitz, M.; Schakel, K. Controlling the pro-inflammatory function of 6-sulfo LacNAc (slan) dendritic cells with dimethylfumarate. J. Dermatol. Sci. 2017, 87, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Thaci, D.; Mrowietz, U.; Kamps, A.; Neureither, M.; Luger, T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis—A retrospective study (future). J. Dtsch. Dermatol. Ges. 2009, 7, 603–611. [Google Scholar] [CrossRef]
- Anstey, A.V. Fumaric acid esters in the treatment of psoriasis. Br. J. Dermatol. 2010, 162, 237–238. [Google Scholar] [CrossRef]
- Carboni, I.; De Felice, C.; De Simoni, I.; Soda, R.; Chimenti, S. Fumaric acid esters in the treatment of psoriasis: An italian experience. J. Dermatol. Treat. 2004, 15, 23–26. [Google Scholar] [CrossRef]
- Heelan, K.; Markham, T. Fumaric acid esters as a suitable first-line treatment for severe psoriasis: An irish experience. Clin. Exp. Dermatol. 2012, 37, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Kokelj, F.; Plozzer, C.; Avian, A.; Trevisan, G. Fumaric acid and its derivatives in the treatment of psoriasis vulgaris: Our experience in forty-one patients. Acta Dermatovenerol. Croat. 2009, 17, 170–175. [Google Scholar]
- Agency, E.M. Assessment Report: Skilarence; European Medicines Agency: London, UK, 2017.
- Altmeyer, P.J.; Matthes, U.; Pawlak, F.; Hoffmann, K.; Frosch, P.J.; Ruppert, P.; Wassilew, S.W.; Horn, T.; Kreysel, H.W.; Lutz, G.; et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J. Am. Acad. Dermatol. 1994, 30, 977–981. [Google Scholar] [CrossRef]
- Fallah Arani, S.; Neumann, H.; Hop, W.C.; Thio, H.B. Fumarates vs. Methotrexate in moderate to severe chronic plaque psoriasis: A multicentre prospective randomized controlled clinical trial. Br. J. Dermatol. 2011, 164, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, H.; Altmeyer, P.; Kaufmann, R.; Ring, J.; Christophers, E.; Pavel, S.; Ziegler, J. Topical calcipotriol plus oral fumaric acid is more effective and faster acting than oral fumaric acid monotherapy in the treatment of severe chronic plaque psoriasis vulgaris. Dermatology 2002, 205, 46–53. [Google Scholar] [CrossRef]
- Nieboer, C.; de Hoop, D.; Langendijk, P.N.; van Loenen, A.C.; Gubbels, J. Fumaric acid therapy in psoriasis: A double-blind comparison between fumaric acid compound therapy and monotherapy with dimethylfumaric acid ester. Dermatologica 1990, 181, 33–37. [Google Scholar] [CrossRef]
- Nugteren-Huying, W.M.; van der Schroeff, J.G.; Hermans, J.; Suurmond, D. Fumaric acid therapy for psoriasis: A randomized, double-blind, placebo-controlled study. J. Am. Acad. Dermatol. 1990, 22, 311–312. [Google Scholar] [CrossRef]
- Schafer, P.H.; Parton, A.; Gandhi, A.K.; Capone, L.; Adams, M.; Wu, L.; Bartlett, J.B.; Loveland, M.A.; Gilhar, A.; Cheung, Y.F.; et al. Apremilast, a camp phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br. J. Pharmacol. 2010, 159, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Oehrl, S.; Prakash, H.; Ebling, A.; Trenkler, N.; Wolbing, P.; Kunze, A.; Dobel, T.; Schmitz, M.; Enk, A.; Schakel, K. The phosphodiesterase 4 inhibitor apremilast inhibits th1 but promotes th17 responses induced by 6-sulfo LacNAc (slan) dendritic cells. J. Dermatol. Sci. 2017, 87, 110–115. [Google Scholar] [CrossRef]
