
Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes

 , and
, and 
Abstract
1. Introduction
2. Definitions
2.1. World Health Organization
2.2. International Census Classification
3. Diagnosis
3.1. Presentation
3.2. Immunohistochemistry and Flow Cytometry
3.3. Cytogenetic Characteristics
4. Molecular Biology and Genomic Features of AEL
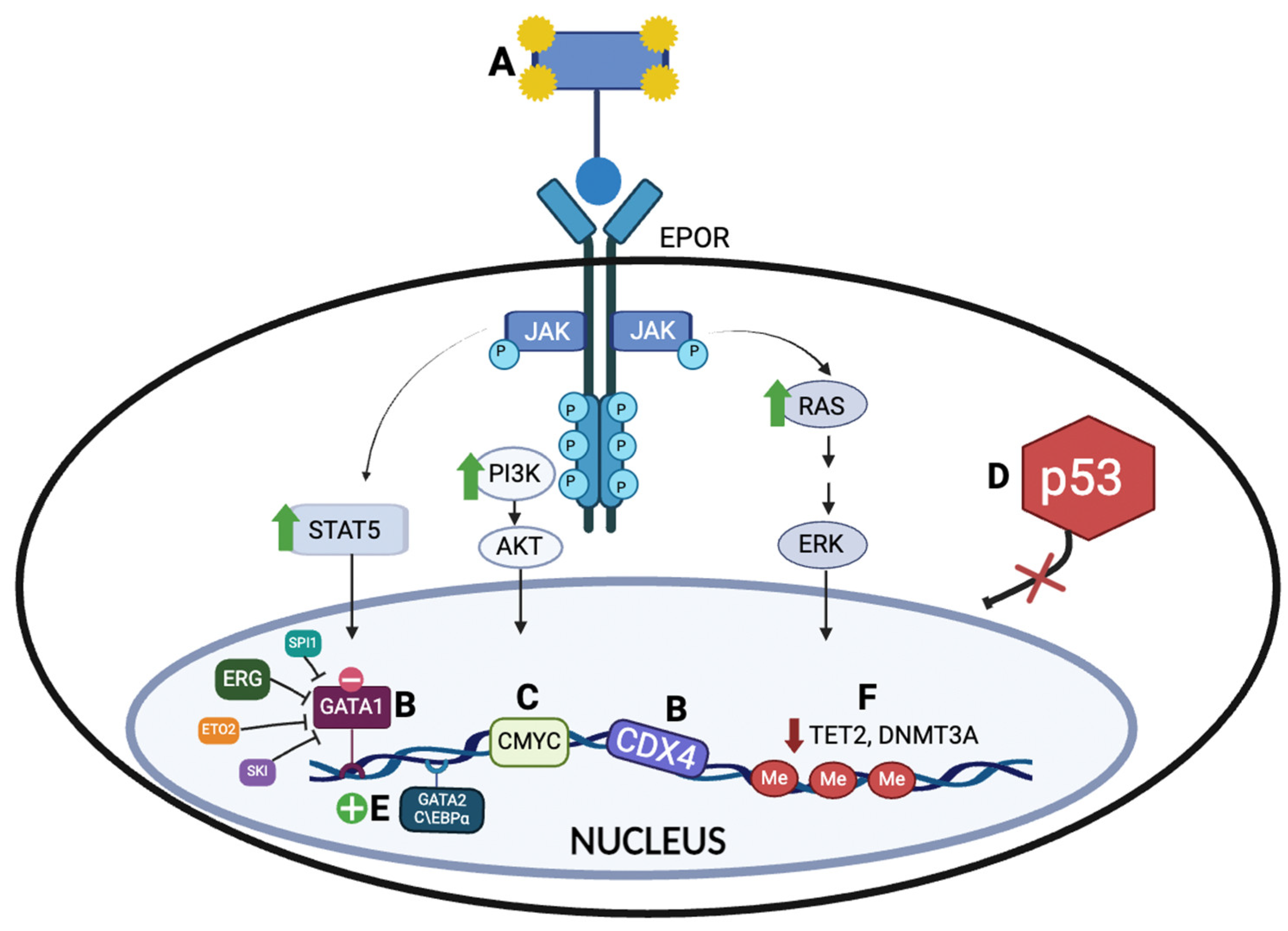
4.1. Erythropoietin Receptor (EPOR) Activation and Downstream JAK2 Signaling Pathway
4.2. Erythroid Transcriptional Regulators
4.2.1. GATA Binding Protein 1 (GATA1)
4.2.2. ETS Transcription Factors (ERG, SPI1, and FLI1)
4.2.3. Caudal-Type Homeobox 4 (CDX4)
4.3. Expression of Master Oncogenes
4.4. Impaired TP53 Activity in AEL Biology
4.5. Impaired C/EBPα Function
GATA2 and C\EBPα
4.6. Epigenetic Dysregulation in Erythroleukemia
4.7. Other Less Frequent Genetic Alterations
5. Treatments and Clinical Outcomes
5.1. Intensive Chemotherapy
5.2. Hypomethylating Agents
Venetoclax
5.3. Allogeneic Bone Marrow Transplant
5.4. Chimeric Antigen Receptor T-Cell Therapy
5.5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zuo, Z.; Polski, J.M.; Kasyan, A.; Medeiros, L.J. Acute Erythroid Leukemia. Arch. Pathol. Lab. Med. 2010, 134, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Cervera, N.; Guille, A.; Adélaïde, J.; Hospital, M.-A.; Garciaz, S.; Mozziconacci, M.-J.; Vey, N.; Gelsi-Boyer, V.; Birnbaum, D. Erythroleukemia: Classification. EJHaem 2023, 4, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Prebet, T.; Itzykson, R.; Ramos, F.; Al-Ali, H.; Shammo, J.; Pinto, R.; Maurillo, L.; Wetzel, J.; Musto, P.; et al. Clinical Outcomes of 217 Patients with Acute Erythroleukemia According to Treatment Type and Line: A Retrospective Multinational Study. Int. J. Mol. Sci. 2017, 18, 837. [Google Scholar] [CrossRef] [PubMed]
- Novik, Y.; Marino, P.; Makower, D.F.; Wiernik, P.H. Familial Erythroleukemia: A Distinct Clinical and Genetic Type of Familial Leukemias. Leuk. Lymphoma 1998, 30, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C. A History and Current Understanding of Acute Erythroid Leukemia. Clin. Lymphoma Myeloma Leuk. 2023, 23, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L. 100-Year Old Haematologica Images: Di Guglielmo Disease or Pure Erythroid Leukemia. Haematologica 2020, 105, 525. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Reichard, K.K.; Tefferi, A.; Abdelmagid, M.; Orazi, A.; Alexandres, C.; Haack, J.; Greipp, P.T. Pure (Acute) Erythroid Leukemia: Morphology, Immunophenotype, Cytogenetics, Mutations, Treatment Details, and Survival Data among 41 Mayo Clinic Cases. Blood Cancer J. 2022, 12, 147. [Google Scholar] [CrossRef]
- Kasyan, A.; Medeiros, L.J.; Zuo, Z.; Santos, F.P.; Ravandi-Kashani, F.; Miranda, R.; Vadhan-Raj, S.; Koeppen, H.; Bueso-Ramos, C.E. Acute Erythroid Leukemia as Defined in the World Health Organization Classification Is a Rare and Pathogenetically Heterogeneous Disease. Mod. Pathol. 2010, 23, 1113–1126. [Google Scholar] [CrossRef]
