
Analysis of the Genetic Relationship between Atherosclerosis and Non-Alcoholic Fatty Liver Disease through Biological Interaction Networks

 , , ,
, , ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Data Acquisition and Visualization and Identification of Differentially Expressed Genes
2.2. Protein–Protein Interaction (PPI) Network
2.3. Functional Modules and Hubs
2.4. Checking Key Genes through Public Gene–Disease Association Databases
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Visualization and Identification of Differentially Expressed Genes
4.2. Protein–Protein Interaction (PPI) Network
4.3. Functional Modules and Hubs
4.4. Checking Key Genes through Public Gene–Disease Association Databases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, H.; Yu, X.-H.; Ou, X.; Ouyang, X.-P.; Tang, C.-K. Hepatic Cholesterol Transport and Its Role in Non-Alcoholic Fatty Liver Disease and Atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J.; American Gastroenterological Association; American Association for the Study of Liver Diseases. The Diagnosis and Management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic Fatty Liver Disease: From Steatosis to Cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, J.; Gallego-Durán, R.; Romero-Gómez, M. Association of NAFLD with Subclinical Atherosclerosis and Coronary-Artery Disease: Meta-Analysis. Rev. Esp. Enferm. Dig. 2015, 107, 10–16. [Google Scholar]
- Angulo, P. Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Clouston, A.D.; Powell, E.E. Nonalcoholic Fatty Liver Disease: Is All the Fat Bad? Intern. Med. J. 2004, 34, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Gofton, C.; Upendran, Y.; Zheng, M.-H.; George, J. MAFLD: What Is Different from NAFLD? Clin. Mol. Hepatol. 2022. [Google Scholar] [CrossRef]
- Hassen, G.; Singh, A.; Belete, G.; Jain, N.; De la Hoz, I.; Camacho-Leon, G.P.; Dargie, N.K.; Carrera, K.G.; Alemu, T.; Jhaveri, S.; et al. Nonalcoholic Fatty Liver Disease: An Emerging Modern-Day Risk Factor for Cardiovascular Disease. Cureus 2022, 14, e25495. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.-J.; Ji, Y.-X.; Zhang, P.; She, Z.-G.; Li, H. Nonalcoholic Fatty Liver Disease Pandemic Fuels the Upsurge in Cardiovascular Diseases. Circ. Res. 2020, 126, 679–704. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.-H.; Kim, S.U.; Kim, H.C. Metabolic Dysfunction-Associated Fatty Liver Disease and Incident Cardiovascular Disease Risk: A Nationwide Cohort Study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2021, 19. [Google Scholar] [CrossRef] [PubMed]
- Salgado Alvarez, G.A.; Pinto Galvez, S.M.; Garcia Mora, U.; Cano Contreras, A.D.; Durán Rosas, C.; Priego-Parra, B.A.; Triana Romero, A.; Amieva Balmori, M.; Roesch Dietlen, F.; Martinez Vazquez, S.E.; et al. Higher Cardiovascular Risk Scores and Liver Fibrosis Risk Estimated by Biomarkers in Patients with Metabolic-Dysfunction-Associated Fatty Liver Disease. World J. Hepatol. 2022, 14, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Hoebinger, C.; Rajcic, D.; Hendrikx, T. Oxidized Lipids: Common Immunogenic Drivers of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 824481. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the Road Ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int (accessed on 29 January 2023).
- Zhang, L.; She, Z.-G.; Li, H.; Zhang, X.-J. Non-Alcoholic Fatty Liver Disease: A Metabolic Burden Promoting Atherosclerosis. Clin. Sci. 2020, 134, 1775–1799. [Google Scholar] [CrossRef]
- Gaudio, E.; Nobili, V.; Franchitto, A.; Onori, P.; Carpino, G. Nonalcoholic Fatty Liver Disease and Atherosclerosis. Intern. Emerg. Med. 2012, 7 (Suppl. S3), S297–S305. [Google Scholar] [CrossRef]
- Abdallah, L.R.; de Matos, R.C.; E Souza, Y.P.D.M.; Vieira-Soares, D.; Muller-Machado, G.; Pollo-Flores, P. Non-Alcoholic Fatty Liver Disease and Its Links with Inflammation and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 7. [Google Scholar] [CrossRef]
