
Computational Drug Repurposing Based on a Recommendation System and Drug–Drug Functional Pathway Similarity


Abstract
:1. Introduction
2. Results and Discussion
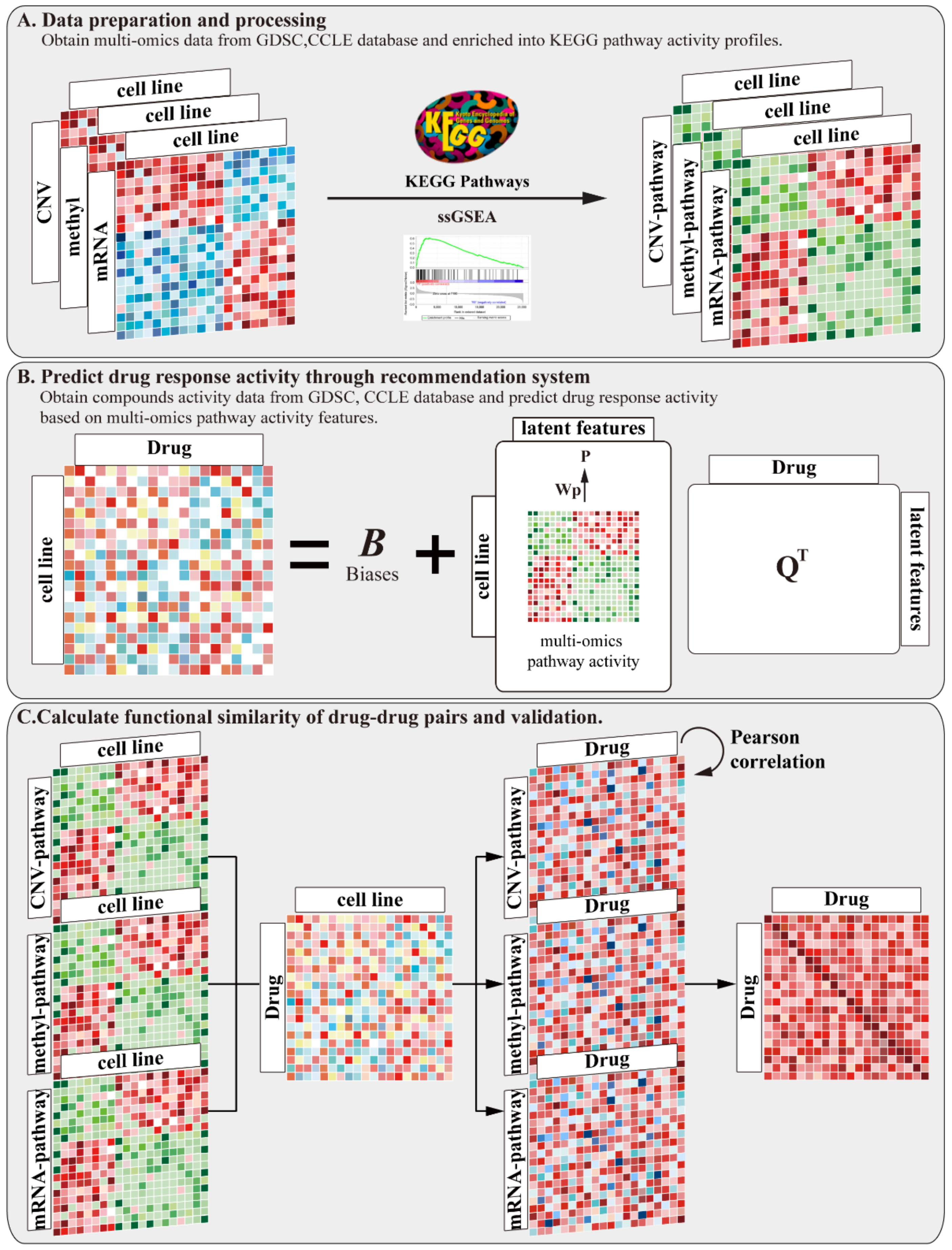
2.1. Workflow Overview
2.2. Evaluation of Predictive Drug Response Results
2.3. Drugs’ Effects on Biological Pathway Levels
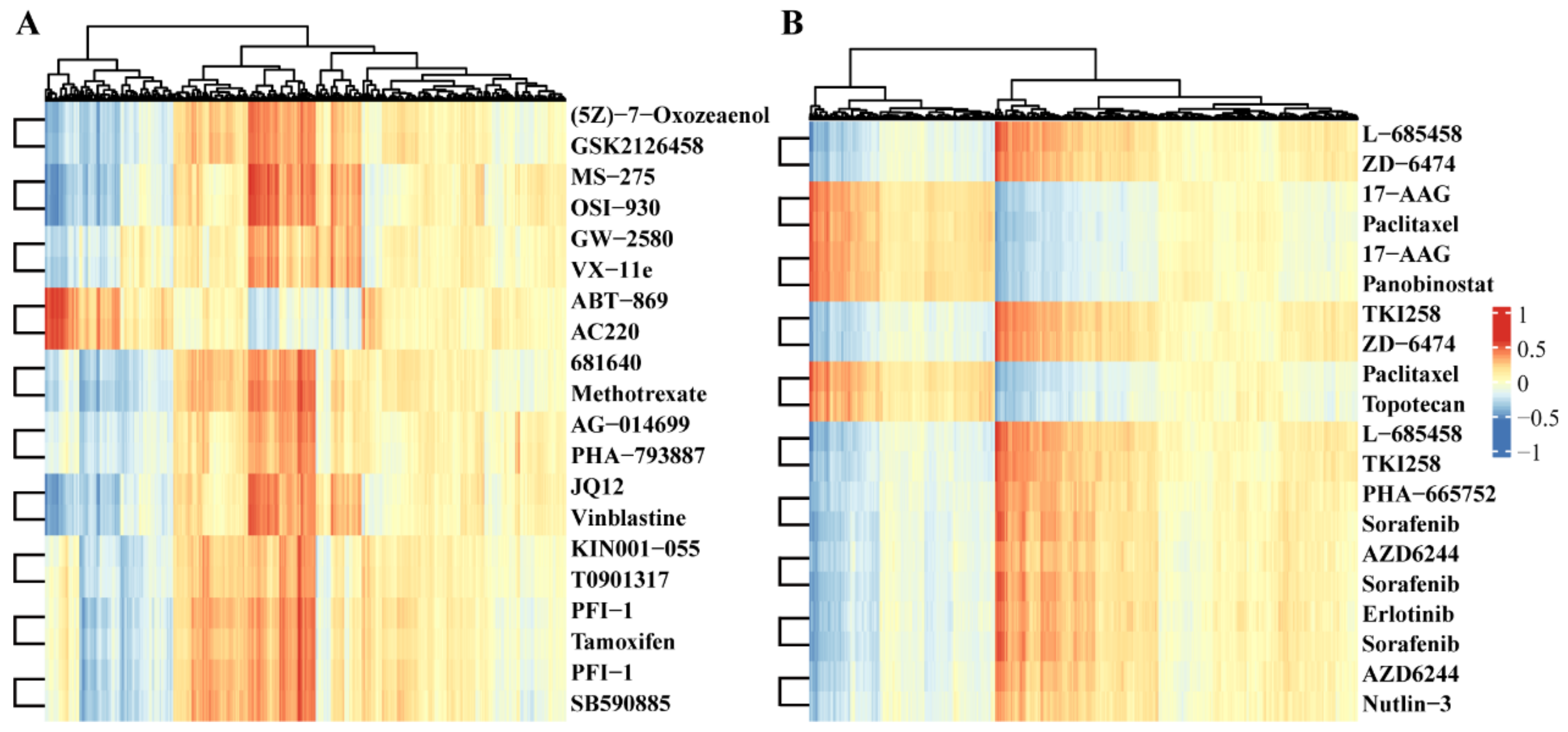
2.4. Comparison of Drug Pair Similarities
2.5. Case Study
3. Materials and Methods
3.1. Data Sources and Data Processing
3.1.1. Chemical Compounds Activity Data
3.1.2. Multi-Omics Expression Data
3.2. Inferring Multi-Omics Pathway Activity Profiles
3.3. Predict Drug Response Activity through Recommendation System Based on Multi-Omics Pathway Activity Profiles
3.4. Calculate the Functional Similarity of Drug–Drug Pairs Based on Multi-Omics Pathways Profiles
3.5. Molecular Structural Similarities and Drug Target Similarities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zahan, T.; Das, P.K.; Akter, S.F.; Habib, R.; Rahman, H.; Karim, R.; Islam, F. Therapy resistance in cancers: Phenotypic, metabolic, epigenetic and tumour microenvironmental perspectives. Anti-Cancer Agents Med. Chem. 2020, 20, 2190–2206. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug-disease relationships for computational drug repositioning. Briefings Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Pijl, H.; Ohashi, S.; Matsuda, M.; Miyazaki, Y.; Mahankali, A.; Kumar, V.; Pipek, R.; Iozzo, P.; Lancaster, J.L.; Cincotta, A.H.; et al. Bromocriptine: A novel approach to the treatment of type 2 diabetes. Diabetes Care 2000, 23, 1154–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; So, H.