- Acharya, S.; Kala, P.S. Role of CD71 in Acute Leukemia- An Immunophenotypic Marker for Erythroid Lineage or Proliferation? Indian J. Pathol. Microbiol. 2019, 62, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Kaumeyer, B.A.; Fidai, S.S.; Thakral, B.; Wang, S.A.; Arber, D.A.; Cheng, J.X.; Gurbuxani, S.; Venkataraman, G. GLUT1 Immunohistochemistry Is a Highly Sensitive and Relatively Specific Marker for Erythroid Lineage in Benign and Malignant Hematopoietic Tissues. Am. J. Clin. Pathol. 2022, 158, 228–234. [Google Scholar] [CrossRef]
- Fang, H.; Wang, S.A.; Khoury, J.D.; El Hussein, S.; Kim, D.H.; Tashakori, M.; Tang, Z.; Li, S.; Hu, Z.; Jelloul, F.Z.; et al. Pure Erythroid Leukemia Is Characterized by Biallelic TP53 Inactivation and Abnormal P53 Expression Patterns in de Novo and Secondary Cases. Haematologica 2022, 107, 2232–2237. [Google Scholar] [CrossRef] [PubMed]
- Gera, K.; Martir, D.; Xue, W.; Wingard, J.R. Survival after Pure (Acute) Erythroid Leukemia in the United States: A SEER-Based Study. Cancers 2023, 15, 3941. [Google Scholar] [CrossRef]
- Campo, E.; Cymbalista, F.; Ghia, P.; Jäger, U.; Pospisilova, S.; Rosenquist, R.; Schuh, A.; Stilgenbauer, S. TP53 Aberrations in Chronic Lymphocytic Leukemia: An Overview of the Clinical Implications of Improved Diagnostics. Haematologica 2018, 103, 1956–1968. [Google Scholar] [CrossRef]
- Mazzella, F.M.; Kowal-Vern, A.; Shrit, M.A.; Wibowo, A.L.; Rector, J.T.; Cotelingam, J.D.; Collier, J.; Mikhael, A.; Cualing, H.; Schumacher, H.R. Acute Erythroleukemia: Evaluation of 48 Cases with Reference to Classification, Cell Proliferation, Cytogenetics, and Prognosis. Am. J. Clin. Pathol. 1998, 110, 590–598. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Zuo, Z.; Garcia, C.; Tang, G.; Kasyan, A.; Luthra, R.; Abruzzo, L.V.; Kantarjian, H.M.; Medeiros, L.J.; Wang, S.A. Acute Erythroid Leukemia: A Reassessment Using Criteria Refined in the 2008 WHO Classification. Blood 2010, 115, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Neaga, A.; Jimbu, L.; Mesaros, O.; Bota, M.; Lazar, D.; Cainap, S.; Blag, C.; Zdrenghea, M. Why Do Children with Acute Lymphoblastic Leukemia Fare Better Than Adults? Cancers 2021, 13, 3886. [Google Scholar] [CrossRef]
- Grossmann, V.; Bacher, U.; Haferlach, C.; Schnittger, S.; Pötzinger, F.; Weissmann, S.; Roller, A.; Eder, C.; Fasan, A.; Zenger, M.; et al. Acute Erythroid Leukemia (AEL) Can Be Separated into Distinct Prognostic Subsets Based on Cytogenetic and Molecular Genetic Characteristics. Leukemia 2013, 27, 1940–1943. [Google Scholar] [CrossRef]
- Linu, J.A.; Udupa, M.N.; Madhumathi, D.S.; Lakshmaiah, K.C.; Babu, K.G.; Lokanatha, D.; Babu, M.S.; Lokesh, K.N.; Rajeev, L.K.; Rudresha, A.H. Study of Clinical, Haematological and Cytogenetic Profile of Patients with Acute Erythroid Leukaemia. Ecancermedicalscience 2017, 11, 712. [Google Scholar] [CrossRef] [PubMed]
- Fagnan, A.; Piqué-Borràs, M.-R.; Tauchmann, S.; Mercher, T.; Schwaller, J. Molecular Landscapes and Models of Acute Erythroleukemia. HemaSphere 2021, 5, e558. [Google Scholar] [CrossRef] [PubMed]
