
Inner Amino Acid Contacts Are Key Factors of Multistage Structural Rearrangements of DNA and Affect Substrate Specificity of Apurinic/Apyrimidinic Endonuclease APE1
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of Amino Acid Residues Facilitating the Widening of the Active-Site Pocket
2.2. Comparison of F-Site Cleavage Efficiency by Mutant zAPE1 Enzymes
2.3. Efficiency of Cleavage of Damaged-Base-Containing DNA Substrates
2.4. Stopped-Flow Fluorescence Measurement
2.5. EPR Measurements
3. Materials and Methods
3.1. Site-Directed Mutagenesis and Protein Purification
3.2. DNA Substrates
3.3. DEER Sample Preparation
3.4. EPR Measurements
3.5. DEER Data Processing
3.6. PAGE Analysis of the DNA Cleavage
3.7. Stopped-Flow Fluorescence Detection of Interactions with DNA Substrates
3.8. Global Fitting of the Stopped-Flow Data
3.9. MST Titration
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lesiak, K.B.; Wheeler, K.T. Formation of Alpha-Deoxyadenosine in Polydeoxynucleotides Exposed to Ionizing Radiation under Anoxic Conditions. Radiat Res. 1990, 121, 328–337. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Laval, J.; Boiteux, S. Substrate Specificity of the Escherichia Coli Endonuclease III: Excision of Thymine- and Cytosine-Derived Lesions in DNA Produced by Radiation-Generated Free Radicals. Biochemistry 1993, 32, 12105–12111. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Keynote: Past, Present, and Future Aspects of Base Excision Repair. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 17–30. [Google Scholar] [CrossRef]
- Ide, H.; Tedzuka, K.; Shimzu, H.; Kimura, Y.; Purmal, A.A.; Wallace, S.S.; Kow, Y.W. Alpha.-Deoxyadenosine, a Major Anoxic Radiolysis Product of Adenine in DNA, Is a Substrate for Escherichia Coli Endonuclease IV. Biochemistry 1994, 33, 7842–7847. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Ide, H.; Ramotar, D.; Nevinsky, G.; Saparbaev, M. Alpha-Anomeric Deoxynucleotides, Anoxic Products of Ionizing Radiation, Are Substrates for the Endonuclease IV-Type AP Endonucleases. Biochemistry 2004, 43, 15210–15216. [Google Scholar] [CrossRef] [PubMed]
- Hang, B. Differential Cleavage of Oligonucleotides Containing the Benzene- Derived Adduct, 1,N6-Benzetheno-DA, by the Major Human AP Endonuclease HAP1 and Escherichia Coli Exonuclease III and Endonuclease IV. Carcinogenesis 1998, 19, 1339–1343. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.M. Properties of and Substrate Determinants for the Exonuclease Activity of Human Apurinic Endonuclease Ape1. J. Mol. Biol. 2003, 330, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Dyrkheeva, N.S.; Khodyreva, S.N.; Lavrik, O.I. Multifunctional Human Apurinic/Apyrimidinic Endonuclease 1: Role of Additional Functions. Mol. Biol. 2007, 41, 402–416. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′-5′ Exonuclease Activity of Human AP-Endonuclease APE1. Molecules 2018, 23, 2101. [Google Scholar] [CrossRef] [Green Version]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Comparative Analysis of Exo-and Endonuclease Activities of APE1-like Enzymes. Int. J. Mol. Sci. 2022, 23, 2869. [Google Scholar] [CrossRef]