- Choe, Y.G.; Jin, W.; Cho, Y.K.; Chung, W.G.; Kim, H.J.; Jeon, W.K.; Kim, B.I. Apolipoprotein B/AI Ratio Is Independently Associated with Non-Alcoholic Fatty Liver Disease in Nondiabetic Subjects. J. Gastroenterol. Hepatol. 2013, 28, 678–683. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.; Katsiki, N.; Montalto, G.; Isenovic, E.R.; Mikhailidis, D.P.; Rizzo, M. Lipoprotein Subfractions in Metabolic Syndrome and Obesity: Clinical Significance and Therapeutic Approaches. Nutrients 2013, 5, 928–948. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.S.J.; Beigneux, A.P.; Barnes, R.H.; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyrén, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 Is Responsible for the Entry of Lipoprotein Lipase into Capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atef, M.E.; Anand-Srivastava, M.B. Role of PKCδ in Enhanced Expression of Gqα/PLCβ1 Proteins and VSMC Hypertrophy in Spontaneously Hypertensive Rats. PLoS ONE 2016, 11, e0157955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, K.; Schirm, S.; Johns, A.; Morser, J.; Light, D.R. FXa-Induced Responses in Vascular Wall Cells Are PAR-Mediated and Inhibited by ZK-807834. Thromb. Res. 2001, 103, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Zernecke, A. The Hemostatic System as a Regulator of Inflammation in Atherosclerosis. IUBMB Life 2014, 66, 735–744. [Google Scholar] [CrossRef]
- Marin, V.; Farnarier, C.; Grès, S.; Kaplanski, S.; Su, M.S.; Dinarello, C.A.; Kaplanski, G. The P38 Mitogen-Activated Protein Kinase Pathway Plays a Critical Role in Thrombin-Induced Endothelial Chemokine Production and Leukocyte Recruitment. Blood 2001, 98, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Shu, B.; Yang, Y.; Qian, M. The phenotypic switching of vascular smooth muscle cells induced by cholesterol. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2014, 30, 725–728, 731. [Google Scholar]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular Smooth Muscle Cell in Atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef]
- Provost, P.; Lam, J.Y.; Lacoste, L.; Merhi, Y.; Waters, D. Endothelium-Derived Nitric Oxide Attenuates Neutrophil Adhesion to Endothelium under Arterial Flow Conditions. Arterioscler. Thromb. 1994, 14, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, Y.; Bai, Y.; Fiscus, R.R. Mechanism of SNAP Potentiating Antiproliferative Effect of Calcitonin Gene-Related Peptide in Cultured Vascular Smooth Muscle Cells. J. Mol. Cell. Cardiol. 1999, 31, 1599–1606. [Google Scholar] [CrossRef]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling Endothelial Dysfunction by Modulating NOS Uncoupling: New Insights into Its Pathogenesis and Therapeutic Possibilities. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [Green Version]
- Rekka, E.A.; Chrysselis, M.C. Nitric Oxide in Atherosclerosis. Mini Rev. Med. Chem. 2002, 2, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, H.C. Natural History and Histological Classification of Atherosclerotic Lesions: An Update. Arterioscler. Thromb Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, Y.; Xu, J.; Wu, J.; Chen, Q.; Zeng, G.; Zhao, G. TG and VLDL Cholesterol Activate NLRP1 Inflammasome by Nuclear Factor-ΚB in Endothelial Cells. Int. J. Cardiol. 2017, 234, 103. [Google Scholar] [CrossRef]
- Kawakami, A.; Aikawa, M.; Alcaide, P.; Luscinskas, F.W.; Libby, P.; Sacks, F.M. Apolipoprotein CIII Induces Expression of Vascular Cell Adhesion Molecule-1 in Vascular Endothelial Cells and Increases Adhesion of Monocytic Cells. Circulation 2006, 114, 681–687. [Google Scholar] [CrossRef]
- Bisgaard, L.S.; Mogensen, C.K.; Rosendahl, A.; Cucak, H.; Nielsen, L.B.; Rasmussen, S.E.; Pedersen, T.X. Bone Marrow-Derived and Peritoneal Macrophages Have Different Inflammatory Response to OxLDL and M1/M2 Marker Expression—Implications for Atherosclerosis Research. Sci. Rep. 2016, 6, 35234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Stringer, S.E.; Hamilton, A.; Charlton-Menys, V.; Götting, C.; Müller, B.; Aeschlimann, D.; Alexander, M.Y. Decorin GAG Synthesis and TGF-β Signaling Mediate Ox-LDL-Induced Mineralization of Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.L.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.H.; McGilvray, I.; Fischer, S.E.; et al. Altered Hepatic Gene Expression in Nonalcoholic Fatty Liver Disease Is Associated with Lower Hepatic n-3 and n-6 Polyunsaturated Fatty Acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Steenman, M.; Espitia, O.; Maurel, B.; Guyomarch, B.; Heymann, M.-F.; Pistorius, M.-A.; Ory, B.; Heymann, D.; Houlgatte, R.; Gouëffic, Y.; et al. Identification of Genomic Differences among Peripheral Arterial Beds in Atherosclerotic and Healthy Arteries. Sci. Rep. 2018, 8, 3940. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-Z.; Ning, L.-F.; Xu, X.; Jiang, W.-Y.; Xing, C.; Jia, W.-P.; Chen, X.-L.; Tang, Q.-Q.; Huang, H.-Y. The MiR-181d-Regulated Metalloproteinase Adamts1 Enzymatically Impairs Adipogenesis via ECM Remodeling. Cell Death Differ. 2016, 23, 1778–1791. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.C.; Riley, G.P. ADAMTS Proteinases: A Multi-Domain, Multi-Functional Family with Roles in Extracellular Matrix Turnover and Arthritis. Arthritis Res. Ther. 2005, 7, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Salter, R.C.; Ashlin, T.G.; Kwan, A.P.L.; Ramji, D.P. ADAMTS Proteases: Key Roles in Atherosclerosis? J. Mol. Med. 2010, 88, 1203–1211. [Google Scholar] [CrossRef]