- Ney, P.A.; D’Andrea, A.D. Friend Erythroleukemia Revisited. Blood 2000, 96, 3675–3680. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Longmore, G.; Lodish, H.F. Point Mutation in the Exoplasmic Domain of the Erythropoietin Receptor Resulting in Hormone-Independent Activation and Tumorigenicity. Nature 1990, 348, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Qu, C.; Varotto, E.; Janke, L.J.; Yang, X.; Seth, A.; Shelat, A.; Friske, J.D.; Fukano, R.; Yu, J.; et al. Modeling and Targeting of Erythroleukemia by Hematopoietic Genome Editing. Blood 2021, 137, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Rimmelé, P.; Kosmider, O.; Mayeux, P.; Moreau-Gachelin, F.; Guillouf, C. Spi-1/PU.1 Participates in Erythroleukemogenesis by Inhibiting Apoptosis in Cooperation with Epo Signaling and by Blocking Erythroid Differentiation. Blood 2007, 109, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Denis, N.; Lacout, C.; Vainchenker, W.; Dubreuil, P.; Moreau-Gachelin, F. Kit-Activating Mutations Cooperate with Spi-1/PU.1 Overexpression to Promote Tumorigenic Progression during Erythroleukemia in Mice. Cancer Cell 2005, 8, 467–478. [Google Scholar] [CrossRef]
- Takeda, J. Molecular pathogenesis and therapeutic targets in acute erythroid leukemia. Rinsho Ketsueki 2022, 63, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.; Yoshida, K.; Nakagawa, M.M.; Nannya, Y.; Yoda, A.; Saiki, R.; Ochi, Y.; Zhao, L.; Okuda, R.; Qi, X.; et al. Amplified EPOR/JAK2 Genes Define a Unique Subtype of Acute Erythroid Leukemia. Blood Cancer Discov. 2022, 3, 410–427. [Google Scholar] [CrossRef]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 Levels in Erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef]
- Fagnan, A.; Bagger, F.O.; Piqué-Borràs, M.-R.; Ignacimouttou, C.; Caulier, A.; Lopez, C.K.; Robert, E.; Uzan, B.; Gelsi-Boyer, V.; Aid, Z.; et al. Human Erythroleukemia Genetics and Transcriptomes Identify Master Transcription Factors as Functional Disease Drivers. Blood 2020, 136, 698–714. [Google Scholar] [CrossRef]
- Moreau-Gachelin, F.; Wendling, F.; Molina, T.; Denis, N.; Titeux, M.; Grimber, G.; Briand, P.; Vainchenker, W.; Tavitian, A. Spi-1/PU.1 Transgenic Mice Develop Multistep Erythroleukemias. Mol. Cell. Biol. 1996, 16, 2453–2463. [Google Scholar] [CrossRef]
- Carmichael, C.L.; Metcalf, D.; Henley, K.J.; Kruse, E.A.; Di Rago, L.; Mifsud, S.; Alexander, W.S.; Kile, B.T. Hematopoietic Overexpression of the Transcription Factor Erg Induces Lymphoid and Erythro-Megakaryocytic Leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 15437–15442. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, H.; Liu, T.; Zacksenhaus, E.; Ben-David, Y. The Ets Transcription Factor Fli-1 in Development, Cancer and Disease. Oncogene 2015, 34, 2022–2031. [Google Scholar] [CrossRef]
- Shimizu, R.; Kuroha, T.; Ohneda, O.; Pan, X.; Ohneda, K.; Takahashi, S.; Philipsen, S.; Yamamoto, M. Leukemogenesis Caused by Incapacitated GATA-1 Function. Mol. Cell. Biol. 2004, 24, 10814–10825. [Google Scholar] [CrossRef] [PubMed]