-C. Using drug expression profiles and machine learning approach for drug repurposing. Methods Mol. Biol. 2019, 1903, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Dai, E.; Song, Q.; Ma, X.; Meng, Q.; Jiang, Y.; Jiang, W. In silico drug repositioning based on drug-miRNA associations. Brief. Bioinform. 2020, 21, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Peyvandipour, A.; Saberian, N.; Shafi, A.; Donato, M.; Draghici, S. A novel computational approach for drug repurposing using systems biology. Bioinformatics 2018, 34, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Chen, Q.; Bei, M.; Shao, M.; Xu, J. Characterizing the tumor RBP-ncRNA circuits by integrating transcriptomics, interactomics and clinical data. Comput. Struct. Biotechnol. J. 2021, 19, 5235–5245. [Google Scholar] [CrossRef]
- Mehrabad, E.M.; Hassanzadeh, R.; Eslahchi, C. PMLPR: A novel method for predicting subcellular localization based on recommender systems. Sci. Rep. 2018, 8, 12006. [Google Scholar] [CrossRef]
- Corrado, G.; Tebaldi, T.; Costa, F.; Frasconi, P.; Passerini, A. RNAcommender: Genome-wide recommendation of RNA–protein interactions. Bioinformatics 2016, 32, 3627–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Lim, H.; Cheng, S.-Y.; Xie, L. ANTENNA, a multi-rank, multi-layered recommender system for inferring reliable drug-gene-disease associations: Repurposing diazoxide as a targeted anti-cancer therapy. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1960–1967. [Google Scholar] [CrossRef]