- Ashlin, T.G.; Kwan, A.P.L.; Ramji, D.P. Regulation of ADAMTS-1, -4 and -5 Expression in Human Macrophages: Differential Regulation by Key Cytokines Implicated in Atherosclerosis and Novel Synergism between TL1A and IL-17. Cytokine 2013, 64, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Manzaneque, J.C.; Carpizo, D.; Plaza-Calonge, M. del C.; Torres-Collado, A.X.; Thai, S.N.-M.; Simons, M.; Horowitz, A.; Iruela-Arispe, M.L. Cleavage of Syndecan-4 by ADAMTS1 Provokes Defects in Adhesion. Int. J. Biochem. Cell Biol. 2009, 41, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Esselens, C.; Malapeira, J.; Colomé, N.; Casal, C.; Rodríguez-Manzaneque, J.C.; Canals, F.; Arribas, J. The Cleavage of Semaphorin 3C Induced by ADAMTS1 Promotes Cell Migration. J. Biol. Chem. 2010, 285, 2463–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque, A.; Carpizo, D.R.; Iruela-Arispe, M.L. ADAMTS1/METH1 Inhibits Endothelial Cell Proliferation by Direct Binding and Sequestration of VEGF165. J. Biol. Chem. 2003, 278, 23656–23665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuno, K.; Okada, Y.; Kawashima, H.; Nakamura, H.; Miyasaka, M.; Ohno, H.; Matsushima, K. ADAMTS-1 Cleaves a Cartilage Proteoglycan, Aggrecan. FEBS Lett. 2000, 478, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Q.; Lei, J.; Wang, X.; Chen, X.; Ding, Y. MiR-362-3p Inhibits the Proliferation and Migration of Vascular Smooth Muscle Cells in Atherosclerosis by Targeting ADAMTS1. Biochem. Biophys. Res. Commun. 2017, 493, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Makin, K.; Akbareian, S.; Johnson, R.; Alghamdi, A.A.A.; Robinson, S.D.; Edwards, D.R. ADAMTS-1 and Syndecan-4 Intersect in the Regulation of Cell Migration and Angiogenesis. J. Cell Sci. 2020, 133, jcs235762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudijanto, A. The Role of Vascular Smooth Muscle Cells on the Pathogenesis of Atherosclerosis. Acta Med. Indones. 2007, 39, 86–93. [Google Scholar] [PubMed]
- Xiao, C.; Chen, S.; Yang, C.; Liu, J.; Yu, M. Identification of Polyunsaturated Fatty Acids Related Key Modules and Genes in Metabolic Dysfunction-Associated Fatty Liver Disease Using WGCNA Analysis. Front. Genet. 2022, 13, 951224. [Google Scholar] [CrossRef]
- Torres-Collado, A.X.; Kisiel, W.; Iruela-Arispe, M.L.; Rodríguez-Manzaneque, J.C. ADAMTS1 Interacts with, Cleaves, and Modifies the Extracellular Location of the Matrix Inhibitor Tissue Factor Pathway Inhibitor-2. J. Biol. Chem. 2006, 281, 17827–17837. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.P.; Sukhova, G.K.; Kisiel, W.; Foster, D.; Kehry, M.R.; Libby, P.; Schönbeck, U. Tissue Factor Pathway Inhibitor-2 Is a Novel Inhibitor of Matrix Metalloproteinases with Implications for Atherosclerosis. J. Clin. Investig. 2001, 107, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaysen, G.A.; Eiserich, J.P. The Role of Oxidative Stress-Altered Lipoprotein Structure and Function and Microinflammation on Cardiovascular Risk in Patients with Minor Renal Dysfunction. J. Am. Soc. Nephrol. 2004, 15, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C.; Goldstein, J.L.; Brown, M.S.; Russell, D.W. The LDL Receptor Gene: A Mosaic of Exons Shared with Different Proteins. Science 1985, 228, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Brown, A.J.; Sun, L.; Feramisco, J.D.; Brown, M.S.; Goldstein, J.L. Cholesterol Addition to ER Membranes Alters Conformation of SCAP, the SREBP Escort Protein That Regulates Cholesterol Metabolism. Mol. Cell 2002, 10, 237–245. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Sun, L.-P.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Direct Binding of Cholesterol to the Purified Membrane Region of SCAP: Mechanism for a Sterol-Sensing Domain. Mol. Cell 2004, 15, 259–268. [Google Scholar] [CrossRef]