- Senchurova, S.I.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The Kinetic Mechanism of 3′-5′ Exonucleolytic Activity of AP Endonuclease Nfo from E. coli. Cells 2022, 11, 2998. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Gavrilova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces Cerevisiae. Int. J. Mol. Sci 2022, 23, 14404. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 Protein Has a 3′-5′ Exonuclease Activity That Acts Preferentially on Mismatched Base Pairs. Nucleic. Acids. Res 2006, 34, 2508–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, B.D.; Berman, Z.; Mueller, G.A.; Lin, Y.; Chang, T.; Andres, S.N.; Wojtaszek, J.L.; DeRose, E.F.; Appel, C.D.; London, R.E.; et al. APE2 Zf-GRF Facilitates 3′-5′ Resection of DNA Damage Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 2017, 114, 304–309. [Google Scholar] [CrossRef]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. APE1 Senses DNA Single-Strand Breaks for Repair and Signaling. Nucleic. Acids. Res 2020, 48, 1925–1940. [Google Scholar] [CrossRef] [Green Version]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 Is Required for ATR-Chk1 Checkpoint Activation in Response to Oxidative Stress. Proc. Natl. Acad. Sci. USA 2013, 48, 1925–1940. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Cupello, S.; Hossain, M.A.; Deem, B.; McLeod, M.; Raj, J.; Yan, S. APE2 Promotes DNA Damage Response Pathway from a Single-Strand Break. Nucleic. Acids. Res 2018, 46, 2479–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilay, G.; Walker, L.J.; Robson, C.N.; Hickson, I.D. Site-Directed Mutagenesis of the Human DNA Repair Enzyme HAP1: Identification of Residues Important for AP Endonuclease and RNase H Activity. Nucleic. Acids. Res. 1995, 23, 1544–1550. [Google Scholar] [CrossRef] [Green Version]
- Berquist, B.R.; McNeill, D.R.; Wilson, D.M., 3rd. Characterization of Abasic Endonuclease Activity of Human Ape1 on Alternative Substrates, as Well as Effects of ATP and Sequence Context on AP Site Incision. J. Mol. Biol. 2008, 379, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.A.; Gavrilova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of APE1 Endoribonuclease Activity. Mol. Biol 2021, 55, 211–224. [Google Scholar] [CrossRef]
- Kim, S.E.; Gorrell, A.; Rader, S.D.; Lee, C.H. Endoribonuclease Activity of Human Apurinic/Apyrimidinic Endonuclease 1 Revealed by a Real-Time Fluorometric Assay. Anal. Biochem. 2010, 398, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 Toward Damaged DNA and Native RNA With Non-Canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Effect of the Substrate Structure and Metal Ions on the Hydrolysis of Undamaged RNA by Human AP Endonuclease APE1. Acta Nat. 2020, 12, 74–85. [Google Scholar] [CrossRef]