- Di Genua, C.; Valletta, S.; Buono, M.; Stoilova, B.; Sweeney, C.; Rodriguez-Meira, A.; Grover, A.; Drissen, R.; Meng, Y.; Beveridge, R.; et al. C/EBPα and GATA-2 Mutations Induce Bilineage Acute Erythroid Leukemia through Transformation of a Neomorphic Neutrophil-Erythroid Progenitor. Cancer Cell 2020, 37, 690–704. [Google Scholar] [CrossRef]
- Iacobucci, I.; Wen, J.; Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds, S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et al. Genomic Subtyping and Therapeutic Targeting of Acute Erythroleukemia. Nat. Genet. 2019, 51, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Micci, F.; Thorsen, J.; Haugom, L.; Zeller, B.; Tierens, A.; Heim, S. Translocation t(1;16)(P31;Q24) Rearranging CBFA2T3 Is Specific for Acute Erythroid Leukemia. Leukemia 2011, 25, 1510–1512. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Micci, F.; Thorsen, J.; Haugom, L.; Buechner, J.; Kerndrup, G.; Tierens, A.; Zeller, B.; Heim, S. Fusion of ZMYND8 and RELA Genes in Acute Erythroid Leukemia. PLoS ONE 2013, 8, e63663. [Google Scholar] [CrossRef]
- Micci, F.; Thorsen, J.; Panagopoulos, I.; Nyquist, K.B.; Zeller, B.; Tierens, A.; Heim, S. High-Throughput Sequencing Identifies an NFIA/CBFA2T3 Fusion Gene in Acute Erythroid Leukemia with t(1;16)(P31;Q24). Leukemia 2013, 27, 980–982. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Aisaki, K.; Yamamura, Y.; Noda, M.; Ikawa, Y. Induction of Erythroid Differentiation by Inhibition of Ras/ERK Pathway in a Friend Murine Leukemia Cell Line. Oncogene 2000, 19, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Linnik, Y.; Pastakia, D.; Dryden, I.; Head, D.R.; Mason, E.F. Primary Central Nervous System Erythroid Sarcoma with NFIA-CBFA2T3 Translocation: A Rare but Distinct Clinicopathologic Entity. Am. J. Hematol. 2020, 95, E299–E301. [Google Scholar] [CrossRef] [PubMed]
- Ducker, C.; Shaw, P.E. Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development. Int. J. Mol. Sci. 2021, 22, 5119. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Thoms, J.A.I.; Perera, D.; Schütte, J.; Unnikrishnan, A.; Knezevic, K.; Kinston, S.J.; Wilson, N.K.; O’Brien, T.A.; Göttgens, B.; et al. Genome-Wide Analysis of Transcriptional Regulators in Human HSPCs Reveals a Densely Interconnected Network of Coding and Noncoding Genes. Blood 2013, 122, e12–e22. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.K.; Foster, S.D.; Wang, X.; Knezevic, K.; Schütte, J.; Kaimakis, P.; Chilarska, P.M.; Kinston, S.; Ouwehand, W.H.; Dzierzak, E.; et al. Combinatorial Transcriptional Control In Blood Stem/Progenitor Cells: Genome-Wide Analysis of Ten Major Transcriptional Regulators. Cell Stem Cell 2010, 7, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.J.; Rehn, M.; Hasemann, M.S.; Rapin, N.; Bagger, F.O.; Ohlsson, E.; Willer, A.; Frank, A.-K.; Søndergaard, E.; Jendholm, J.; et al. ERG Promotes the Maintenance of Hematopoietic Stem Cells by Restricting Their Differentiation. Genes Dev. 2015, 29, 1915–1929. [Google Scholar] [CrossRef] [PubMed]
