
Purine Metabolism Dysfunctions: Experimental Methods of Detection and Diagnostic Potential
 ,
,  ,
,  ,
,
Abstract
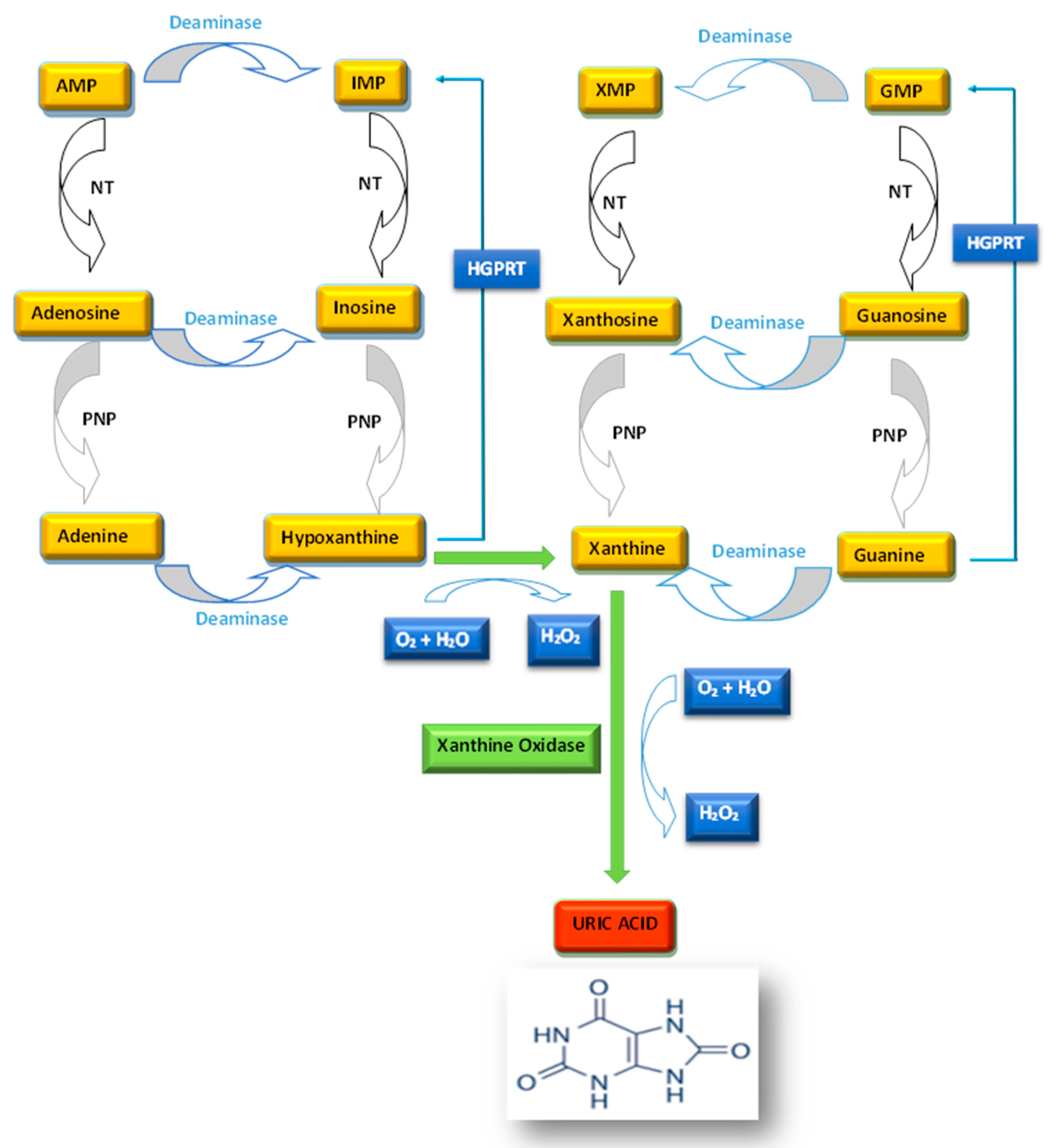
:1. Introduction
2. Hyperuricemia and Hypouricemia
3. Uric Acid as Antioxidant
4. The Key Role of Xanthine Oxidase as a Biomarker of Metabolic Disorders
5. Methods of Uric Acid Detection
6. Detection of Uric Acid in Urine
7. Uric Acid Leaching in Saliva
8. Evaluation of Xanthine Oxidoreductase (XOR) Activity
9. Possible Uses of Purine Metabolism Markers in the Assessment of Oxidative Stress
10. Main Remarks and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaudhary, K.; Malhotra, K.; Sowers, J.; Aroor, A. Uric Acid-key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal Med. 2013, 3, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Yang, F.; Yang, I.; Yin, Y.; Luo, J.J.; Wang, H.; Yang, X.F. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. Landmark Ed. 2012, 17, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Pasalic, D.; Marinkovic, N.; Feher-Turkovic, L. Uric acid as one of the important factors in multifactorial disorders—Facts and controversies. Biochem. Med. 2012, 22, 63–75. [Google Scholar] [CrossRef]
- García-Nieto, V.M.; Claverie-Martín, F.; Moraleda-Mesa, T.; Perdomo-Ramírez, A.; Tejera-Carreño, P.; Cordoba-Lanus, E.; Luis-Yanes, M.I.; Ramos-Trujillo, E.; RenalTube Group. Gout associated with reduced renal excretion of uric acid. Renal tubular disorder that nephrologists do not treat. Nefrología (Engl. Ed.) 2022, 42, 273–279. [Google Scholar] [CrossRef]
- Cicero, A.F.; Rosticci, M.; Fogacci, F.; Grandi, E.; D’Addato, S.; Borghi, C.; Brisighella Heart Study Group. High serum uric acid is associated to poorly controlled blood pressure and higher arterial stiffness in hypertensive subjects. Eur. J. Intern. Med. 2017, 37, 38–42. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Giovannini, M.; Grandi, E.; D’Addato, S.; Borghi, C.; Brisighella Heart Study Group. Interaction between low-density lipoprotein-cholesterolaemia, serum uric level and incident hypertension: Data from the Brisighella Heart Study. J. Hypertens. 2019, 37, 728–731. [Google Scholar] [CrossRef]
- Mortada, I. Hyperuricemia, Type 2 Diabetes Mellitus, and Hypertension: An Emerging Association. Curr. Hypertens. Rep. 2017, 19, 69. [Google Scholar] [CrossRef]
- Masulli, M.; D’Elia, L.; Angeli, F.; Barbagallo, C.M.; Bilancio, G.; Bombelli, M.; Bruno, B.; Casiglia, E.; Cianci, R.; Cicero, A.F.; et al. Serum uric acid levels threshold for mortality in diabetic individuals: The URic acid Right for heArt Health (URRAH) project. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1245–1252. [Google Scholar] [CrossRef]
- Palatini, P.; Virdis, A.; Masi, S.; Mengozzi, A.; Casiglia, E.; Tikhonoff, V.; Cicero, A.F.; Ungar, A.; Parati, G.; Rivasi, G.; et al. Hyperuricemia increases the risk of cardiovascular mortality associated with very high HdL-cholesterol level. Nutr. Metab. Cardiovasc. Dis. 2023, 33, 323–330. [Google Scholar] [CrossRef]