- Piergentili, R.; Gullo, G.; Basile, G.; Gulia, C.; Porrello, A.; Cucinella, G.; Marinelli, E.; Zaami, S. Circulating MiRNAs as a Tool for Early Diagnosis of Endometrial Cancer–Implications for the Fertility-Sparing Process: Clinical, Biological and Legal Aspects. Int. J. Mol. Sci. 2023, 24, 11356. [Google Scholar] [CrossRef]
- Barnes, T.; Kim, W.C.; Mantha, A.K.; Kim, S.E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of Apurinic/Apyrimidinic Endonuclease 1 (APE1) as the Endoribonuclease That Cleaves c-Myc MRNA. Nucleic. Acids. Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuninger, D.T.; Izumi, T.; Papaconstantinou, J.; Mitra, S. Human AP-Endonuclease 1 and HnRNP-L Interact with a NCaRE-like Repressor Element in the AP-Endonuclease 1 Promoter. Nucleic. Acids. Res. 2002, 30, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Rossbach, O.; Hung, L.-H.; Schreiner, S.; Grishina, I.; Heiner, M.; Hui, J.; Bindereif, A. Auto- and Cross-Regulation of the HnRNP L Proteins by Alternative Splicing. Mol. Cell Biol. 2009, 29, 1442–1451. [Google Scholar] [CrossRef] [Green Version]
- Redrejo-Rodríguez, M.; Vigouroux, A.; Mursalimov, A.; Grin, I.; Alili, D.; Koshenov, Z.; Akishev, Z.; Maksimenko, A.; Bissenbaev, A.K.; Matkarimov, B.T.; et al. Structural Comparison of AP Endonucleases from the Exonuclease III Family Reveals New Amino Acid Residues in Human AP Endonuclease 1 That Are Involved in Incision of Damaged DNA. Biochimie 2016, 128–129, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Davletgildeeva, A.T.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The Enigma of Substrate Recognition and Catalytic Efficiency of APE1-Like Enzymes. Front. Cell Dev. Biol. 2021, 9, 617161. [Google Scholar] [CrossRef]
- Bulygin, A.A.; Fedorova, O.S.; Kuznetsov, N. Insights into Mechanisms of Damage Recognition and Catalysis by APE1-like Enzymes. Int. J. Mol. Sci. 2022, 23, 4361. [Google Scholar] [CrossRef]
- Bulygin, A.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Active-Site Plasticity in Damaged-Nucleotide Recognition by Human Apurinic/Apyrimidinic Endonuclease APE1. Molecules 2020, 25, 3940. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.C.; Loeb, L.A. Mutations in the A8 Loop of Human APE1 Alter Binding and Cleavage of DNA Containing an Abasic Site. J. Biol. Chem. 2003, 278, 46994–47001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, T.; Schein, C.H.; Oezguen, N.; Feng, Y.; Braun, W. Effects of Backbone Contacts 3′ to the Abasic Site on the Cleavage and the Product Binding by Human Apurinic/Apyrimidinic Endonuclease (APE1). Biochemistry 2004, 43, 684–689. [Google Scholar] [CrossRef]