- Baldus, C.D.; Burmeister, T.; Martus, P.; Schwartz, S.; Gökbuget, N.; Bloomfield, C.D.; Hoelzer, D.; Thiel, E.; Hofmann, W.K. High Expression of the ETS Transcription Factor ERG Predicts Adverse Outcome in Acute T-Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2006, 24, 4714–4720. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, M.J.; Crispino, J.D. ETS2 and ERG Promote Megakaryopoiesis and Synergize with Alterations in GATA-1 to Immortalize Hematopoietic Progenitor Cells. Blood 2009, 113, 3337–3347. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Somoza, C.; Shigematsu, H.; Duprez, E.A.; Iwasaki-Arai, J.; Mizuno, S.-I.; Arinobu, Y.; Geary, K.; Zhang, P.; Dayaram, T.; et al. Distinctive and Indispensable Roles of PU.1 in Maintenance of Hematopoietic Stem Cells and Their Differentiation. Blood 2005, 106, 1590–1600. [Google Scholar] [CrossRef]
- Athanasiou, M.; Mavrothalassitis, G.; Sun-Hoffman, L.; Blair, D.G. FLI-1 Is a Suppressor of Erythroid Differentiation in Human Hematopoietic Cells. Leukemia 2000, 14, 439–445. [Google Scholar] [CrossRef]
- Torchia, E.C.; Boyd, K.; Rehg, J.E.; Qu, C.; Baker, S.J. EWS/FLI-1 Induces Rapid Onset of Myeloid/Erythroid Leukemia in Mice. Mol. Cell. Biol. 2007, 27, 7918–7934. [Google Scholar] [CrossRef] [PubMed]
- Lengerke, C.; Daley, G.Q. Caudal Genes in Blood Development and Leukemia. Ann. N. Y. Acad. Sci. 2012, 1266, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Scholl, C.; Fröhling, S.; McDowell, E.; Lee, B.H.; Döhner, K.; Ernst, P.; Davidson, A.J.; Daley, G.Q.; Zon, L.I.; et al. Cdx4 Dysregulates Hox Gene Expression and Generates Acute Myeloid Leukemia Alone and in Cooperation with Meis1a in a Murine Model. Proc. Natl. Acad. Sci. USA 2006, 103, 16924–16929. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Huntly, B.J.; Wang, Y.; Chen, J.; Brumme, K.; Ball, B.; McKinney-Freeman, S.L.; Yabuuchi, A.; Scholl, C.; Bansal, D.; et al. Cdx4 Is Dispensable for Murine Adult Hematopoietic Stem Cells but Promotes MLL-AF9-Mediated Leukemogenesis. Haematologica 2010, 95, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Lachman, H.M. C-Myc Protooncogene Expression in Mouse Erythroleukemia Cells. Environ. Health Perspect. 1989, 80, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Lachman, H.M.; Skoultchi, A.I. Expression of C-Myc Changes during Differentiation of Mouse Erythroleukaemia Cells. Nature 1984, 310, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Skoda, R.C.; Tsai, S.F.; Orkin, S.H.; Leder, P. Expression of C-MYC under the Control of GATA-1 Regulatory Sequences Causes Erythroleukemia in Transgenic Mice. J. Exp. Med. 1995, 181, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Robert-Lézénès, J.; Meneceur, P.; Ray, D.; Moreau-Gachelin, F. Protooncogene Expression in Normal, Preleukemic, and Leukemic Murine Erythroid Cells and Its Relationship to Differentiation and Proliferation. Cancer Res. 1988, 48, 3972–3976. [Google Scholar] [PubMed]
- Tennant, R.W.; Stasiewicz, S.; Eastin, W.C.; Mennear, J.H.; Spalding, J.W. The Tg.AC (v-Ha-Ras) Transgenic Mouse: Nature of the Model. Toxicol. Pathol. 2001, 29, 51–59. [Google Scholar] [CrossRef]
