Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2


Abstract
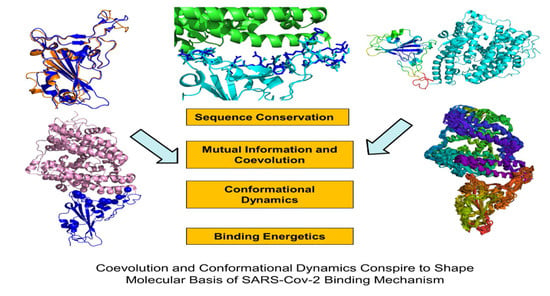
:
1. Introduction
2. Results and Discussion
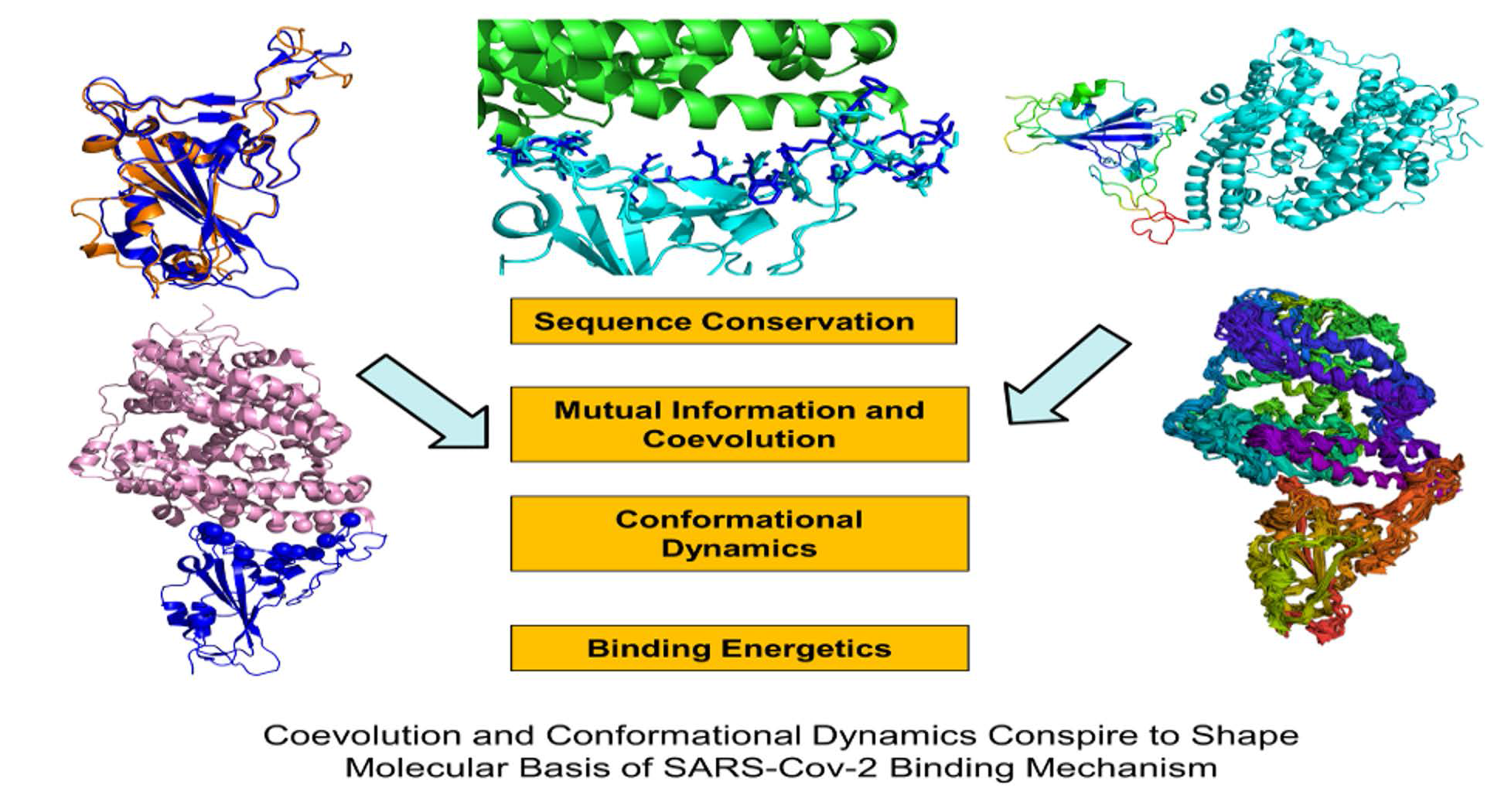
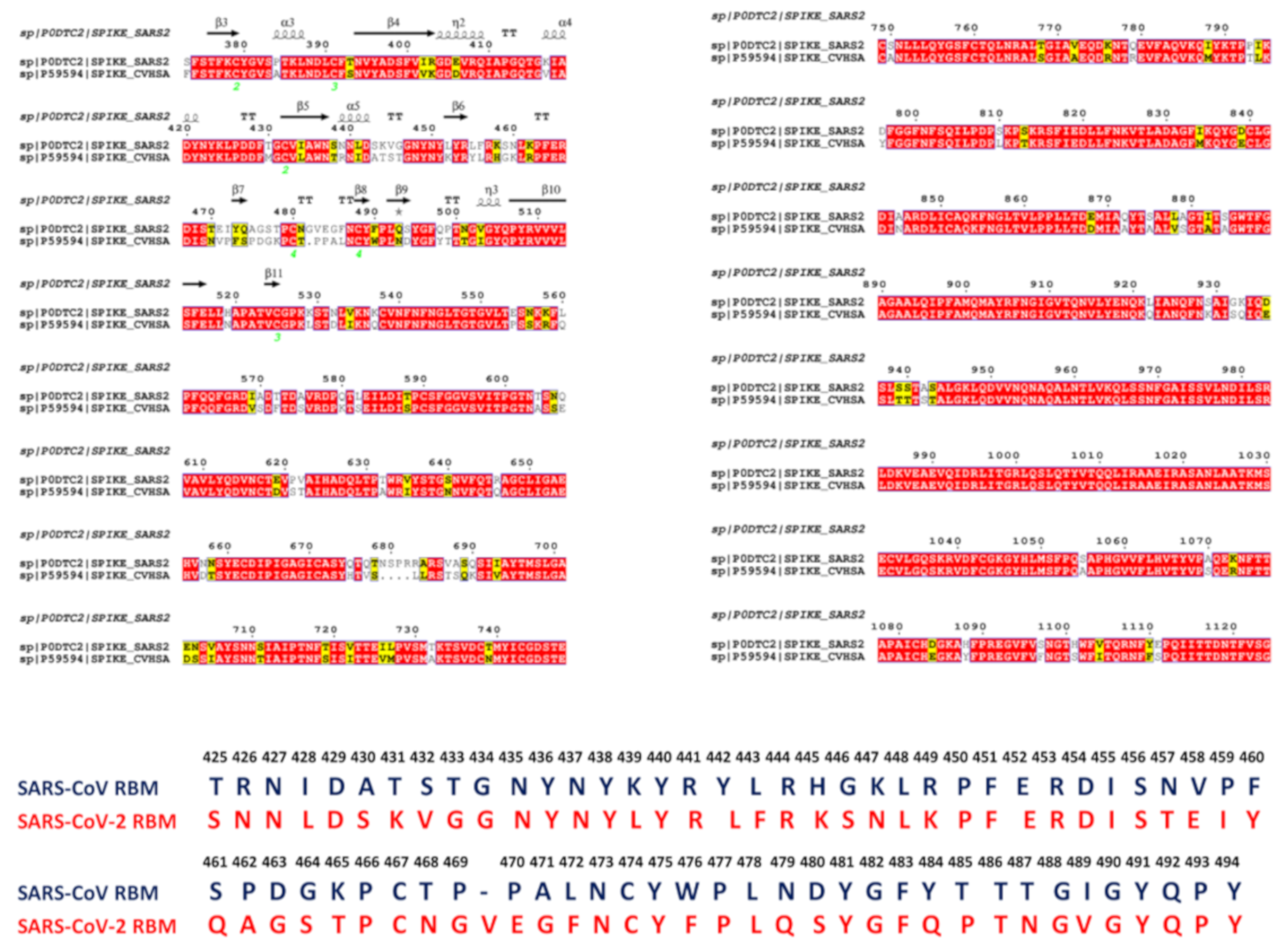
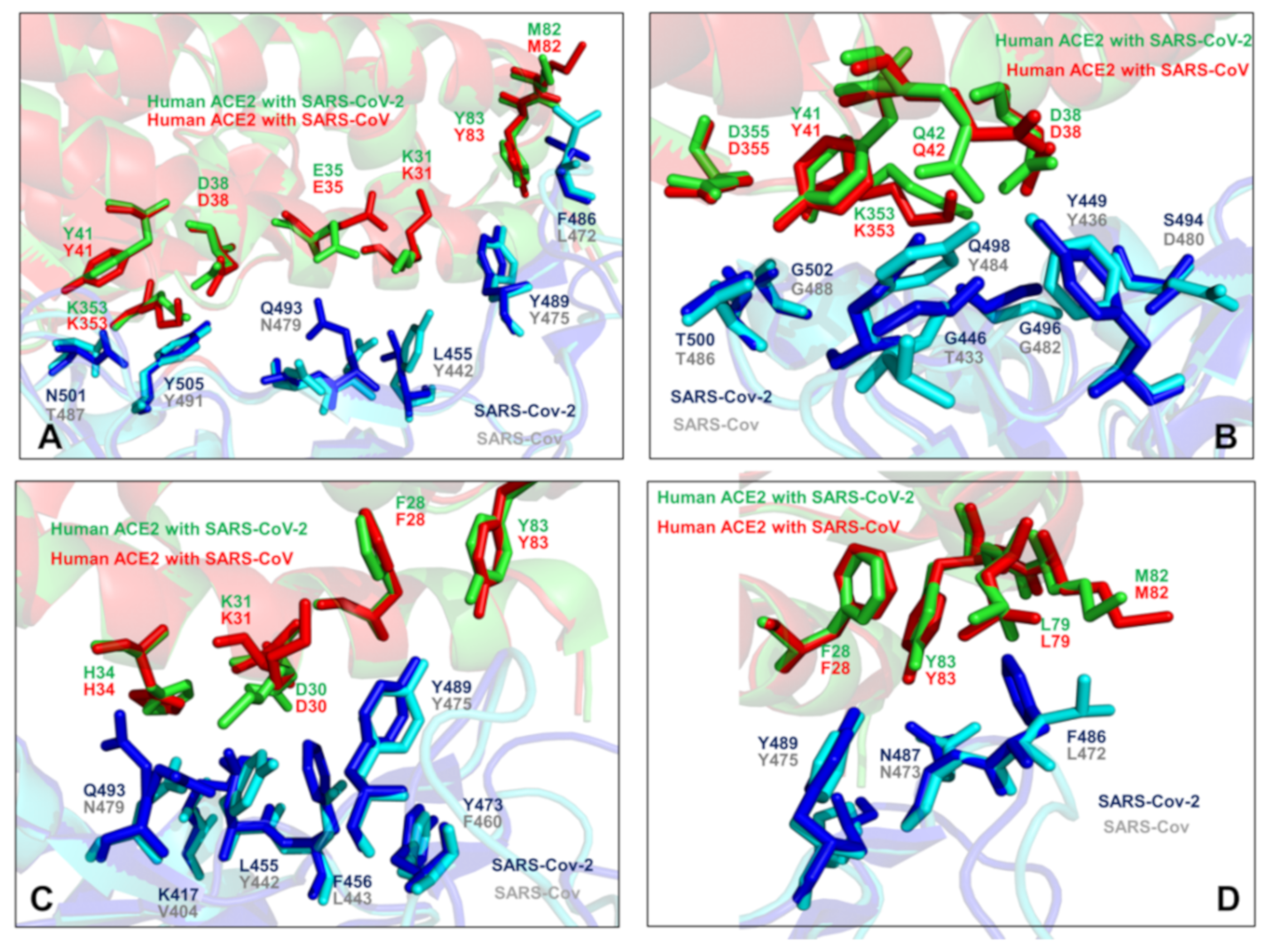
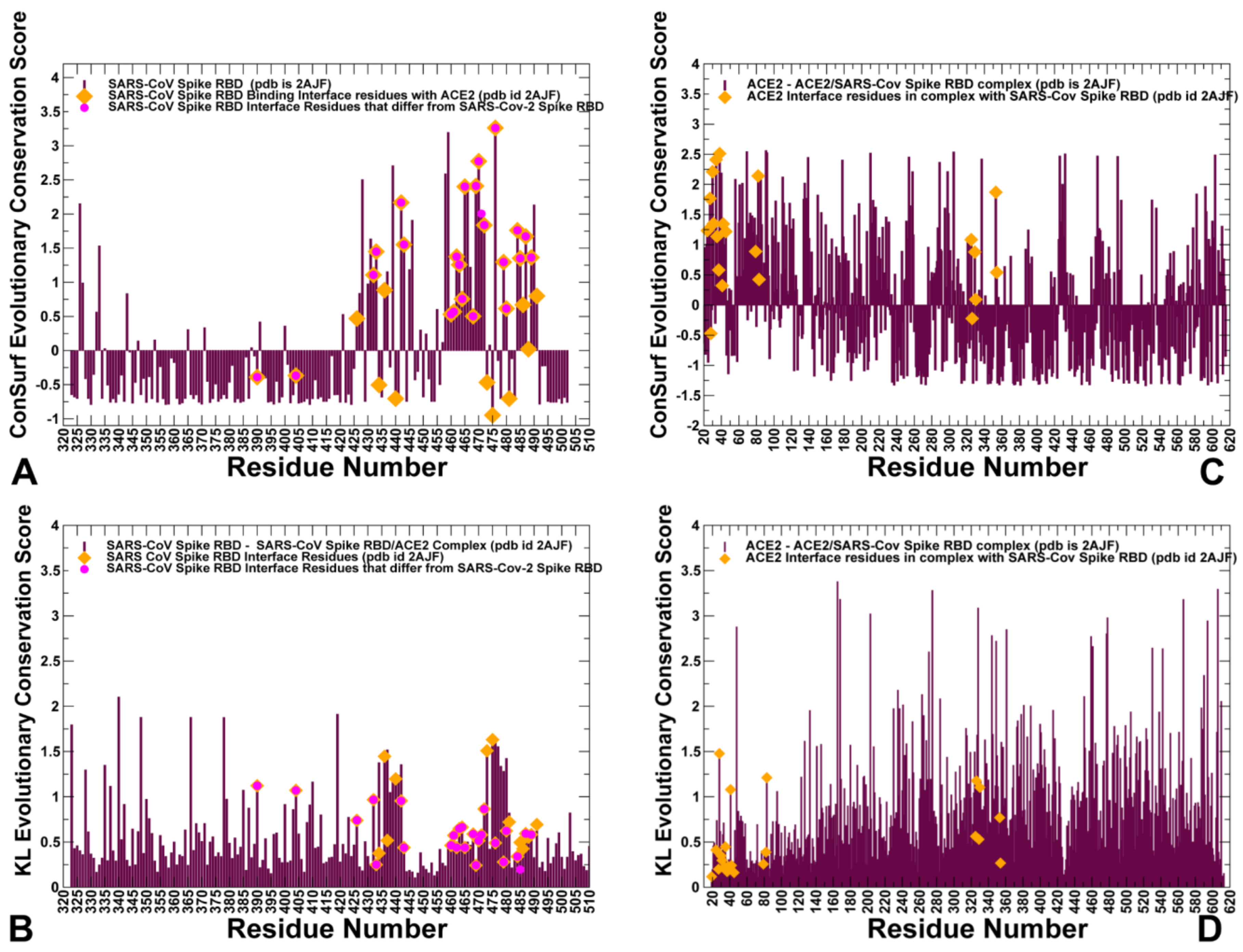
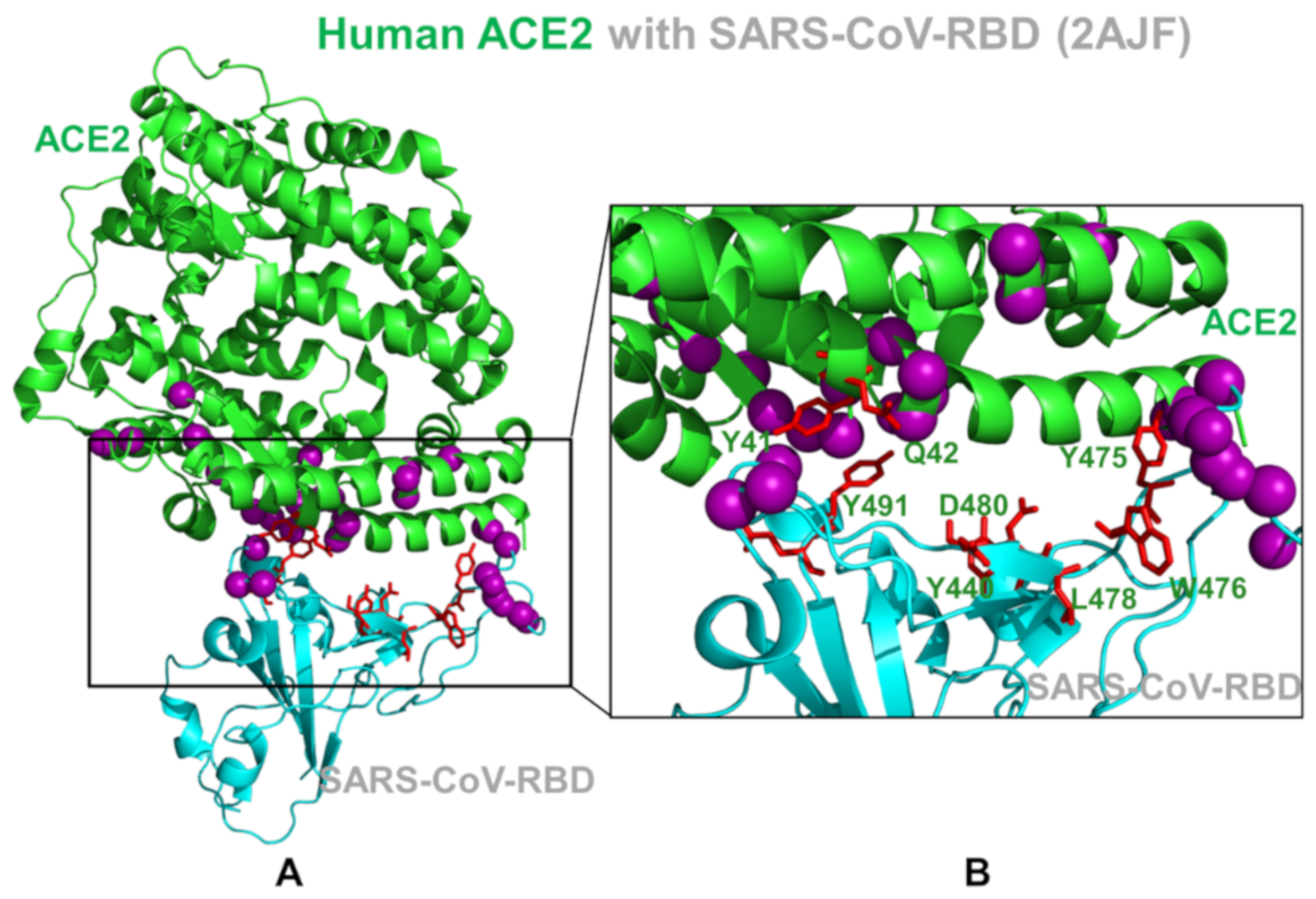
2.1. Sequence Analysis Links Evolutionary Patterns in SARS-CoV Spike Proteins with Shared Conserved Hotspots at the Binding Interface
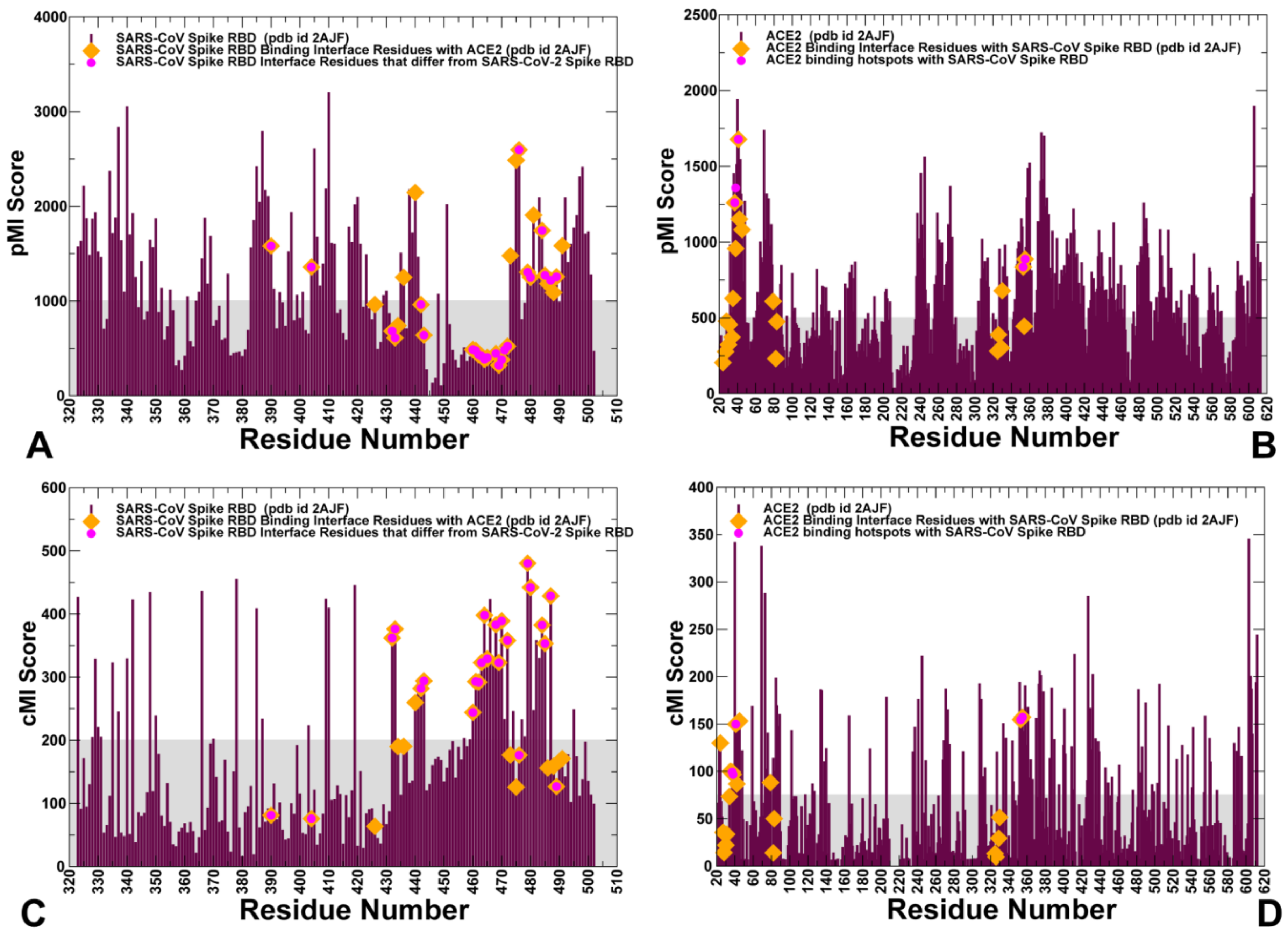
2.2. Coevolution and Mutual Information Interdependences of the SARS-CoV-RBD and ACE2 Suggest Potential Regulator and Transmitter Sites of Binding and Communication
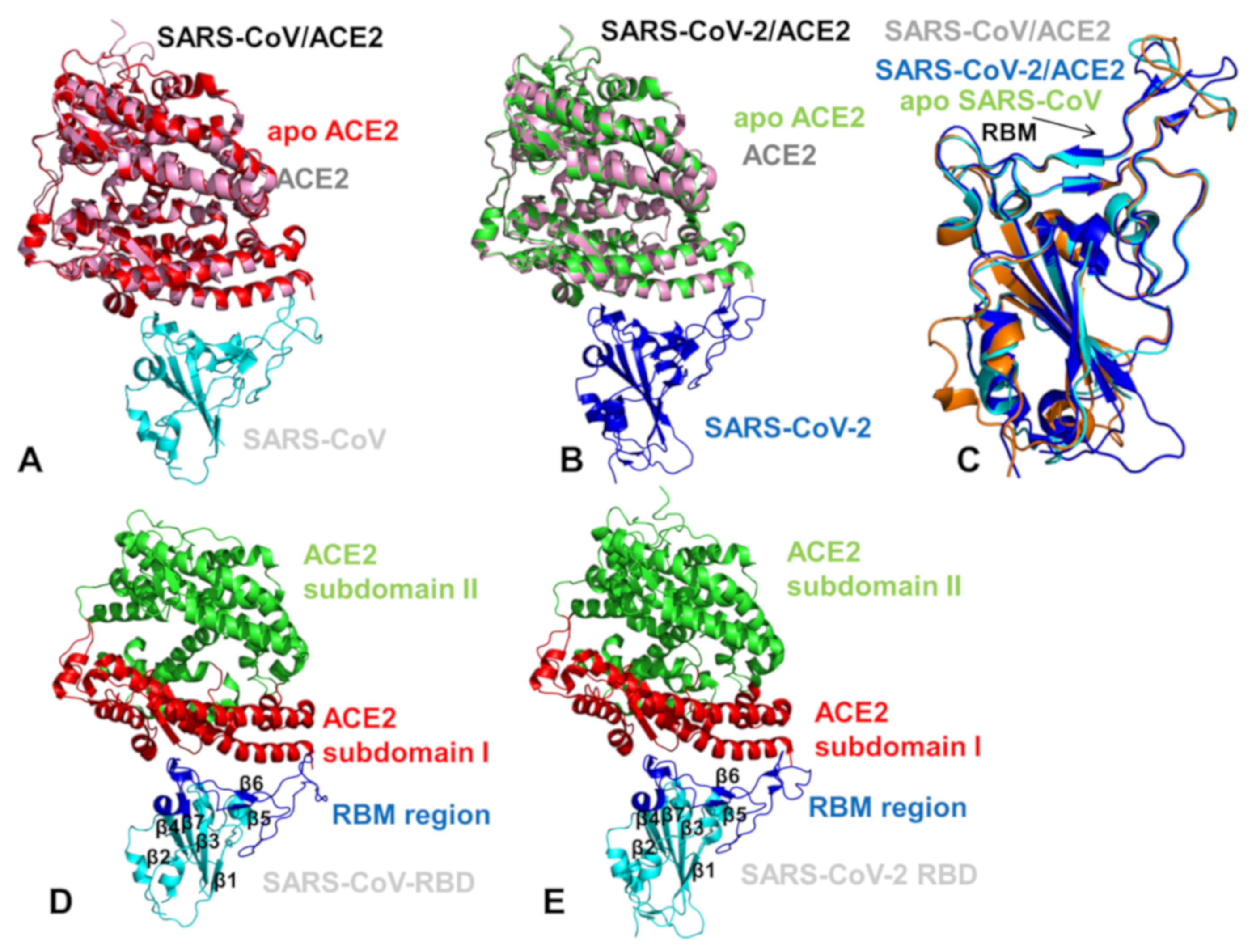
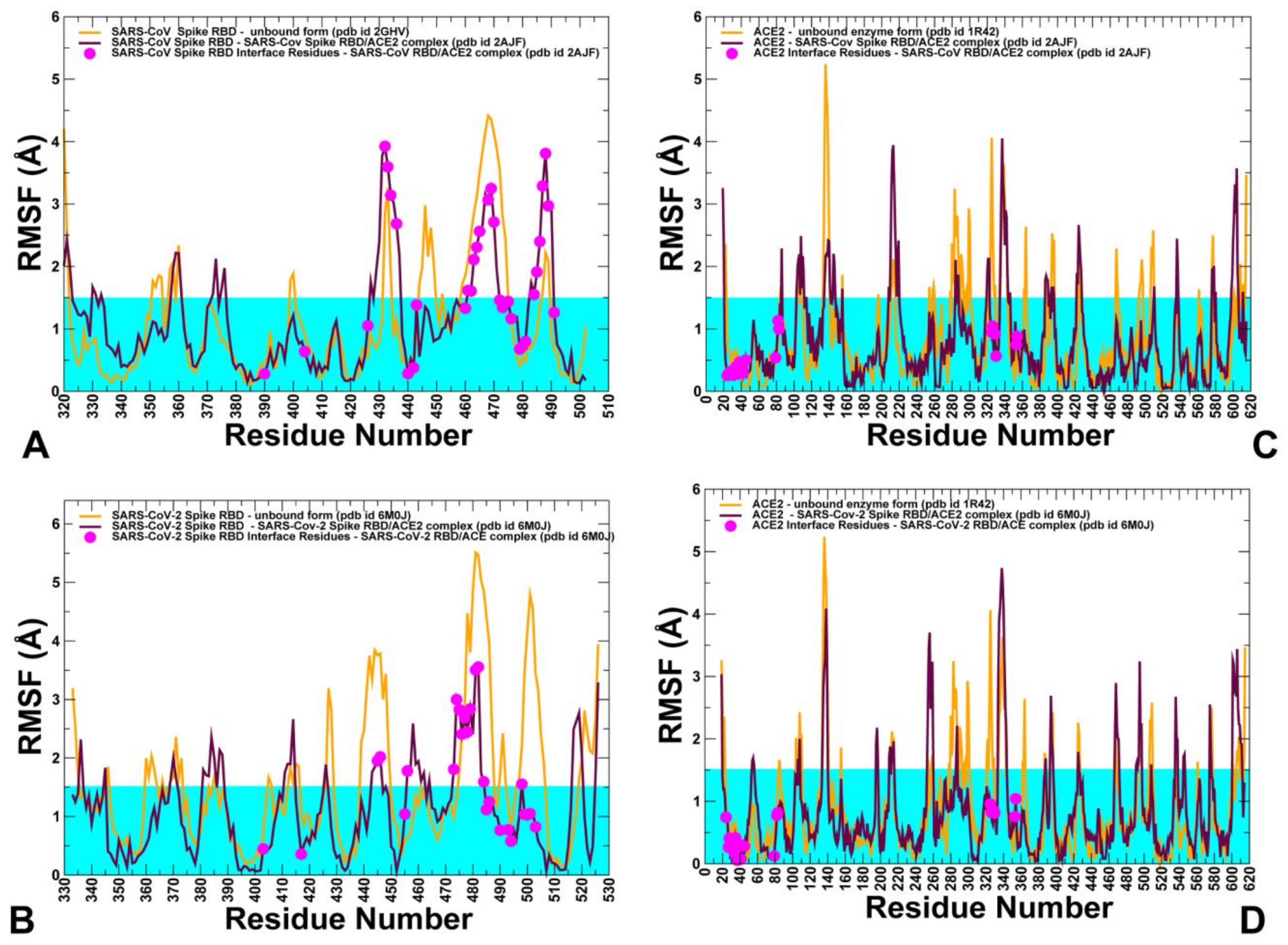
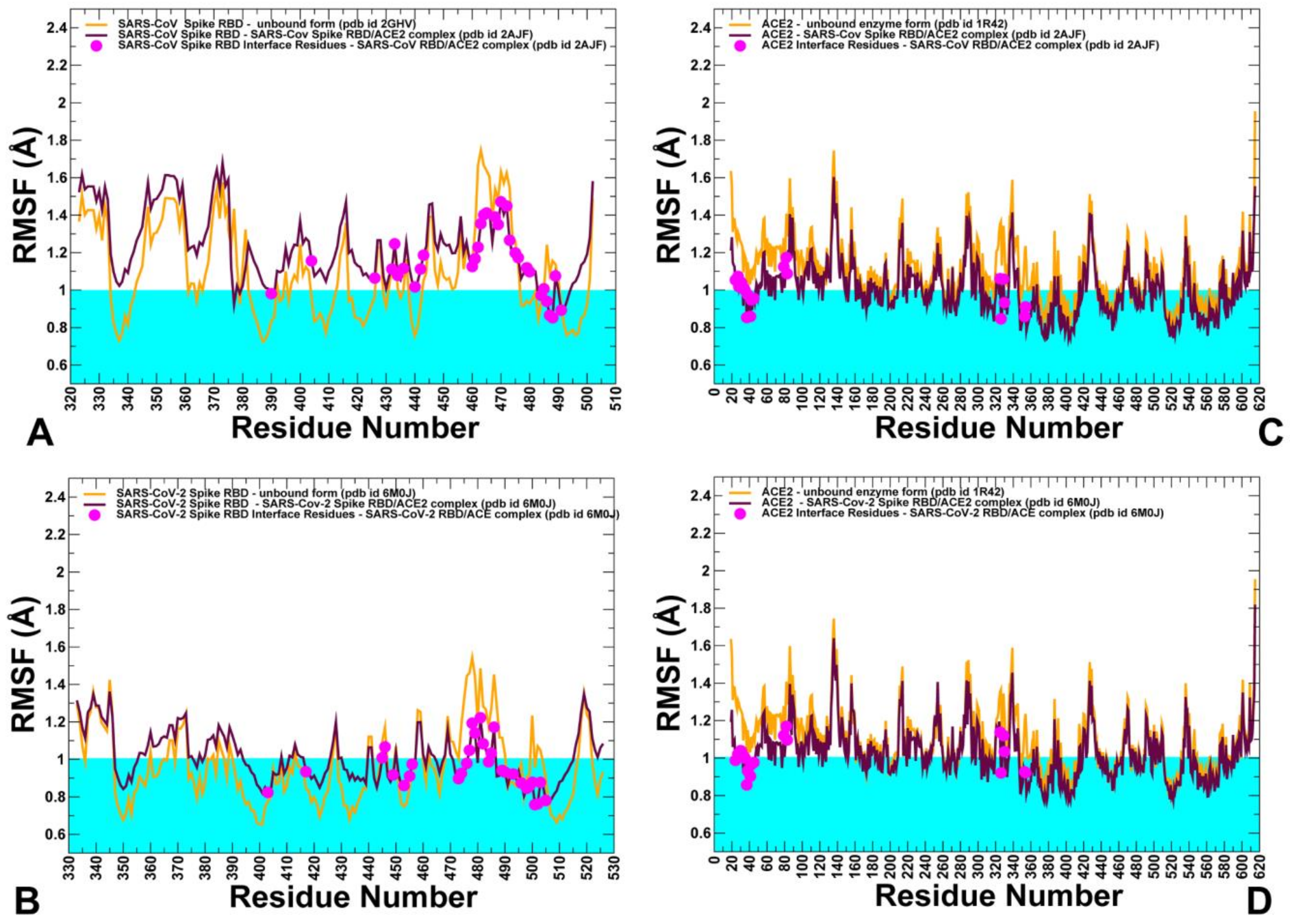
2.3. Conformational Dynamics of the SARS-CoV and SARS-CoV-2 RBD Proteins: A Comparative Analysis of ACE2-Induced Protein Mobility and Dynamics-Driven Allostery
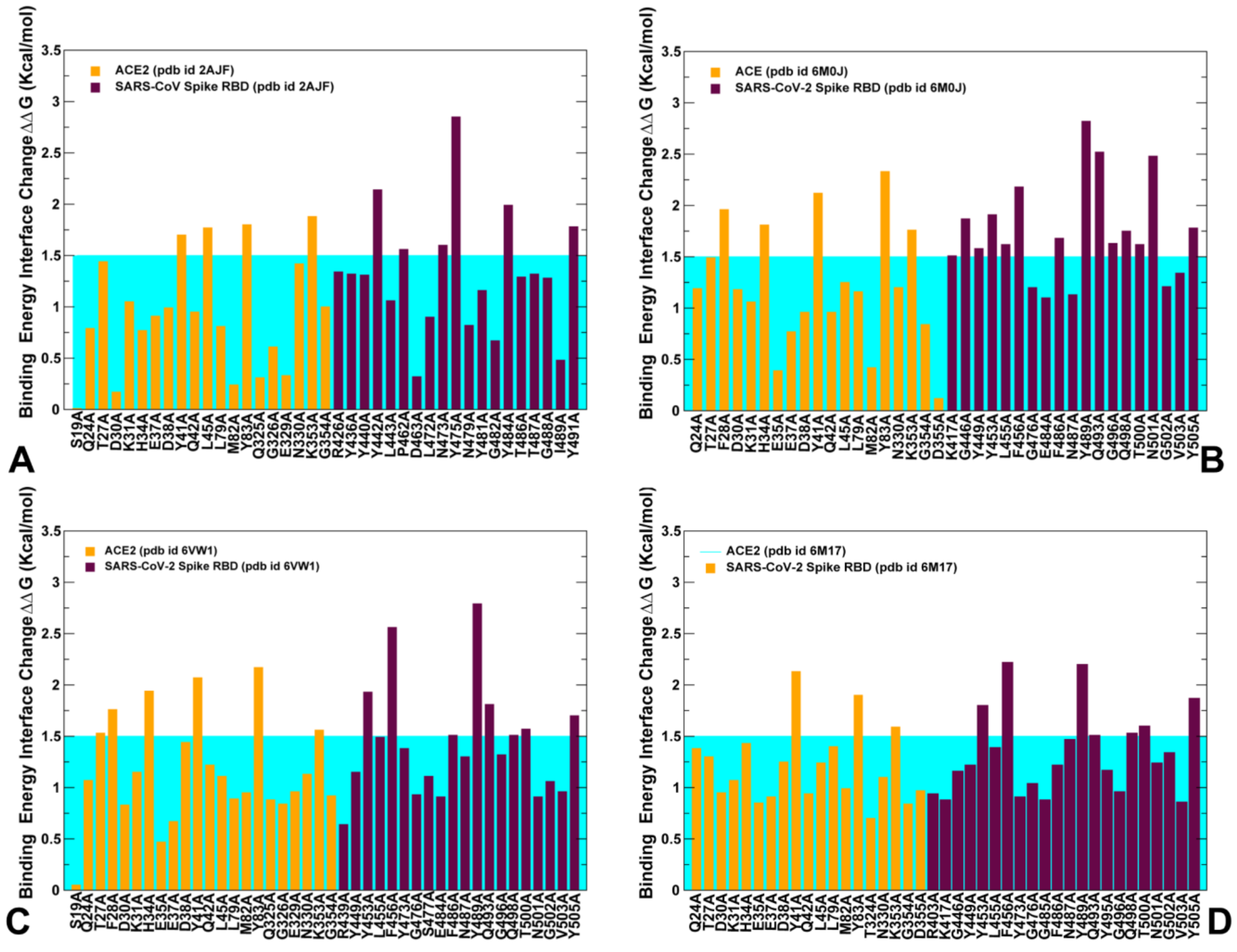
2.4. Mutational Scanning and Energetic Analysis of the Binding Interfaces: A Cooperative Effect of Multiple Residues Drives SARS-CoV-2 Binding with Host Receptor
3. Materials and Methods
3.1. Sequence Conservation and Coevolutionary Analyses
3.2. Coarse-Grained Molecular Simulations
3.3. Structure Preparation and All-Atom Molecular Dynamics Simulations