- Casiglia, E.; Tikhonoff, V.; Virdis, A.; Grassi, G.; Angeli, F.; Barbagallo, C.M.; Bombelli, M.; Cicero, A.F.; Cirillo, M.; Cirillo, P.; et al. Serum uric acid/serum creatinine ratio as a predictor of cardiovascular events. Detection of prognostic cardiovascular cut-off values. J. Hypertens. 2023, 41, 180–186. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Kuwabara, M.; Borghi, C. Therapeutic Strategies for the Treatment of Chronic Hyperuricemia: An Evidence-Based Update. Medicina 2021, 57, 58. [Google Scholar] [CrossRef]
- Strilchuk, L.; Fogacci, F.; Cicero, A.F. Safety and tolerability of available urate-lowering drugs: A critical review. Expert Opin. Drug Saf. 2019, 18, 261–271. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Cincione, R.I.; Tocci, G.; Borghi, C. Clinical Effects of Xanthine Oxidase Inhibitors in Hyperuricemic Patients. Med. Princ. Pract. 2021, 30, 122–130. [Google Scholar] [CrossRef]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef]
- Zhang, J.; Dierckx, R.; Mohee, K.; Clark, A.L.; Cleland, J.G. Xanthine oxidase inhibition for the treatment of cardiovascular disease: An updated systematic review and meta-analysis. ESC Heart Fail. 2017, 4, 40–45. [Google Scholar] [CrossRef]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef]
- Chrysant, S.G. Association of hyperuricemia with cardiovascular diseases: Current evidence. Hosp. Pract. 2023, 51, 54–63. [Google Scholar] [CrossRef]
- Wong, C.K.; Chen, Y.; Ho, L.M.; Zhen, Z.; Siu, C.W.; Tse, H.F.; Yiu, K.H. The effects of hyperuricaemia on flow-mediated and nitroglycerin-mediated dilatation in high-risk patients. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Corry, D.B.; Eslami, P.; Yamamoto, K.; Nyby, M.D.; Makino, H.; Tuck, M.L. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J. Hypertens. 2008, 26, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Role of insulin resistance in human disease (syndrome X): An expanded definition. Annu. Rev. Med. 1993, 44, 121–131. [Google Scholar] [CrossRef]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Curhan, G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: The health professionals follow-up study. Arch. Intern. Med. 2005, 165, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Gaffo, A.L.; Edwards, N.L.; Saag, K.G. Gout. Hyperuricemia and cardiovascular disease: How strong is the evidence for a causal link? Arthritis Res. Ther. 2009, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef]
- Sebesta, I.; Stiburkova, B.; Krijt, J. Hereditary xanthinuria is not so rare disorder of purine metabolism. Nucleosides Nucleotides Nucleic Acids 2018, 37, 324–328. [Google Scholar] [CrossRef]
- Kubihal, S.; Goyal, A.; Singla, R.; Khadgawat, R. Urolithiasis due to Hereditary Xanthinuria Type II: A Long-term Follow-up report. Indian Pediatr. 2020, 57, 468–469. [Google Scholar] [CrossRef]
- McDonagh, E.M.; Thorn, C.F.; Callaghan, J.T.; Altman, R.B.; Klein, T.E. PharmGKB summary: Uric acid-lowering drugs pathway, pharmacodynamics. Pharmacogenet. Genom. 2014, 24, 464–476. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef]