- Ando, W.; Yokomori, H.; Tsutsui, N.; Yamanouchi, E.; Suzuki, Y.; Oda, M.; Inagaki, Y.; Otori, K.; Okazaki, I. Serum Matrix Metalloproteinase-1 Level Represents Disease Activity as Opposed to Fibrosis in Patients with Histologically Proven Nonalcoholic Steatohepatitis. Clin. Mol. Hepatol. 2018, 24, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokomori, H.; Oda, M.; Ando, W.; Inagaki, Y.; Okazaki, I. Hepatic Progenitor Cell Expansion in Early-Stage Nonalcoholic Steatohepatitis: Evidence from Immunohistochemistry and Immunoelectron Microscopy of Matrix Metalloproteinase-1. Med. Mol. Morphol. 2017, 50, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Kuno, K.; Kanada, N.; Nakashima, E.; Fujiki, F.; Ichimura, F.; Matsushima, K. Molecular Cloning of a Gene Encoding a New Type of Metalloproteinase-Disintegrin Family Protein with Thrombospondin Motifs as an Inflammation Associated Gene. J. Biol. Chem. 1997, 272, 556–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Baena, F.J.; Redondo-García, S.; Peris-Torres, C.; Martino-Echarri, E.; Fernández-Rodríguez, R.; Plaza-Calonge, M.D.C.; Anderson, P.; Rodríguez-Manzaneque, J.C. ADAMTS1 Protease Is Required for a Balanced Immune Cell Repertoire and Tumour Inflammatory Response. Sci. Rep. 2018, 8, 13103. [Google Scholar] [CrossRef] [PubMed]
- Moayedfard, Z.; Sani, F.; Alizadeh, A.; Bagheri Lankarani, K.; Zarei, M.; Azarpira, N. The Role of the Immune System in the Pathogenesis of NAFLD and Potential Therapeutic Impacts of Mesenchymal Stem Cell-Derived Extracellular Vesicles. Stem. Cell Res. Ther. 2022, 13, 242. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- McLaren, J.E.; Calder, C.J.; McSharry, B.P.; Sexton, K.; Salter, R.C.; Singh, N.N.; Wilkinson, G.W.G.; Wang, E.C.Y.; Ramji, D.P. The TNF-like Protein 1A-Death Receptor 3 Pathway Promotes Macrophage Foam Cell Formation in Vitro. J. Immunol. 2010, 184, 5827–5834. [Google Scholar] [CrossRef] [Green Version]
- Jönsson-Rylander, A.-C.; Nilsson, T.; Fritsche-Danielson, R.; Hammarström, A.; Behrendt, M.; Andersson, J.-O.; Lindgren, K.; Andersson, A.-K.; Wallbrandt, P.; Rosengren, B.; et al. Role of ADAMTS-1 in Atherosclerosis: Remodeling of Carotid Artery, Immunohistochemistry, and Proteolysis of Versican. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Hwang, I.; Park, C.-S.; Lee, H.; Park, D.-W.; Kang, S.-J.; Lee, S.-H.; Kim, Y.-H.; Park, S.-W.; Park, S.-J. Comparison of ADAMTS-1, -4 and -5 Expression in Culprit Plaques between Acute Myocardial Infarction and Stable Angina. J. Clin. Pathol. 2011, 64, 399–404. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, W.; Yu, C.; Zhong, F.; Li, G.; Kong, W.; Zheng, J. A Disintegrin and Metalloproteinase with Thrombospondin Motif 1 (ADAMTS1) Expression Increases in Acute Aortic Dissection. Sci. China Life Sci. 2016, 59, 59–67. [Google Scholar] [CrossRef]
- Laurent, M.-A.; Bonnier, D.; Théret, N.; Tufféry, P.; Moroy, G. In Silico Characterization of the Interaction between LSKL Peptide, a LAP-TGF-Beta Derived Peptide, and ADAMTS1. Comput. Biol. Chem. 2016, 61, 155–161. [Google Scholar] [CrossRef]
- Bourd-Boittin, K.; Bonnier, D.; Leyme, A.; Mari, B.; Tuffery, P.; Samson, M.; Ezan, F.; Baffet, G.; Theret, N. Protease Profiling of Liver Fibrosis Reveals the ADAM Metallopeptidase with Thrombospondin Type 1 Motif, 1 as a Central Activator of Transforming Growth Factor Beta. Hepatology 2011, 54, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Y.; Cheng, X.; Wu, D.; Huang, X.; Chen, B.; Ren, Y.; Jiang, W.; Tang, X.; Bai, T.; et al. Targeting Epigenetically Maladapted Vascular Niche Alleviates Liver Fibrosis in Nonalcoholic Steatohepatitis. Sci. Transl. Med. 2021, 13, eabd1206. [Google Scholar] [CrossRef]
- Ambardekar, A.V.; Stratton, M.S.; Dobrinskikh, E.; Hunter, K.S.; Tatman, P.D.; Lemieux, M.E.; Cleveland, J.C.; Tuder, R.M.; Weiser-Evans, M.C.M.; Moulton, K.S.; et al. Matrix-Degrading Enzyme Expression and Aortic Fibrosis during Continuous-Flow Left Ventricular Mechanical Support. J. Am. Coll. Cardiol. 2021, 78, 1782–1795. [Google Scholar] [CrossRef]
- Prokesch, A.; Hackl, H.; Hakim-Weber, R.; Bornstein, S.R.; Trajanoski, Z. Novel Insights into Adipogenesis from Omics Data. Curr. Med. Chem. 2009, 16, 2952–2964. [Google Scholar] [CrossRef] [Green Version]