3.4. Protein Stability Analysis and Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Lagniton, P.N.P.; Ye, S.; Li, E.; Xu, R.H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Indwiani Astuti, Y. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039.e15. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Chakraborti, S.; Prabakaran, P.; Xiao, X.; Dimitrov, D.S. The SARS coronavirus S glycoprotein receptor binding domain: Fine mapping and functional characterization. Virol. J. 2005, 2, 73. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Lu, H.; Siddiqui, P.; Zhou, Y.; Jiang, S. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J. Immunol. 2005, 174, 4908–4915. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Crawford, K.H.D.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; Veesler, D.; Bloom, J.D. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Procko, E. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Brielle, E.S.; Schneidman-Duhovny, D.; Linial, M. The SARS-CoV-2 Exerts a Distinctive Strategy for Interacting with the ACE2 Human Receptor. Viruses 2020, 12, 497. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Saltalamacchia, A.; Magistrato, A. Is the Rigidity of SARS-CoV-2 Spike Receptor-Binding Motif the Hallmark for Its Enhanced Infectivity? Insights from All-Atom Simulations. J. Phys. Chem. Lett. 2020, 11, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 525, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, G.K.; Thunuguntla, V.B.S.C.; Bobbillapati, J.; Bondili, J.S. Structural and simulation analysis of hotspot residues interactions of SARS-CoV 2 with human ACE2 receptor. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 10, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Di Paola, L.; Hadi-Alijanvand, H.; Song, X.; Hu, G.; Giuliani, A. The Discovery of a Putative Allosteric Site in the SARS-CoV-2 Spike Protein Using an Integrated Structural/Dynamic Approach. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Sequence variation of SARS-CoV-2 spike protein may facilitate stronger interaction with ACE2 promoting high infectivity. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Armijos-Jaramillo, V.; Yeager, J.; Muslin, C.; Perez-Castillo, Y. SARS-CoV-2, an evolutionary perspective of interaction with human ACE2 reveals undiscovered amino acids necessary for complex stability. Evol. Appl. 2020, 13, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Marino Buslje, C.; Teppa, E.; Di Domenico, T.; Delfino, J.M.; Nielsen, M. Networks of high mutual information define the structural proximity of catalytic sites: Implications for catalytic residue identification. PLoS Comput. Biol. 2010, 6, e1000978. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Marino Buslje, C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillier, E.R.M.; Lui, T.W.H. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics 2003, 19, 750–755. [Google Scholar] [CrossRef]
- Martin, L.C.; Gloor, G.B.; Dunn, S.D.; Wahl, L.M. Using information theory to search for co-evolving residues in proteins. Bioinformatics 2005, 21, 4116–4124. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.D.; Wahl, L.M.; Gloor, G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 2008, 24, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Macours, N.; Poels, J.; Hens, K.; Francis, C.; Huybrechts, R. Structure, evolutionary conservation, and functions of angiotensin- and endothelin-converting enzymes. Int. Rev. Cytol. 2004, 239, 47–97. [Google Scholar] [PubMed]
- Warner, F.J.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme-2: A molecular and cellular perspective. Cell. Mol. Life Sci. 2004, 61, 2704–2713. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Li, W.; Choe, H.; Farzan, M. Angiotensin-converting enzyme 2: A functional receptor for SARS coronavirus. Cell. Mol. Life Sci. 2004, 61, 2738–2743. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Park, K.; Kim, D. Analysis of the residue-residue coevolution network and the functionally important residues in proteins. Proteins 2008, 72, 863–872. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Panchenko, A.R. Coevolution in defining the functional specificity. Proteins 2009, 75, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Panchenko, A.R. Structural and functional roles of coevolved sites in proteins. PLoS ONE 2010, 5, e8591. [Google Scholar] [CrossRef] [Green Version]