- Papp, K.; Reich, K.; Leonardi, C.L.; Kircik, L.; Chimenti, S.; Langley, R.G.; Hu, C.; Stevens, R.M.; Day, R.M.; Gordon, K.B.; et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: Results of a phase III, randomized, controlled trial (efficacy and safety trial evaluating the effects of apremilast in psoriasis [esteem] 1). J. Am. Acad. Dermatol. 2015, 73, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Haydey, R.; Rosoph, L.A.; Lynde, C.W.; Bukhalo, M.; Fowler, J.F.; Delorme, I.; Gagne-Henley, A.; Gooderham, M.; Poulin, Y.; et al. Apremilast for the treatment of moderate-to-severe palmoplantar psoriasis: Results from a double-blind, placebo-controlled, randomized study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.; Gooderham, M.; Bachelez, H.; Goncalves, J.; Day, R.M.; Chen, R.; Crowley, J. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with difficult-to-treat nail and scalp psoriasis: Results of 2 phase iii randomized, controlled trials (ESTEEM 1 and ESTEEM 2). J. Am. Acad. Dermatol. 2016, 74, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Lucka, T.C.; Pathirana, D.; Sammain, A.; Bachmann, F.; Rosumeck, S.; Erdmann, R.; Schmitt, J.; Orawa, H.; Rzany, B.; Nast, A. Efficacy of systemic therapies for moderate-to-severe psoriasis: A systematic review and meta-analysis of long-term treatment. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1331–1344. [Google Scholar] [CrossRef]
- Pasut, G. Pegylation of biological molecules and potential benefits: Pharmacological properties of certolizumab pegol. BioDrugs 2014, 28 (Suppl. 1), S15–S23. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Gordon, K.B.; Fakharzadeh, S.; Yeilding, N.; Szapary, P.O.; Schenkel, B.; Guzzo, C.; Li, S.; Papp, K.A. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis: Results from the phoenix 1 trial through up to 3 years. Br. J. Dermatol. 2012, 166, 861–872. [Google Scholar] [CrossRef]
- Gniadecki, R.; Bang, B.; Bryld, L.E.; Iversen, L.; Lasthein, S.; Skov, L. Comparison of long-term drug survival and safety of biologic agents in patients with psoriasis vulgaris. Br. J. Dermatol. 2015, 172, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Van den Reek, J.M.; Zweegers, J.; Kievit, W.; Otero, M.E.; van Lumig, P.P.; Driessen, R.J.; Ossenkoppele, P.M.; Njoo, M.D.; Mommers, J.M.; Koetsier, M.I.; et al. ‘Happy’ drug survival of adalimumab, etanercept and ustekinumab in psoriasis in daily practice care: Results from the BioCAPTURE network. Br. J. Dermatol. 2014, 171, 1189–1196. [Google Scholar] [CrossRef]
- Warren, R.B.; Smith, C.H.; Yiu, Z.Z.N.; Ashcroft, D.M.; Barker, J.; Burden, A.D.; Lunt, M.; McElhone, K.; Ormerod, A.D.; Owen, C.M.; et al. Differential drug survival of biologic therapies for the treatment of psoriasis: A prospective observational cohort study from the British association of dermatologists biologic interventions register (Badbir). J. Investig. Dermatol. 2015, 135, 2632–2640. [Google Scholar] [CrossRef]
- Lynch, M.; Roche, L.; Horgan, M.; Ahmad, K.; Hackett, C.; Ramsay, B. Peritoneal tuberculosis in the setting of ustekinumab treatment for psoriasis. JAAD Case Rep. 2017, 3, 230–232. [Google Scholar] [CrossRef]
- Tsai, T.F.; Ho, J.C.; Song, M.; Szapary, P.; Guzzo, C.; Shen, Y.K.; Li, S.; Kim, K.J.; Kim, T.Y.; Choi, J.H.; et al. Efficacy and safety of ustekinumab for the treatment of moderate-to-severe psoriasis: A phase III, randomized, placebo-controlled trial in Taiwanese and Korean patients (PEARL). J. Dermatol. Sci. 2011, 63, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Kulig, P.; Musiol, S.; Freiberger, S.N.; Schreiner, B.; Gyulveszi, G.; Russo, G.; Pantelyushin, S.; Kishihara, K.; Alessandrini, F.; Kundig, T.; et al. IL-12 protects from psoriasiform skin inflammation. Nat. Commun. 2016, 7, 13466. [Google Scholar] [CrossRef]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase iii, double-blinded, placebo- and active comparator-controlled voyage 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar] [PubMed]