- Dinour, D.; Gray, N.K.; Campbell, S.; Shu, X.; Sawyer, L.; Richardson, W.; Rechavi, G.; Amariglio, N.; Ganon, L.; Sela, B.-A.; et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J. Am. Soc. Nephrol. 2010, 21, 64–72. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Davies, K.J.; Sevanian, A.; Muakkassah-Kelly, S.F.; Hochstein, P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem. J. 1986, 235, 747–754. [Google Scholar] [CrossRef]
- Sánchez-Lozada, L.G.; Lanaspa, M.A.; Cristóbal-García, M.; García-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.A.; Kang, D.H.; Johnson, R.J. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp. Nephrol. 2012, 121, e71–e78. [Google Scholar] [CrossRef]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef]
- Farquharson, C.A.; Butler, R.; Hill, A.; Belch, J.J.; Struthers, A.D. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation 2002, 106, 221–226. [Google Scholar] [CrossRef]
- Ostendorf, B.N.; Blau, O.; Uharek, L.; Blau, I.W.; Penack, O. Association between low uric acid levels and acute graftversus-host disease. Ann. Hematol. 2015, 94, 139–144. [Google Scholar] [CrossRef]
- Fang, P.; Li, X.; Luo, J.J.; Wang, H.; Yang, X.F. A double-edged sword: Uric acid and neurological disorders. Brain Disord. Ther. 2013, 2, 109. [Google Scholar]
- Bobulescu, I.A.; Moe, O.W. Renal transport of uric acid: Evolving concepts and uncertainties. Adv. Chronic Kidney Dis. 2012, 19, 358–371. [Google Scholar] [CrossRef]
- Hediger, M.A.; Johnson, R.J.; Miyazaki, H.; Endou, H. Molecular physiology of urate transport. Physiology 2005, 20, 125–133. [Google Scholar] [CrossRef]
- Preitner, F.; Bonny, O.; Laverrière, A.; Rotman, S.; Firsov, D.; Da Costa, A.; Metref, S.; Thorens, B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc. Natl. Acad. Sci. USA 2009, 106, 15501–15506. [Google Scholar] [CrossRef]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Jutabha, P.; Anzai, N.; Kitamura, K.; Taniguchi, A.; Kaneko, S.; Yan, K.; Yamada, H.; Shimada, H.; Kimura, T.; Katada, T.; et al. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J. Biol. Chem. 2010, 285, 35123–35132. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jo, Y.I.; Lee, J.H. Renal effects of uric acid: Hyperuricemia and hypouricemia. Korean J. Intern. Med. 2020, 35, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; O’Reilly, E.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Plasma urate and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 166, 561–567. [Google Scholar] [CrossRef]
- Gao, X.; O’Reilly, É.J.; Schwarzschild, M.A.; Ascherio, A. Prospective study of plasma urate and risk of Parkinson disease in men and women. Neurology 2016, 86, 520–526. [Google Scholar] [CrossRef]
- Gedeon, C.; Behravan, J.; Koren, G.; Piquette-Miller, M. Transport of glyburide by placental ABC transporters: Implications in fetal drug exposure. Placenta 2006, 27, 1096–1102. [Google Scholar] [CrossRef]
- Zhao, J.; Guo, S.; Schrodi, S.J.; He, D. Trends in the Contribution of Genetic Susceptibility Loci to Hyperuricemia and Gout and Associated Novel Mechanisms. Front. Cell Dev. Biol. 2022, 10, 937855. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K. Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout. J. Biol. Inorg. Chem. 2015, 20, 195–207. [Google Scholar] [CrossRef]