- Horodyska, J.; Reyer, H.; Wimmers, K.; Trakooljul, N.; Lawlor, P.G.; Hamill, R.M. Transcriptome Analysis of Adipose Tissue from Pigs Divergent in Feed Efficiency Reveals Alteration in Gene Networks Related to Adipose Growth, Lipid Metabolism, Extracellular Matrix, and Immune Response. Mol. Genet. Genom. 2019, 294, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Hsu, C.-H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPalpha Induces Adipogenesis through PPARgamma: A Unified Pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Inukai, K.; Katagiri, H.; Awata, T.; Oka, Y.; Katayama, S. Regulation of PPAR Gamma Transcriptional Activity in 3T3-L1 Adipocytes. Biochem. Biophys. Res. Commun. 2003, 300, 429–436. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From Stem Cell to Adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding Adipocyte Differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadrich, F.; Sayadi, S. Apigetrin Inhibits Adipogenesis in 3T3-L1 Cells by Downregulating PPARγ and CEBP-α. Lipids Health Dis. 2018, 17, 95. [Google Scholar] [CrossRef] [Green Version]
- Simão, J.J.; Cruz, M.M.; Abdala, F.M.; Bolsoni-Lopes, A.; Armelin-Correa, L.; Alonso-Vale, M.I.C. Palmitoleic Acid Acts on Adipose-Derived Stromal Cells and Promotes Anti-Hypertrophic and Anti-Inflammatory Effects in Obese Mice. Pharmaceuticals 2022, 15, 1194. [Google Scholar] [CrossRef]
- Zhou, J.; Li, H.; Xia, X.; Herrera, A.; Pollock, N.; Reebye, V.; Sodergren, M.H.; Dorman, S.; Littman, B.H.; Doogan, D.; et al. Anti-Inflammatory Activity of MTL-CEBPA, a Small Activating RNA Drug, in LPS-Stimulated Monocytes and Humanized Mice. Mol. Ther. 2019, 27, 999–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in Chronic Inflammatory Diseases. Cell. Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Josefs, T.; Barrett, T.J.; Brown, E.J.; Quezada, A.; Wu, X.; Voisin, M.; Amengual, J.; Fisher, E.A. Neutrophil Extracellular Traps Promote Macrophage Inflammation and Impair Atherosclerosis Resolution in Diabetic Mice. JCI Insight 2020, 5, e134796. [Google Scholar] [CrossRef] [Green Version]
- Van Avondt, K.; Maegdefessel, L.; Soehnlein, O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb. Haemost. 2019, 119, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in Modulating Acute and Chronic Inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil Secretion Products Pave the Way for Inflammatory Monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil Extracellular Traps License Macrophages for Cytokine Production in Atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of Luminal Neutrophil Extracellular Traps in Atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and Neutrophils Potentiate Endothelial Stress, Apoptosis and Detachment: Implications for Superficial Erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristol, J.A.; Morrison, T.E.; Kenney, S.C. CCAAT/Enhancer Binding Proteins Alpha and Beta Regulate the Tumor Necrosis Factor Receptor 1 Gene Promoter. Mol. Immunol. 2009, 46, 2706–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene Expression of Tumor Necrosis Factor Alpha and TNF-Receptors, P55 and P75, in Nonalcoholic Steatohepatitis Patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Daniele, A.; DE Luca, R.; Mazzocca, A.; Ruggieri, E.; Savino, E.; Casamassima, P.; Simone, M.; Sabba, C.; Paradiso, A. Synergism of Adipocytokine Profile and ADIPOQ/TNF-α Polymorphisms in NAFLD-Associated MetS Predict Colorectal Liver Metastases Outgrowth. Cancer Genom. Proteom. 2019, 16, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Wandrer, F.; Liebig, S.; Marhenke, S.; Vogel, A.; John, K.; Manns, M.P.; Teufel, A.; Itzel, T.; Longerich, T.; Maier, O.; et al. TNF-Receptor-1 Inhibition Reduces Liver Steatosis, Hepatocellular Injury and Fibrosis in NAFLD Mice. Cell Death Dis. 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Ding, A.; Kim, H.-J.; Zheng, H.; Wei, F.; Ma, X. Progranulin Controls Sepsis via C/EBPα-Regulated Il10 Transcription and Ubiquitin Ligase/Proteasome-Mediated Protein Degradation. J. Immunol. 2016, 197, 3393–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.H.; Jun, D.W.; Jang, K.; Saeed, W.K.; Lee, J.S.; Kang, H.T.; Chae, Y.J. Granulocyte Colony Stimulating Factor Treatment in Non-Alcoholic Fatty Liver Disease: Beyond Marrow Cell Mobilization. Oncotarget 2017, 8, 97965–97976. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, K.; Chen, D.; Chen, H.; Sun, K.; Ju, X.; Lan, J.; Zhou, Y.; Wang, W.; Pang, L. The Effect of Granulocyte Colony-Stimulating Factor on the Progression of Atherosclerosis in Animal Models: A Meta-Analysis. Biomed. Res. Int. 2017, 2017, 6705363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Holze, H.; Kirsch, R.; Nastou, K.C.; Cuesta-Astroz, Y.; Rattei, T.; Szklarczyk, D.; von Mering, C.; Jensen, L.J. Cytoscape StringApp 2.0: Analysis and Visualization of Heterogeneous Biological Networks. J. Proteome Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov/ (accessed on 29 January 2023).