- Gordon, K.B.; Duffin, K.C.; Bissonnette, R.; Prinz, J.C.; Wasfi, Y.; Li, S.; Shen, Y.K.; Szapary, P.; Randazzo, B.; Reich, K. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N. Engl. J. Med. 2015, 373, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaci, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (resurface 1 and resurface 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Papp, K.; Thaci, D.; Reich, K.; Riedl, E.; Langley, R.G.; Krueger, J.G.; Gottlieb, A.B.; Nakagawa, H.; Bowman, E.P.; Mehta, A.; et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIB randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Thaci, D.; Blauvelt, A.; Reich, K.; Tsai, T.F.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Hugot, S.; You, R.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: Clear, a randomized controlled trial. J. Am. Acad. Dermatol. 2015, 73, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Reich, K.; Tsai, T.F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the clear study. J. Am. Acad. Dermatol. 2017, 76, 60–69.e9. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Colombel, J.F.; Hardin, D.S. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2016, 375, 2102. [Google Scholar] [CrossRef]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Puig, L. Brodalumab: The first anti-IL-17 receptor agent for psoriasis. Drugs Today 2017, 53, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Bagel, J.; Duffin, K.C.; Moore, A.; Ferris, L.K.; Siu, K.; Steadman, J.; Kianifard, F.; Nyirady, J.; Lebwohl, M. The effect of secukinumab on moderate-to-severe scalp psoriasis: Results of a 24-week, randomized, double-blind, placebo-controlled phase 3b study. J. Am. Acad. Dermatol. 2017, 77, 667–674. [Google Scholar] [CrossRef]
- Cantini, F.; Nannini, C.; Niccoli, L.; Petrone, L.; Ippolito, G.; Goletti, D. Risk of tuberculosis reactivation in patients with rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis receiving non-anti-TNF-targeted biologics. Mediat. Inflamm. 2017, 2017, 8909834. [Google Scholar] [CrossRef] [PubMed]
- National Psoriasis Foundation. Available online: https://www.Psoriasis.Org/drug-pipeline (accessed on 2 October 2018).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target Genes | Tissue/Cell Type (Human) | Function |
|---|---|---|---|
| miR-21 | TIMP3, TPM1, PDCD4, PTEN, IL12A, RECK, RTN4, NFIB | Skin, PBMCs | Keratinocyte differentiation and proliferation, T cell activation, inflammation [136] |
| miR-31 | FIH-1, STK40 | Skin | NF-κB activity, keratinocyte differentiation and proliferation [135] |
| miR-135b | COL4A3 | Skin | Keratinocyte differentiation and proliferation [137] |
| miR-146a | IRAK1, TRAF6, EGFR | Skin | Hematopoiesis, inflammation, and keratinocyte proliferation [142,143] |
| miR-155 | CTLA-4 | Skin | Inflammation [144] |
| miR-203 | TNF-α, IL-8, IL-24, SOCS-3, SOCS-6 | Skin | STAT3 signaling, keratinocyte differentiation and proliferation, and inflammation [145] |
| miR-210 | FOXP3 | PBMCs | Regulatory T cell activation Induction of Th17 and Th1 differentiation [139,146] |
| miR-221/222 | TIMP3, c-KIT | Skin | Immune cell activation Keratinocyte proliferation [138] |
| miR-424 | MEK1, Cyclin E1 | Skin | Keratinocyte differentiation and proliferation [147] |
| Study | Sample (n) | Method | Psoriasis | Healthy Skin | Comments |
|---|---|---|---|---|---|
| Gao et al., 2008 [153] | Skin swabs (six psoriatic patients) | broad range PCR | ↑ diversity ↑ Firmicutes | ↑ Actinobacteria ↑ Proteobacteria | Healthy controls taken from previous study [158]. |