- Leder, A.; Kuo, A.; Cardiff, R.D.; Sinn, E.; Leder, P. V-Ha-Ras Transgene Abrogates the Initiation Step in Mouse Skin Tumorigenesis: Effects of Phorbol Esters and Retinoic Acid. Proc. Natl. Acad. Sci. USA 1990, 87, 9178–9182. [Google Scholar] [CrossRef]
- Trempus, C.S.; Ward, S.; Farris, G.; Malarkey, D.; Faircloth, R.S.; Cannon, R.E.; Mahler, J.F. Association of V-Ha-Ras Transgene Expression with Development of Erythroleukemia in Tg.AC Transgenic Mice. Am. J. Pathol. 1998, 153, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Trainor, C.D.; Mas, C.; Archambault, P.; Di Lello, P.; Omichinski, J.G. GATA-1 Associates with and Inhibits P53. Blood 2009, 114, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; Leduc, M.; Gautier, E.-F.; Guyot, B.; et al. P53 Activation during Ribosome Biogenesis Regulates Normal Erythroid Differentiation. Blood 2021, 137, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Liu, Y.; Bae, N.; Nimer, S.D. The P53 Tumor Suppressor Protein Regulates Hematopoietic Stem Cell Fate. J. Cell. Physiol. 2011, 226, 2215–2221. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional Diversity of P53 Proteins in Adult Acute Myeloid Leukemia: Projections on Diagnostic Workup and Therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.K.; Schenone, M.; Ferreira, M.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Germing, U.; Platzbecker, U.; et al. Rps14 Haploinsufficiency Causes a Block in Erythroid Differentiation Mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kong, G.; Rajagopalan, A.; Lu, L.; Song, J.; Hussaini, M.; Zhang, X.; Ranheim, E.A.; Liu, Y.; Wang, J.; et al. P53−/− Synergizes with Enhanced NrasG12D Signaling to Transform Megakaryocyte-Erythroid Progenitors in Acute Myeloid Leukemia. Blood 2017, 129, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zuber, J.; Diaz-Flores, E.; Lintault, L.; Kogan, S.C.; Shannon, K.; Lowe, S.W. P53 Loss Promotes Acute Myeloid Leukemia by Enabling Aberrant Self-Renewal. Genes Dev. 2010, 24, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta-Kishino, T.; Koya, J.; Kataoka, K.; Narukawa, K.; Sumitomo, Y.; Kobayashi, H.; Sato, T.; Kurokawa, M. Loss of P53 Induces Leukemic Transformation in a Murine Model of Jak2 V617F-Driven Polycythemia Vera. Oncogene 2017, 36, 3300–3311. [Google Scholar] [CrossRef]
- Piqué-Borràs, M.-R.; Jevtic, Z.; Bagger, F.O.; Seguin, J.; Sivalingam, R.; Bezerra, M.F.; Louwagie, A.; Juge, S.; Nellas, I.; Ivanek, R.; et al. The NFIA-ETO2 Fusion Blocks Erythroid Maturation and Induces Pure Erythroid Leukemia in Cooperation with Mutant TP53. Blood 2023, 141, 2245–2260. [Google Scholar] [CrossRef]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and Functional Analysis of Leukemic Transformation of Myeloproliferative Neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed]