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A Cytoscape Plugin for Centrality Analysis and Evaluation of Protein Interaction Networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Iny Stein, T.; Levitt, J.; Gershoni, M.; Morrey, C.P.; Safran, M.; Lancet, D. MalaCards: An Amalgamated Human Disease Compendium with Diverse Clinical and Genetic Annotation and Structured Search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Clyne, M.; Khoury, M.J.; Gwinn, M. Phenopedia and Genopedia: Disease-Centered and Gene-Centered Views of the Evolving Knowledge of Human Genetic Associations. Bioinformatics 2010, 26, 145–146. [Google Scholar] [CrossRef] [Green Version]
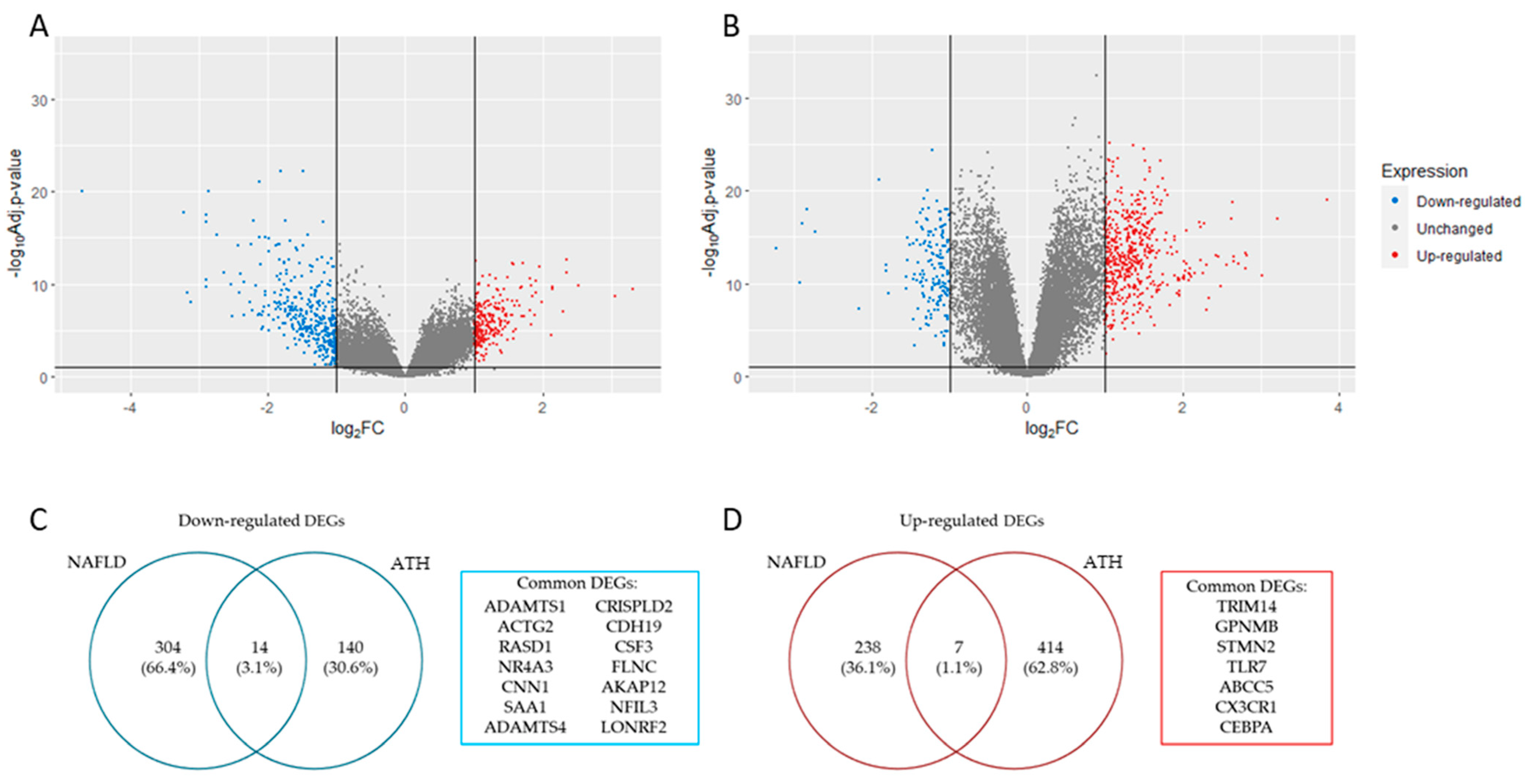
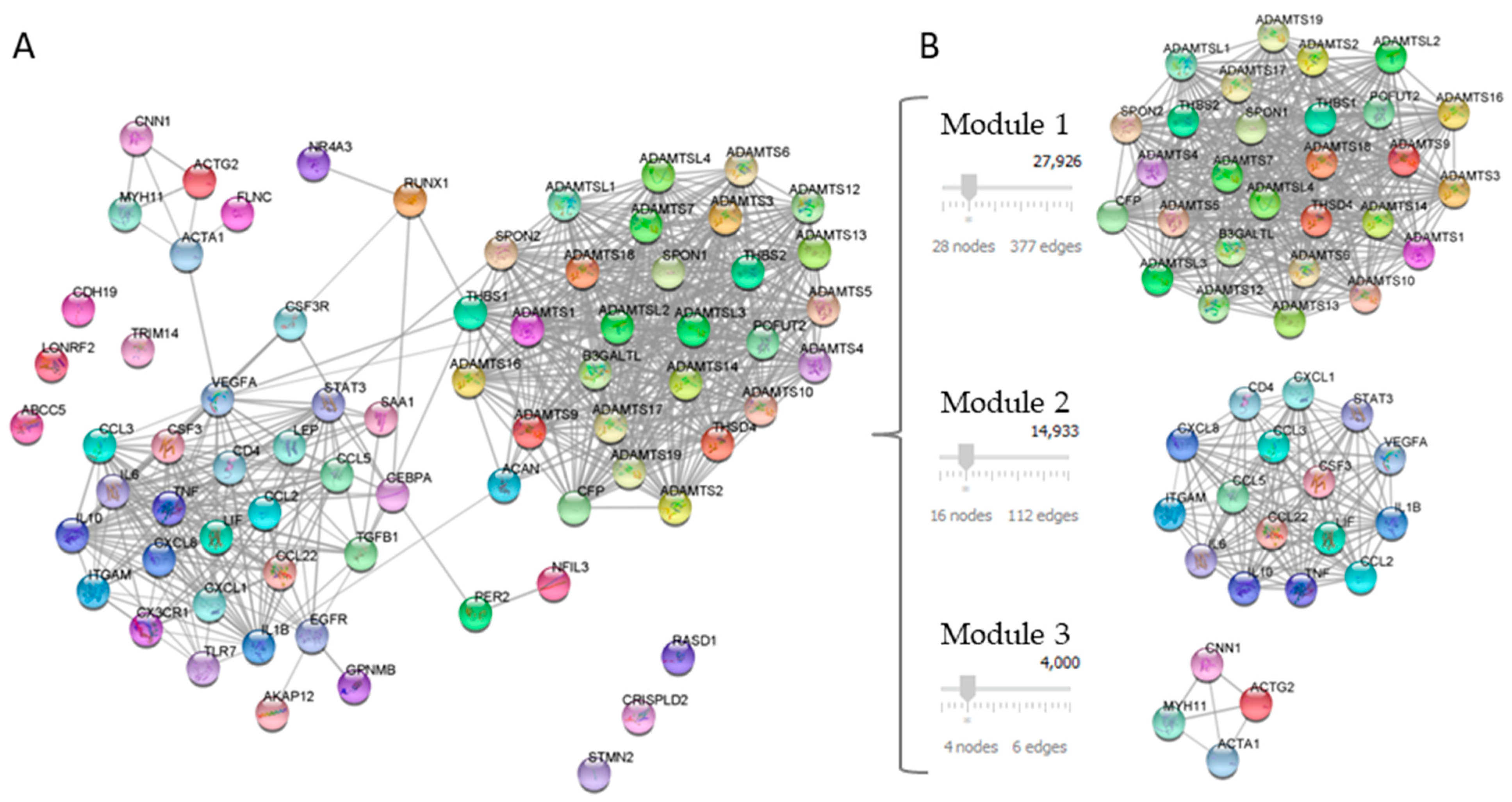


{kind=link}
{kind=link}
{kind=link}
| Category | ID | Term | Count | p-Value | FDR |
|---|---|---|---|---|---|
| Reactome pathway | R-HSA-5173214 | O-glycosylation of TSR domain-containing proteins | 28 | 2.78 × 10−54 | 9.50 × 10−52 |
| Reactome pathway | R-HSA-5083635 | Defective B3GALTL causes PpS | 27 | 5.38 × 10−52 | 9.20 × 10−50 |
| Reactome pathway | R-HSA-3906995 | Diseases associated with O-glycosylation of proteins | 27 | 2.46 × 10−42 | 2.80 × 10−40 |
| Reactome pathway | R-HSA-5173105 | O-linked glycosylation | 28 | 8.67 × 10−38 | 7.42 × 10−36 |
| Reactome pathway | R-HSA-3781865 | Diseases of glycosylation | 28 | 1.65 × 10−34 | 1.13 × 10−32 |
| GO-Term CC | GO:0031012 | extracellular matrix | 28 | 1.54 × 10−33 | 1.84 × 10−31 |
| GO-Term BP | GO:0030198 | extracellular matrix organization | 23 | 4.30 × 10−29 | 4.57 × 10−26 |
| Reactome pathway | R-HSA-5668914 | Diseases of metabolism | 28 | 1.14 × 10−27 | 6.50 × 10−26 |
| GO-Term MF | GO:0004222 | metalloendopeptidase activity | 21 | 1.46 × 10−27 | 2.55 × 10−25 |
| GO-Term CC | GO:0005576 | Extracellular region | 41 | 6.39 × 10−22 | 3.80 × 10−20 |
| Reactome pathway | R-HSA-1643685 | Disease | 38 | 8.84 × 10−14 | 3.78 × 10−12 |
| GO-Term CC | GO:0005615 | Extracellular space | 28 | 5.48 × 10−11 | 2.17 × 10−9 |
| Reactome pathway | R-HSA-597592 | Post-translational protein modification | 30 | 7.31 × 10−10 | 2.27 × 10−8 |
| Reactome pathway | R-HSA-392499 | Metabolism of proteins | 33 | 1.38 × 10−8 | 3.92 × 10−7 |