- Stevens, C.R.; Millar, T.M.; Clinch, J.G.; Kanczler, J.M.; Bodamyali, T.; Blake, D.R. Antibacterial properties of xanthine oxidase in human milk. Lancet 2000, 356, 829–830. [Google Scholar] [CrossRef]
- Murase, T.; Nampei, M.; Oka, M.; Miyachi, A.; Nakamura, T. A highly sensitive assay of human plasma xanthine oxidoreductase activity using stable isotope-labeled xanthine and LC/TQMS. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci. 2016, 1039, 51–58. [Google Scholar] [CrossRef]
- Furuhashi, M.; Matsumoto, M.; Tanaka, M.; Moniwa, N.; Murase, T.; Nakamura, T.; Ohnishi, H.; Saitoh, S.; Shimamoto, K.; Miura, T. Plasma Xanthine Oxidoreductase Activity as a Novel Biomarker of Metabolic Disorders in a General Population. Circ. J. 2018, 82, 1892–1899. [Google Scholar] [CrossRef]
- Furuhashi, M. Fatty Acid-Binding Protein 4 in Cardiovascular and Metabolic Diseases. J. Atheroscler. Thromb. 2019, 26, 216–232. [Google Scholar] [CrossRef]
- Furuhashi, M.; Matsumoto, M.; Murase, T.; Nakamura, T.; Higashiura, Y.; Koyama, M.; Tanaka, M.; Moniwa, N.; Ohnishi, H.; Saitoh, S.; et al. Independent links between plasma xanthine oxidoreductase activity and levels of adipokines. J. Diabetes Investig. 2019, 10, 1059–1067. [Google Scholar] [CrossRef]
- Furuhashi, M.; Koyama, M.; Matsumoto, M.; Murase, T.; Nakamura, T.; Higashiura, Y.; Tanaka, M.; Moniwa, N.; Ohnishi, H.; Saitoh, S.; et al. Annual change in plasma xanthine oxidoreductase activity is associated with changes in liver enzymes and body weight. Endocr. J. 2019, 66, 777–786. [Google Scholar] [CrossRef]
- Patel, G.K.; Taylor, W.; Singh, S.; Khushman, M.; Singh, A.P. Revisiting the Clinical Importance of DPYD*9A (c.85T>C) Variant of Dihydropyrimidine Dehydrogenase (DPYD) Gene in Patients Treated with Fluoropyrimidine-Based Chemotherapy. J. Mol. Biomark. Diagn. 2018, 9, e129. [Google Scholar] [CrossRef]
- Nakamura, T.; Murase, T.; Satoh, E.; Hibi, C.; Nakayama, Y.; Katoh, N.; Yokoyama, H.; Tomita, H. Establishment of the process in blood sampling and sample handling as a biomarker of hypoxia-inducible diseases; plasma hypoxanthine and xanthine measurement. J. Mol. Biomark. Diagn. 2018, 9, 1000404. [Google Scholar] [CrossRef]
- Furuhashi, M.; Koyama, M.; Higashiura, Y.; Murase, T.; Nakamura, T.; Matsumoto, M.; Sakai, A.; Ohnishi, H.; Tanaka, M.; Saitoh, S.; et al. Differential regulation of hypoxanthine and xanthine by obesity in a general population. J. Diabetes Investig. 2020, 11, 878–887. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Pacheco de Andrade, M.; Hirata, R.D.; Sandrini, F.; Largura, A.; Hirata, M.H. Uric acid biorhythm, a feature of long-term variation in a clinical laboratory database. Clin. Chem. Lab. Med. 2012, 50, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Freiburghaus, K.; Leichtle, A.B.; Nakas, C.T.; Fiedler, G.M.; Largiadèr, C.R. Effects of Freezing and Thawing Procedures on Selected Clinical Chemistry Parameters in Plasma. Biopreservation Biobanking 2020, 18, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Singla, P.; Manocha, A.; Kankra, M.; Sharma, A.; Ahirwar, A.; Ralhan, R.; Thapliyal, U.; Mehra, P. The Hemolyzed Sample: To Analyse or Not to Analyse. Indian J. Clin. Biochem. 2020, 35, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Erden, P.E.; Kılıç, E. A review of enzymatic uric acid biosensors based on amperometric detection. Talanta 2013, 107, 312–323. [Google Scholar] [CrossRef]