- Ping, N.; Sun, A.; Song, Y.; Wang, Q.; Yin, J.; Cheng, W.; Xu, Y.; Wen, L.; Yao, H.; Ma, L.; et al. Exome Sequencing Identifies Highly Recurrent Somatic GATA2 and CEBPA Mutations in Acute Erythroid Leukemia. Leukemia 2017, 31, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Jeromin, S.; Grossmann, V.; Eder, C.; Weissmann, S.; Dicker, F.; Kohlmann, A.; Schindela, S.; et al. The Role of Different Genetic Subtypes of CEBPA Mutated AML. Leukemia 2014, 28, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Belizaire, R.; Wong, W.J.; Robinette, M.L.; Ebert, B.L. Clonal Haematopoiesis and Dysregulation of the Immune System. Nat. Rev. Immunol. 2023, 23, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; Yuita, H.; Rao, A. Dysregulation of the TET Family of Epigenetic Regulators in Lymphoid and Myeloid Malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.-H.; Rao, A.; et al. DNMT3A and TET2 Compete and Cooperate to Repress Lineage-Specific Transcription Factors in Hematopoietic Stem Cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 Gene Fusions and Hematopoietic Malignancies: Common Themes and New Biologic Insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, K.M.; Heerema-McKenney, A.E.; Choi, J.K.; Smith, J.; Ries, R.E.; Hirsch, B.A.; Raimondi, S.C.; Alonzo, T.A.; Wang, Y.-C.; Aplenc, R.; et al. Acute Erythroid Leukemia Is Enriched in NUP98 Fusions: A Report from the Children’s Oncology Group. Blood Adv. 2020, 4, 6000–6008. [Google Scholar] [CrossRef]
- Alkhateeb, H.B.; Damlaj, M.; Hefazi, M.; Dias, A.; Hashmi, S.K.; Hogan, W.J.; Litzow, M.R.; Patnaik, M.S. Allogeneic Hematopoietic Stem Cell Transplant Outcomes in Patients with Acute Erythroleukemia. Biol. Blood Marrow Transplant. 2016, 22, S195–S196. [Google Scholar] [CrossRef]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating Agents (HMA) for the Treatment of Acute Myeloid Leukemia and Myelodysplastic Syndromes: Mechanisms of Resistance and Novel HMA-Based Therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Kuusanmäki, H.; Dufva, O.; Vähä-Koskela, M.; Leppä, A.-M.; Huuhtanen, J.; Vänttinen, I.; Nygren, P.; Klievink, J.; Bouhlal, J.; Pölönen, P.; et al. Erythroid/Megakaryocytic Differentiation Confers BCL-XL Dependency and Venetoclax Resistance in Acute Myeloid Leukemia. Blood 2023, 141, 1610–1625. [Google Scholar] [CrossRef] [PubMed]
- Gottschlich, A.; Thomas, M.; Grünmeier, R.; Lesch, S.; Rohrbacher, L.; Igl, V.; Briukhovetska, D.; Benmebarek, M.-R.; Vick, B.; Dede, S.; et al. Single-Cell Transcriptomic Atlas-Guided Development of CAR-T Cells for the Treatment of Acute Myeloid Leukemia. Nat. Biotechnol. 2023, 41, 1618–1632. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.S.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Morales-Martínez, M.; Vega, M.I. Roles and Regulation of BCL-xL in Hematological Malignancies. Int. J. Mol. Sci. 2022, 23, 2193. [Google Scholar] [CrossRef]
- Poeta, G.; Bruno, A.; Del Principe, M.; Venditti, A.; Maurillo, L.; Buccisano, F.; Stasi, R.; Neri, B.; Luciano, F.; Siniscalchi, A.; et al. Deregulation of the Mitochondrial Apoptotic Machinery and Development of Molecular Targeted Drugs in Acute Myeloid Leukemia. Curr. Cancer Drug Targets 2008, 8, 207–222. [Google Scholar] [CrossRef]