| Reactome pathway | R-HSA-1280215 | Cytokine Signaling in Immune system | 20 | 3.16 × 10−8 | 8.32 × 10−7 |
| Reactome pathway | R-HSA-168256 | Immune System | 23 | 2.81 × 10−3 | 5.33 × 10−2 |
| Reactome pathway | R-HSA-162582 | Signal Transduction | 22 | 8.14 × 10−2 | 7.52 × 10−1 |
| Reactome pathway | R-HSA-5173214 | O-glycosylation of TSR domain-containing proteins | 28 | 2.78 × 10−54 | 9.50 × 10−52 |
| Reactome pathway | R-HSA-5083635 | Defective B3GALTL causes PpS | 27 | 5.38 × 10−52 | 9.20 × 10−50 |
| Reactome pathway | R-HSA-3906995 | Diseases associated with O-glycosylation of proteins | 27 | 2.46 × 10−42 | 2.80 × 10−40 |
| Ranking | Gen Symbol | Uniprot:ID * | Description | Betweenness | Degree |
|---|---|---|---|---|---|
| 1 | VEGFA | Q9H1W9 | Vascular endothelial growth factor A | 1208,552 | 19.0 |
| 2 | THBS1 | P07996 | Thrombospondin 1 | 969,255 | 29.0 |
| 3 | ACTA1 | P68133 | Actin alpha 1 | 478,000 | 5.0 |
| 4 | ADAMTS1 | Q9UHI8 | ADAM Metallopeptidase with Thrombospondin Type 1 Motif 1 | 459,444 | 29.0 |
| 5 | SPON2 | Q9BUD6 | Spondin 2 | 314,440 | 28.0 |
| 6 | ITGAM | P11215 | Integrin Subunit Alpha M | 304,704 | 14.0 |
| 7 | CEBPA | P49715 | CCAAT Enhancer Binding Protein Alpha | 292,120 | 8.0 |
| 8 | TGFB1 | P01137 | Transforming Growth Factor Beta 1 | 287,228 | 12.0 |
| 9 | RUNX1 | Q16285 | RUNX Family Transcription Factor 1 | 256,626 | 4.0 |
| 10 | EGFR | Q9H2C9 | Epidermal Growth Factor Receptor | 247,008 | 11.0 |
| Gen | Group | Disgenet | MalaCards | HuGE Genopedia |
|---|---|---|---|---|
| ADAMTS1 | Liver-related diseases | Liver Carcinoma | Liver Cirrhosis | Fatty Liver |
| Fibrosis, Liver | ||||
| Cardiovascular Diseases | Atherosclerosis | Aortic Aneurysm | Coronary Disease | |
| Arteriosclerosis | Atherosclerosis Susceptibility | Cardiovascular Diseases | ||
| Coronary Arteriosclerosis | Ischemia | |||
| Coronary Artery Disease | ||||
| ADAMTS4 | Liver-related diseases | Liver Carcinoma | Hepatocellular Carcinoma | |
| Cardiovascular Diseases | Atherosclerosis | Cerebrovascular Disease | Aortic Aneurysm, Thoracic | |
| Arteriosclerosis | Cardiovascular System Disease | |||
| Aortic Aneurysm | Carotid Stenosis | |||
| Coronary Arteriosclerosis | Atherosclerosis Susceptibility | |||