- Teppa, E.; Wilkins, A.D.; Nielsen, M.; Buslje, C.M. Disentangling evolutionary signals: Conservation, specificity determining positions and coevolution. Implication for catalytic residue prediction. BMC Bioinform. 2012, 13, 235. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Nam, H.J.; Choi, Y.S.; Yang, J.S.; Hwang, J.; Kim, S. Molecular evolution of protein conformational changes revealed by a network of evolutionarily coupled residues. Mol. Biol. Evol. 2011, 28, 2675–2685. [Google Scholar] [CrossRef] [Green Version]
- Stetz, G.; Verkhivker, G.M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017, 13, e1005299. [Google Scholar] [CrossRef]
- Tse, A.; Verkhivker, G.M. Molecular Determinants Underlying Binding Specificities of the ABL Kinase Inhibitors: Combining Alanine Scanning of Binding Hot Spots with Network Analysis of Residue Interactions and Coevolution. PLoS ONE 2015, 10, e0130203. [Google Scholar] [CrossRef] [Green Version]
- Stetz, G.; Tse, A.; Verkhivker, G.M. Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Sci. Rep. 2018, 8, 6899. [Google Scholar] [CrossRef] [Green Version]
- Popovych, N.; Sun, S.; Ebright, R.H.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. [Google Scholar] [CrossRef]
- Tzeng, S.R.; Kalodimos, C.G. Dynamic activation of an allosteric regulatory protein. Nature 2009, 462, 368–372. [Google Scholar] [CrossRef]
- Huang, C.; Kalodimos, C.G. Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys. 2017, 46, 317–336. [Google Scholar] [CrossRef]