- Alekseeva, I.V.; Kuznetsova, A.A.; Bakman, A.S.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Active-Site Amino Acid Residues in the Cleavage of DNA and RNA Substrates by Human Apurinic/Apyrimidinic Endonuclease APE1. BBA Gen. Subj. 2020, 1864, 129718. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, I.V.; Bakman, A.S.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. Role of Ionizing Amino Acid Residues in the Process of DNA Binding by Human AP Endonuclease 1 and in Its Catalysis. J. Phys. Chem. B 2019, 123, 9546–9556. [Google Scholar] [CrossRef] [PubMed]
- Kuzmic, P. Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Matveeva, A.G.; Milov, A.D.; Vorobjev, Y.N.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. Substrate Specificity of Human Apurinic/Apyrimidinic Endonuclease APE1 in the Nucleotide Incision Repair Pathway. Nucleic Acids Res. 2018, 46, 11454–11465. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D. Spin-Label Electron Paramagnetic Resonance Spectroscopy; SRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Steinhoff, H.J.; Radzwill, N.; Thevis, W.; Lenz, V.; Brandenburg, D.; Antson, A.; Dodson, G.; Wollmeri, A. Determination of Interspin Distances between Spin Labels Attached to Insulin: Comparison of Electron Paramagnetic Resonance Data with the X-Ray Structure. Biophys. J. 1997, 73, 3287. [Google Scholar] [CrossRef]
- Tsvetkov, Y.D.; Milov, A.D.; Maryasov, A.G. Pulsed Electron–Electron Double Resonance (PELDOR) as EPR Spectroscopy in Nanometre Range. Russ. Chem. Rev. 2008, 77, 487–520. [Google Scholar] [CrossRef]
- Milov, A.D.; Salikhov, K.M.; Shirov, M.D. Application of ELDOR in Electron-Spin Echo for Paramagnetic Center Space Distribution in Solids. Fiz. Tverd. Tela 1981, 23, 975–982. [Google Scholar]
- Schiemann, O.; Cekan, P.; Margraf, D.; Prisner, T.F.; Sigurdsson, S.T. Relative Orientation of Rigid Nitroxides by PELDOR: Beyond Distance Measurements in Nucleic Acids. Angew. Chem. Int. Ed. Engl. 2009, 48, 3292–3295. [Google Scholar] [CrossRef] [PubMed]
- Abé, C.; Klose, D.; Dietrich, F.; Ziegler, W.H.; Polyhach, Y.; Jeschke, G.; Steinhoff, H.-J. Orientation Selective DEER Measurements on Vinculin Tail at X-Band Frequencies Reveal Spin Label Orientations. J. Magn. Reson. 2012, 216, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Senchurova, S.I.; Syryamina, V.N.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. The Mechanism of Damage Recognition by Apurinic/Apyrimidinic Endonuclease Nfo from Escherichia Coli. BBA Gen. Subj. 2022, 1866, 130216. [Google Scholar] [CrossRef] [PubMed]
- Milov, A.D.; Maryasov, A.G.; Tsvetkov, Y.D. Pulsed Electron Double Resonance (PELDOR) and Its Applications in Free-Radicals Research. Appl. Magn. Reson. 1998, 15, 107–143. [Google Scholar] [CrossRef]