- Andersson, A.; Ritz, C.; Lindgren, D.; Edén, P.; Lassen, C.; Heldrup, J.; Olofsson, T.; Råde, J.; Fontes, M.; Porwit-MacDonald, A.; et al. Microarray-Based Classification of a Consecutive Series of 121 Childhood Acute Leukemias: Prediction of Leukemic and Genetic Subtype as Well as of Minimal Residual Disease Status. Leukemia 2007, 21, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; An, W.; Wang, H.; Baslan, T.; Mowla, S.; Krishnan, A.; Xiao, W.; Koche, R.P.; Liu, Y.; Cai, S.F.; et al. BMP2/SMAD Pathway Activation in JAK2/P53-Mutant Megakaryocyte/Erythroid Progenitors Promotes Leukemic Transformation. Blood 2022, 139, 3630–3646. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase I Clinical Trial of DNA Methyltransferase Inhibitor Decitabine and PARP Inhibitor Talazoparib Combination Therapy in Relapsed/Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Authors | AEL Definition Criteria | Participants | Treatment | Outcome | Reference Number |
|---|---|---|---|---|---|
| Almeida et al., 2017 | WHO 2008 Criteria for AEL | 217 total patients with AEL pooled from 28 international registries and 8 countries (1998–2014) | HMA or ICT |
| [3] |
| 88 patients with AEL (mean age 69) | HMA
|
| [3] | ||
| 122 patients with AEL (mean age 60) | ICT
idarubicin with cytarabine (n = 25) mitoxantrone with cytarabine (n = 8) |
| [3] | ||
| Gera et al., 2023 | WHO 2001 Criteria for AEL | 968 patients with PEL from the 2000–2019 SEER database (Median Age 68 years old, 62% male) | 65% of patients were treated with ICT |
| [15] |
| 918 Adults > 18 years of age | 559 patients treated with ICT | Adults
| [15] | ||
| 50 Children < 18 years of age | 46 patients treated with ICT | Children
| [15] | ||
| Reichard et al., 2022 | WHO 2016 Criteria for AEL | 41 PEL patients (14 de novo, 12 secondary to MDS, 14 therapy-related) (Mean age 66 years, 71% male) | 29 patients had treatment data recordedHMA (n = 5), HMA with Venetoclax (n = 12), ICT (n = 4), and best supportive care (n = 8) |
| [10] |
| Alkhateeb et al., 2016 | WHO 2008 Criteria for AEL | 43 patients at Mayo Clinic
| Stem Cell Transplant
|
| [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, P.; Waldron, N.; Chatzilygeroudi, T.; Naji, N.S.; Karantanos, T. Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes. Int. J. Mol. Sci. 2024, 25, 6256. https://doi.org/10.3390/ijms25116256
Fernandes P, Waldron N, Chatzilygeroudi T, Naji NS, Karantanos T. Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes. International Journal of Molecular Sciences. 2024; 25(11):6256. https://doi.org/10.3390/ijms25116256
Chicago/Turabian StyleFernandes, Priyanka, Natalie Waldron, Theodora Chatzilygeroudi, Nour Sabiha Naji, and Theodoros Karantanos. 2024. "Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes" International Journal of Molecular Sciences 25, no. 11: 6256. https://doi.org/10.3390/ijms25116256
APA StyleFernandes, P., Waldron, N., Chatzilygeroudi, T., Naji, N. S., & Karantanos, T. (2024). Acute Erythroid Leukemia: From Molecular Biology to Clinical Outcomes. International Journal of Molecular Sciences, 25(11), 6256. https://doi.org/10.3390/ijms25116256