- Jiang, Y.; Kalodimos, C.G. NMR Studies of Large Proteins. J. Mol. Biol. 2017, 429, 2667–2676. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Wu, K.; Chen, L.; Peng, G.; Zhou, W.; Pennell, C.A.; Mansky, L.M.; Geraghty, R.J.; Li, F. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J. Virol. 2011, 85, 5331–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Finn, R.D.; Miller, B.L.; Clements, J.; Bateman, A. iPfam: A database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014, 42, D364–D373. [Google Scholar] [CrossRef]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar] [CrossRef]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.E.; Kloczkowski, A.; Kolinski, A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3496. [Google Scholar] [CrossRef] [Green Version]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef] [Green Version]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2019, 35, 694–695. [Google Scholar] [CrossRef]
- Hwang, W.C.; Lin, Y.; Santelli, E.; Sui, J.; Jaroszewski, L.; Stec, B.; Farzan, M.; Marasco, W.A.; Liddington, R.C. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J. Biol. Chem. 2006, 281, 34610–34616. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Rotkiewicz, P.; Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, L.E.; Marti, M.A.; Capece, L. CG2AA: Backmapping protein coarse-grained structures. Bioinformatics 2016, 32, 1235–1237. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Bünning, P.; Riordan, J.F. The functional role of zinc in angiotensin converting enzyme: Implications for the enzyme mechanism. J. Inorg. Biochem. 1985, 24, 183–198. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.L.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z.; Fuchs, S.; Touyz, R.M. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev. 2012, 65, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Hekkelman, M.L.; Te Beek, T.A.H.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A Protein Structure Bioinformatics Web Service Collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated Modeling of Loops in Protein Structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Fernandez-Fuentes, N.; Zhai, J.; Fiser, A. ArchPRED: A Template Based Loop Structure Prediction Server. Nucleic Acids Res. 2006, 34, W173–W176. [Google Scholar] [CrossRef]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric-Isothermal Ensemble with Ewald Summations for All Long Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A Graphical Interface for the FoldX Force Field. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [Green Version]
- Christensen, N.J.; Kepp, K.P. Accurate Stabilities of Laccase Mutants Predicted With a Modified FoldX Protocol. J. Chem. Inf. Model. 2012, 52, 3028–3042. [Google Scholar] [CrossRef] [Green Version]