- Wang, X.; Yao, Q.; Tang, X.M.; Zhong, H.P.; Qiu, P.; Wang, X.L. A Highly Selective and Sensitive Colorimetric Detection of Uric Acid in Human Serum Based on MoS2-CatalyzedOxidation TMB. Anal. Bioanal. Chem. 2019, 411, 943–952. [Google Scholar] [CrossRef]
- He, Y.; Qi, F.; Niu, X.; Zhang, W.; Zhang, X.; Pan, J. Uricase-free on-demand colorimetric biosensing of uric acid enabled by integrated CoP nanosheet arrays as a monolithic peroxidase mimic. Anal. Chim. Acta. 2018, 1021, 113–120. [Google Scholar] [CrossRef]
- Wang, Q.; Wen, X.; Kong, J. Recent Progress on Uric Acid Detection: A Review. Crit. Rev. Anal. Chem. 2020, 50, 359–375. [Google Scholar] [CrossRef]
- Ferrari, P.; Bonny, O. Diagnosis and prevention of uric acid stones. Ther. Umsch. 2004, 61, 571–574. [Google Scholar] [CrossRef]
- Rodriguez, A.; Gomila, R.M.; Martorell, G.; Costa-Bauza, A.; Grases, F. Quantification of xanthine- and uric acid-related compounds in urine using a “dilute-and-shoot” technique coupling ultra-high-performance liquid chromatography and high-resolution Orbitrap mass spectrometry. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci. 2017, 1067, 53–60. [Google Scholar] [CrossRef]
- Bjornstad, P.; Roncal, C.; Milagres, T.; Pyle, L.; Lanaspa, M.A.; Bishop, F.K.; Snell-Bergeon, J.K.; Johnson, R.J.; Wadwa, R.P.; Maahs, D.M. Hyperfiltration and uricosuria in adolescents with type 1 diabetes. Pediatr. Nephrol. 2016, 31, 787–793. [Google Scholar] [CrossRef]
- Svistounov, D.; Solbu, M.D.; Jenssen, T.G.; Mathisen, U.D.; Hansen, T.; Elgstøen, K.B.P.; Zykova, S.N. Development of quantitative assay for simultaneous measurement of purine metabolites and creatinine in biobanked urine by liquid chromatography-tandem mass spectrometry. Scand. J. Clin. Lab. Investig. 2022, 82, 37–49. [Google Scholar] [CrossRef]
- Bakhtiari, S.; Toosi, P.; Samadi, S.; Bakhshi, M. Assessment of Uric Acid Level in the Saliva of Patients with Oral Lichen Planus. Med. Princ. Pract. 2017, 26, 57–60. [Google Scholar] [CrossRef]
- Jaiswal, A.; Madaan, S.; Acharya, N.; Kumar, S.; Talwar, D.; Dewani, D. Salivary Uric Acid: A Noninvasive Wonder for Clinicians? Cureus 2021, 13, e19649. [Google Scholar] [CrossRef]
- Bukharinova, M.A.; Stozhko, N.Y.; Novakovskaya, E.A.; Khamzina, E.I.; Tarasov, A.V.; Sokolkov, S.V. Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid. Biosensors 2021, 11, 287. [Google Scholar] [CrossRef]
- Vernerová, A.; Kujovská Krčmová, L.; Melichar, B.; Švec, F. Non-invasive determination of uric acid in human saliva in the diagnosis of serious disorders. Clin. Chem. Lab. Med. 2020, 59, 797–812. [Google Scholar] [CrossRef]
- Liu, X.Y.; Luo, Y.; Zhou, C.Y.; Peng, A.; Liu, J.Y. A sensitive and accurate method to simultaneously measure uric acid and creatinine in human saliva by using LC-MS/MS. Bioanalysis 2017, 9, 1751–1760. [Google Scholar] [CrossRef]