| CEBPA | Liver-related diseases | Liver Carcinoma | NAFLD | Liver Neoplasms |
| NAFLD | Fatty Liver Disease | |||
| Fatty liver | Liver Cirrhosis | |||
| Cardiovascular Diseases | Atherosclerosis | Cerebral Artery Occlusion | Myocardial Ischemia | |
| Arteriosclerosis | Peripheral Artery Disease | Peripheral Vascular Diseases | ||
| Cardiovascular Diseases | Ischemia | |||
| Peripheral Arterial Diseases | ||||
| CSF3 | Liver-related diseases | Fatty Liver Disease | NAFLD | Liver Cirrhosis |
| NAFLD | Fatty Liver Disease | |||
| Liver Carcinoma | Liver Disease | |||
| Acute-On-Chronic Liver Failure | Liver Cirrhosis | |||
| Cardiovascular Diseases | Atherosclerosis | Carotid Artery Disease | Cardiovascular Diseases | |
| Arteriosclerosis | Ischemia | Carotid Stenosis | ||
| Arteriosclerosis Obliterans | Vascular Disease | Coronary Artery Disease | ||
| Ischemic cardiomyopathy | Hepatic Vascular Disease | |||
| Coronary Arteriorclerosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andújar-Vera, F.; Ferrer-Millán, M.; García-Fontana, C.; García-Fontana, B.; González-Salvatierra, S.; Sanabria-de la Torre, R.; Martínez-Heredia, L.; Riquelme-Gallego, B.; Muñoz-Torres, M. Analysis of the Genetic Relationship between Atherosclerosis and Non-Alcoholic Fatty Liver Disease through Biological Interaction Networks. Int. J. Mol. Sci. 2023, 24, 4124. https://doi.org/10.3390/ijms24044124
Andújar-Vera F, Ferrer-Millán M, García-Fontana C, García-Fontana B, González-Salvatierra S, Sanabria-de la Torre R, Martínez-Heredia L, Riquelme-Gallego B, Muñoz-Torres M. Analysis of the Genetic Relationship between Atherosclerosis and Non-Alcoholic Fatty Liver Disease through Biological Interaction Networks. International Journal of Molecular Sciences. 2023; 24(4):4124. https://doi.org/10.3390/ijms24044124
Chicago/Turabian StyleAndújar-Vera, Francisco, María Ferrer-Millán, Cristina García-Fontana, Beatriz García-Fontana, Sheila González-Salvatierra, Raquel Sanabria-de la Torre, Luis Martínez-Heredia, Blanca Riquelme-Gallego, and Manuel Muñoz-Torres. 2023. "Analysis of the Genetic Relationship between Atherosclerosis and Non-Alcoholic Fatty Liver Disease through Biological Interaction Networks" International Journal of Molecular Sciences 24, no. 4: 4124. https://doi.org/10.3390/ijms24044124