| Alekseyenko et al., 2013 [154] | Skin swabs (54 psoriasis patients, 37 controls) | High-throughput 16S rRNA gene sequencing | ↑ Actinobacteria/Firmicutes ↑ Corynebacterium, Propionibacterium, Staphylococcus, Streptococcus↑ Corynebacterium, Streptococcus, Staphylococcus | ↑ Proteobacteria | OTUs Acidobacteria and Schlegella were strongly associated with psoriasis status. Samples were site-matched. |
| Fahlen et al., 2012 [151] | Skin biopsies (10 psoriasis patients, 10 healthy controls) | Pyrosequencing targeting the V3-V4 regions of the 16S rRNA gene | Streptococcus > Staphylococcus ↑ Proteobacteria (trunk skin) ↑Propionibacteria/Staph. (limb skin) | ↑ Actinobacteria | Included dermis and adnexal structures. Bacterial diversity was increased in the control group (unmatched sites), but not statistically significant. Firmicutes, Proteobacteria, and Actinobacteria predominant in healthy and psoriatic skin. |
| Takemoto et al., 2015 [156] | Psoriatic scale samples (12 psoriatic patients, 12 healthy controls) | Pyrosequencing for fungal rRNAgene sequences | ↑ fungal diversity ↓ Malassezia | ↑ Malassezia | Fungal microbiome study Malassezia were the most abundant species in psoriatic and healthy skin. |
| Drug | Mechanism | Application |
|---|---|---|
| Methotrexate | Dihydrofolate reductase inhibition blocks purine biosynthesis; induction of lymphocyte apoptosis | s.c./oral |
| Cyclosporin | Calcineurin inhibition leading to reduced IL-2 | Oral |
| Acitretin | Normalization of keratinocyte proliferation/differentiation through retinoid receptor binding | Oral |
| Fumarate | Intracellular glutathione, modulation of Nrf2, NF-κB, and HIF-1α; promoting a shift from a pro-inflammatory Th1/Th17 response to an anti-inflammatory/regulatory Th2 response. | Oral |
| Apremilast | PDE4 inhibitor increases in tracellular cAMP levels in immune and non-immune cell types modulating inflammation | Oral |
| Etanercept | Dimeric human fusion protein mimicking TNF-αR | s.c. |
| Infliximab | Chimeric IgG1κ monoclonal antibody that binds to soluble and transmembrane forms of TNF-α | i.v. |
| Adalimumab | Human monoclonal antibody against TNF-α | s.c. |
| Certolizumab | Fab portion of humanized monoclonal antibody against TNF-α conjugated to polyethylene glycol | s.c. |
| Ustekinumab | Human IgG1k monoclonal antibody that binds with specificity to the p40 protein subunit used by both the interleukin (IL)-12 and IL-23 cytokines IL-12/IL-23 p40 | s.c. |
| Tildrakizumab | Humanized IgG1κ, which selectively blocks IL-23 by binding to its p19 subunit | s.c. |
| Guselkumab | Human immunoglobulin G1 lambda (IgG1λ) monoclonal antibody that selectively blocks IL-23 by binding to its p19 subunit | s.c. |
| Risankizumab | Humanized IgG1 monoclonal antibody that inhibits interleukin-23 by specifically targeting the p19 subunit | s.c. |
| Secukinumab | Human IgG1κ monoclonal antibody against IL-17A | s.c. |
| Ixekizumab | Humanized, immunoglobulin G4κ monoclonal antibody selectively binds and neutralizes IL-17A | s.c. |
| Brodalumab | Human monoclonal IgG2 antibody directed at the IL-17RA | s.c. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. https://doi.org/10.3390/ijms20061475
Rendon A, Schäkel K. Psoriasis Pathogenesis and Treatment. International Journal of Molecular Sciences. 2019; 20(6):1475. https://doi.org/10.3390/ijms20061475
Chicago/Turabian StyleRendon, Adriana, and Knut Schäkel. 2019. "Psoriasis Pathogenesis and Treatment" International Journal of Molecular Sciences 20, no. 6: 1475. https://doi.org/10.3390/ijms20061475
APA StyleRendon, A., & Schäkel, K. (2019). Psoriasis Pathogenesis and Treatment. International Journal of Molecular Sciences, 20(6), 1475. https://doi.org/10.3390/ijms20061475