- Dzuba, S.A. Conducting a Three-Pulse DEER Experiment without Dead Time: A Review. J. Magn. Reson. Open 2023, 14–15, 100100. [Google Scholar] [CrossRef]
- Milov, A.D.; Grishin, Y.A.; Dzuba, S.A.; Tsvetkov, Y.D. Effect of Pumping Pulse Duration on Echo Signal Amplitude in Four-Pulse PELDOR. Appl. Magn. Reson 2011, 41, 59–67. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Milov, A.D.; Koval, V.V.; Samoilova, R.I.; Grishin, Y.A.; Knorre, D.G.; Tsvetkov, Y.D.; Fedorova, O.S.; Dzuba, S.A. PELDOR Study of Conformations of Double-Spin-Labeled Single- and Double-Stranded DNA with Non-Nucleotide Inserts. Phys. Chem. Chem. Phys. 2009, 11, 6826–6832. [Google Scholar] [CrossRef]
- Milov, A.D.; Tsvetkov, Y.D. No Charge Effect on Relative Distance Distribution of Fremy’s Radical Ions in Frozen Glassy Solution Studied by PELDORTitle. AMR 2000, 18, 217–226. [Google Scholar]
- Milov, A.D.; Tsvetkov, Y.D.; Maryasov, A.G.; Gobbo, M.; Prinzivalli, C.; De Zotti, M.; Toniolo, C. Conformational Properties of the Spin-Labeled Tylopeptin B and Heptaibin Peptaibiotics Based on PELDOR Spectroscopy Data. Appl. Magn. Reson 2013, 44, 495–508. [Google Scholar] [CrossRef]
- Fajer, P.G.; Brown, L.; Song, L. Practical Pulsed Dipolar ESR (DEER). In ESR Spectroscopy in Membrane Biophysics; Springer: Boston, MA, USA, 2007. [Google Scholar]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006 —A Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Magn. Reson 2006, 30, 473–498. [Google Scholar] [CrossRef]
- Dzuba, S.A. The Determination of Pair-Distance Distribution by Double Electron–Electron Resonance: Regularization by the Length of Distance Discretization with Monte Carlo Calculations. J. Magn. Reson. 2016, 269, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, A.G.; Yushkova, Y.V.; Morozov, S.V.; Grygor’ev, I.A.; Dzuba, S.A. Multi-Gaussian Monte Carlo Analysis of PELDOR Data in the Frequency Domain. Z. Fur Phys. Chem. Int. J. Res. Phys. Chem. Chem. Phys. 2017, 231, 671–688. [Google Scholar] [CrossRef]
- Schiemann, O.; Heubach, C.A.; Abdullin, D.; Ackermann, K.; Azarkh, M.; Bagryanskaya, E.G.; Drescher, M.; Endeward, B.; Freed, J.H.; Galazzo, L.; et al. Benchmark Test and Guidelines for DEER/PELDOR Experiments on Nitroxide-Labeled Biomolecules. J. Am. Chem. Soc. 2021, 143, 17875–17890. [Google Scholar] [CrossRef]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. Effects of Mono- and Divalent Metal Ions on DNA Binding and Catalysis of Human Apurinic/Apyrimidinic Endonuclease 1. Mol. BioSyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.A.; Senchurova, S.I.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Common Kinetic Mechanism of Abasic Site Recognition by Structurally Different Apurinic/Apyrimidinic Endonucleases. Int. J. Mol. Sci. 2021, 22, 8874. [Google Scholar] [CrossRef]