- Honeychurch, K.C. The determination of uric acid in human saliva by liquid chromatography with electrochemical detection. J. Anal. Bioanal. Sep. Tech. 2017, 2, 47–51. [Google Scholar] [CrossRef]
- Guan, Y.; Chu, Q.; Ye, J. Determination of uric acid in human saliva by capillary electrophoresis with electrochemical detection: Potential application in fast diagnosis of gout. Anal. Bioanal. Chem. 2004, 380, 913–917. [Google Scholar] [CrossRef]
- Chu, Q.C.; Lin, M.; Geng, C.H.; Ye, J.N. Determination of uric acid in human saliva and urine using miniaturized capillary electrophoresis with amperometric detection. Chromatographia 2007, 65, 179–184. [Google Scholar] [CrossRef]
- Wu, W.C.; Chen, H.T.; Lin, S.C.; Chen, F.R.; Chang, H.T.; Tseng, F.G. Nitrogen-doped carbon nanodots prepared from polyethylenimine for fluorometric determination of salivary uric acid. Mikrochim. Acta 2019, 186, 166. [Google Scholar] [CrossRef]
- Murase, T.; Nampei, M.; Oka, M.; Ashizawa, N.; Matsumoto, K.; Miyachi, A.; Nakamura, T. Xanthine oxidoreductase activity assay in tissues using stable isotope-labeled substrate and liquid chromatography high-resolution mass spectrometry. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci. 2016, 1008, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Oka, M.; Nampei, M.; Miyachi, A.; Nakamura, T. A highly sensitive assay for xanthine oxidoreductase activity using a combination of [13C2,15N2]xanthine and liquid chromatography/triple quadrupole mass spectrometry. J. Label. Comp. Radiopharm. 2016, 59, 214–220. [Google Scholar] [CrossRef]
- Kurajoh, M.; Fukumoto, S.; Emoto, M.; Murase, T.; Nakamura, T.; Ishihara, T.; Go, H.; Yamamoto, K.; Nakatani, S.; Tsuda, A.; et al. Independent association of plasma xanthine oxidoreductase activity with serum uric acid level based on stable isotope-labeled xanthine and liquid chromatography/triple quadrupole mass spectrometry: MedCity21 health examination registry. Clin. Chem. Lab. Med. 2020, 58, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Glantzounis, G.K.; Tsimoyiannis, E.C.; Kappas, A.M.; Galaris, D.A. Uric acid and oxidative stress. Curr. Pharm. Des. 2005, 11, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Yardim-Akaydin, S.; Sepici, A.; Ozkan, Y.; Torun, M.; Simşek, B.; Sepici, V. Oxidation of uric acid in rheumatoid arthritis: Is allantoin a marker of oxidative stress? Free Radic. Res. 2004, 38, 623–628. [Google Scholar] [CrossRef]
- Kand’ár, R.; Záková, P. Allantoin as a marker of oxidative stress in human erythrocytes. Clin. Chem. Lab. Med. 2008, 46, 1270–1274. [Google Scholar] [CrossRef]
- Kand’ár, R.; Záková, P.; Muzáková, V. Monitoring of antioxidant properties of uric acid in humans for a consideration measuring of levels of allantoin in plasma by liquid chromatography. Clin. Chim. Acta 2006, 365, 249–256. [Google Scholar] [CrossRef]
- Zitnanová, I.; Korytár, P.; Aruoma, O.I.; Sustrová, M.; Garaiová, I.; Muchová, J.; Kalnovicová, T.; Pueschel, S.; Duracková, Z. Uric acid and allantoin levels in Down syndrome: Antioxidant and oxidative stress mechanisms? Clin. Chim. Acta 2004, 341, 139–146. [Google Scholar] [CrossRef]