- Christensen, N.J.; Kepp, K.P. Stability Mechanisms of Laccase Isoforms Using a Modified FoldX Protocol Applicable to Widely Different Proteins. J. Chem. Theory Comput. 2013, 9, 3210–3223. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehouck, Y.; Gilis, D.; Rooman, M. A new generation of statistical potentials for proteins. Biophys. J. 2006, 90, 4010–4017. [Google Scholar] [CrossRef]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ConSurf Hotspots | Consurf Score | KL Hotspots | KL Score |
|---|---|---|---|
| K390 | −0.390 | K390 | 1.12 |
| V404 | −0.367 | V404 | 1.07 |
| G434 | −0.505 | Y436 | 1.45 |
| Y440 | −0.705 | Y440 | 1.19 |
| N473 | −0.467 | N473 | 1.51 |
| Y475 | −0.705 | Y475 | 1.63 |
| Y481 | 473 | Y481 | 0.72 |
| SARS-CoV pMI Hotspot Sites | SARS-CoV-2 pMI Hotspot Sites | SARS-CoV cMI Hotspot Sites | SARS-CoV-2 cMI Hotspot Sites |
|---|---|---|---|
| Y436 | Y449 | L472 | F486 |
| Y440 | Y451 | N479 | Q493 |
| Y475 | Y489 | T487 | N501 |
| W476 | F490 | T433 | G446 |
| Y481 | Y495 | T468 | N481 |
| Y484 | Q498 | P470 | E484 |
| N473 | N487 | Y484 | Q498 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkhivker, G. Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2020, 21, 8268. https://doi.org/10.3390/ijms21218268
Verkhivker G. Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2. International Journal of Molecular Sciences. 2020; 21(21):8268. https://doi.org/10.3390/ijms21218268
Chicago/Turabian StyleVerkhivker, Gennady. 2020. "Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2" International Journal of Molecular Sciences 21, no. 21: 8268. https://doi.org/10.3390/ijms21218268