- Bakman, A.S.; Kuznetsova, A.A.; Yanshole, L.V.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Fluorescently Labeled Human Apurinic/Apyrimidinic Endonuclease APE1 Reveals Effects of DNA Polymerase β on the APE1–DNA Interaction. DNA Repair 2023, 123, 103450. [Google Scholar] [CrossRef] [PubMed]










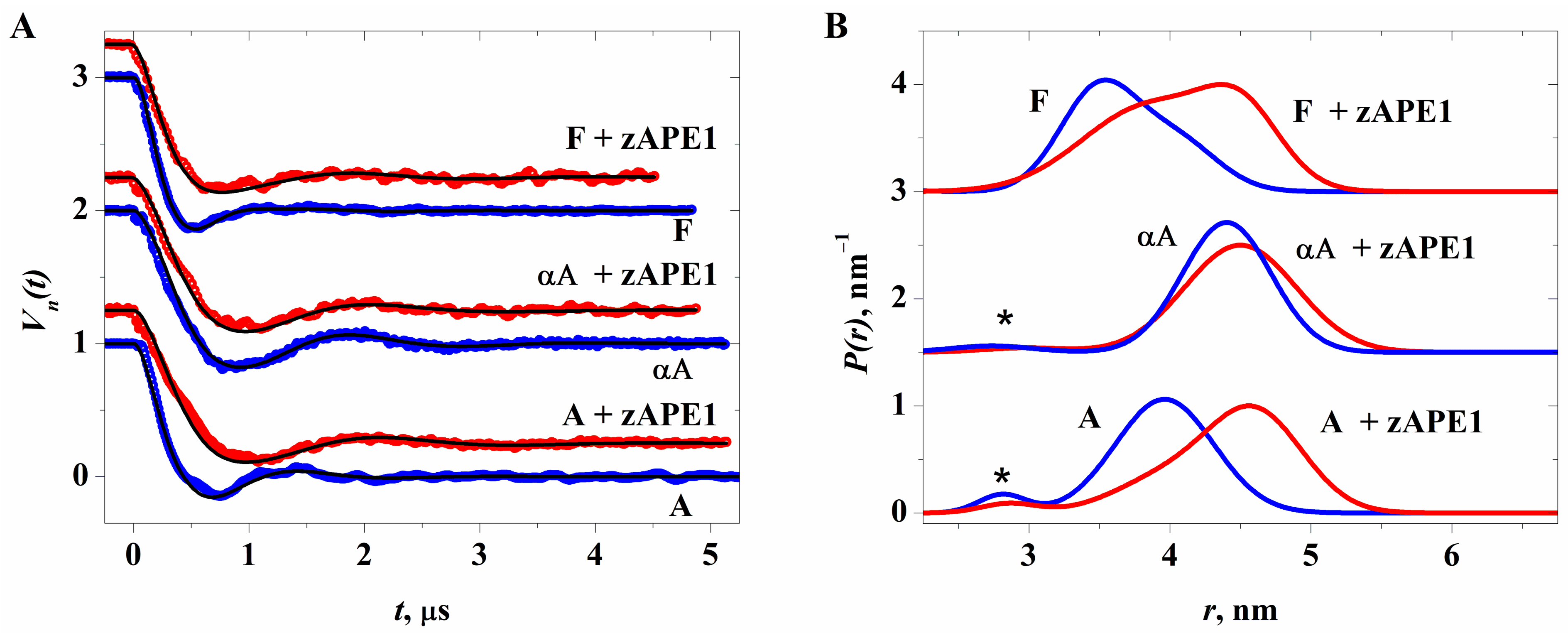

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | WT | N253G | A254G | E260A |
|---|---|---|---|---|
| k1, s−1 | 91 | 85 | N/A | N/A |
| A1 | −0.42 | −0.39 | N/A | N/A |
| k2, s−1 | 20 | 17.4 | 7.5 | 1.6 |
| A2 | 1.63 | 1.6 | 1.2 | 1.0 |
| Ka, µM−1 | 3 ± 0.7 | 2.5 ± 0.5 | 2 ± 0.4 | 0.9 ± 0.2 |
| Constants | N253G | A254G | E260A |
|---|---|---|---|
| k1, μM−1s−1 | 29 ± 2 | 24 ± 3 | 11 ± 2 |
| k−1, s−1 | 12 ± 2 | 12 ± 2 | 13 ± 3 |
| Ka *, μM−1 | 2.5 ± 0.5 | 2.0 ± 0.4 | 0.9 ± 0.2 |
| kcat, s−1 | 13 ± 2 | 8 ± 1 | 4.0 ± 0.5 |
| Kp, μM | 4 ± 1 | 7 ± 1 | 10 ± 2 |
| Sample | A, % | rmax, nm | δr, nm |
|---|---|---|---|
| Undamaged DNA a | 100 | 3.94 ± 0.02 | 0.36 ± 0.02 |
| Undamaged DNA–zAPE1 complex | 24 ± 2/72 ± 2 | 3.94 ± 0.02/4.60 ± 0.02 | 0.36 ± 0.02/0.34 ± 0.02 |
| αA-substrate a | 100 | 4.37 ± 0.03 | 0.33 ± 0.03 |
| αA-substrate–zAPE1 complex | 100 | 4.50 ± 0.02 | 0.40 ± 0.02 |
| F-substrate a | 65 ± 8/35 ± 8 | 3.45 ± 0.05/3.98 ± 0.06 | 0.32 ± 0.04/0.32 ± 0.04 |
| F-substrate–zAPE1 complex | 68 ± 7/31 ± 7 | 3.86 ± 0.04/4.51 ± 0.03 | 0.48 ± 0.05/0.26 ± 0.06 |
 | |
|---|---|
| Shorthand | Sequences of DNA Duplexes |
| X-substrate (FRET) X = C, F-site, DHU, dU, N = G X = αA, εA, N = T | 5′-FAM-GCTCAXGTACAGAGCTG-3′ 3′-CGAGTNCATGTCTCGAC-BHQ1-5′ |
| X-substrate (DEER) X = F-site, αA or A | 5′-CGURCTCTGTACATGURGC-3′ 3′-GCAGAGACXTGTACACG-5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulygin, A.A.; Syryamina, V.N.; Kuznetsova, A.A.; Novopashina, D.S.; Dzuba, S.A.; Kuznetsov, N.A. Inner Amino Acid Contacts Are Key Factors of Multistage Structural Rearrangements of DNA and Affect Substrate Specificity of Apurinic/Apyrimidinic Endonuclease APE1. Int. J. Mol. Sci. 2023, 24, 11474. https://doi.org/10.3390/ijms241411474
Bulygin AA, Syryamina VN, Kuznetsova AA, Novopashina DS, Dzuba SA, Kuznetsov NA. Inner Amino Acid Contacts Are Key Factors of Multistage Structural Rearrangements of DNA and Affect Substrate Specificity of Apurinic/Apyrimidinic Endonuclease APE1. International Journal of Molecular Sciences. 2023; 24(14):11474. https://doi.org/10.3390/ijms241411474
Chicago/Turabian StyleBulygin, Anatoly A., Victoria N. Syryamina, Aleksandra A. Kuznetsova, Darya S. Novopashina, Sergei A. Dzuba, and Nikita A. Kuznetsov. 2023. "Inner Amino Acid Contacts Are Key Factors of Multistage Structural Rearrangements of DNA and Affect Substrate Specificity of Apurinic/Apyrimidinic Endonuclease APE1" International Journal of Molecular Sciences 24, no. 14: 11474. https://doi.org/10.3390/ijms241411474