- Kanďár, R.; Štramová, X.; Drábková, P.; Křenková, J. A monitoring of allantoin, uric acid, and malondialdehyde levels in plasma and erythrocytes after ten minutes of running activity. Physiol. Res. 2014, 63, 753–762. [Google Scholar] [CrossRef]
- Tang, Z.; Ye, W.; Chen, H.; Kuang, X.; Guo, J.; Xiang, M.; Peng, C.; Chen, X.; Liu, H. Role of purines in regulation of metabolic reprogramming. Purinergic Signal. 2019, 15, 423–438. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef]
- Szczurek, P.; Mosiichuk, N.; Woliński, J.; Yatsenko, T.; Grujic, D.; Lozinska, L.; Pieszka, M.; Święch, E.; Pierzynowski, S.G.; Goncharova, K. Oral uricase eliminates blood uric acid in the hyperuricemic pig model. PLoS ONE 2017, 12, e0179195. [Google Scholar] [CrossRef]
- Agnoletti, D.; Cicero, A.F.G.; Borghi, C. The Impact of Uric Acid and Hyperuricemia on Cardiovascular and Renal Systems. Cardiol. Clin. 2021, 39, 365–376. [Google Scholar] [CrossRef]
- Mengozzi, A.; Pugliese, N.R.; Desideri, G.; Masi, S.; Angeli, F.; Barbagallo, C.M.; Bombelli, M.; Cappelli, F.; Casiglia, E.; Cianci, R.; et al. Serum Uric Acid Predicts All-Cause and Cardiovascular Mortality Independently of Hypertriglyceridemia in Cardiometabolic Patients without Established CV Disease: A Sub-Analysis of the URic acid Right for heArt Health (URRAH) Study. Metabolites 2023, 13, 244. [Google Scholar] [CrossRef]
- Czerska, M.; Mikołajewska, K.; Zieliński, M.; Gromadzińska, J.; Wąsowicz, W. Today’s oxidative stress markers. Occup. Med. 2015, 66, 393–405. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Gene | Transporter | Function | Location | Gene Mutation Result |
|---|---|---|---|---|
| SLC22A12 | URAT1 | Reabsorbs glomerular-filtrated UA | Luminal membrane of proximal renal tubule | Hypouricemia |
| SLC2A9 | GLUT9 | Allows intracellular UA to exit through the basolateral side of the cells | Basolateral membrane of proximal renal tubule | Hypouricemia |
| SLC17A3 | NPT4 | UA excretion | Luminal membrane of proximal renal tubule | Hyperuricemia |
| ABCG2 | ABCG2 | UA excretion | Luminal membrane of proximal renal tubule | Hyperuricemia |
| SLC17A1 | NPT1 | UA excretion | Luminal membrane of proximal renal tubule | Hyperuricemia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicero, A.F.G.; Fogacci, F.; Di Micoli, V.; Angeloni, C.; Giovannini, M.; Borghi, C. Purine Metabolism Dysfunctions: Experimental Methods of Detection and Diagnostic Potential. Int. J. Mol. Sci. 2023, 24, 7027. https://doi.org/10.3390/ijms24087027
Cicero AFG, Fogacci F, Di Micoli V, Angeloni C, Giovannini M, Borghi C. Purine Metabolism Dysfunctions: Experimental Methods of Detection and Diagnostic Potential. International Journal of Molecular Sciences. 2023; 24(8):7027. https://doi.org/10.3390/ijms24087027
Chicago/Turabian StyleCicero, Arrigo F. G., Federica Fogacci, Valentina Di Micoli, Cristina Angeloni, Marina Giovannini, and Claudio Borghi. 2023. "Purine Metabolism Dysfunctions: Experimental Methods of Detection and Diagnostic Potential" International Journal of Molecular Sciences 24, no. 8: 7027. https://doi.org/10.3390/ijms24087027