- Suphavilai, C.; Bertrand, D.; Nagarajan, N. Predicting cancer drug response using a recommender system. Bioinformatics 2018, 34, 3907–3914. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.-F.; Roth, M.; Toader, D.; Saeh, J.C.; et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. [Google Scholar] [CrossRef]
- Munster, P.N.; Aggarwal, R.; Hong, D.; Schellens, J.H.; Van Der Noll, R.; Specht, J.M.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.K.; et al. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin. Cancer Res. 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.K.; Li, J.-L.; Murga, M.; Harris, A.L.; Tosato, G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 2006, 107, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M. Erlotinib: Preclinical investigations. Oncology 2003, 17, 11–16. [Google Scholar]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Gao, J.; Chang, Y.S.; Jallal, B.; Viner, J. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 2012, 72, 3–12. [Google Scholar] [CrossRef] [Green Version]
- King, H.; Aleksic, T.; Haluska, P.; Macaulay, V.M. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat. Rev. 2014, 40, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.S.; Cheville, J.C.; Janney, C.A.; Cerhan, J.R. High expression levels of insulin-like growth factor–I receptor predict poor survival among women with clear-cell renal cell carcinomas. Hum. Pathol. 2002, 33, 801–805. [Google Scholar] [CrossRef]
- Spentzos, D.; Cannistra, S.A.; Grall, F.; Levine, D.A.; Pillay, K.; Libermann, T.A.; Mantzoros, C.S. IGF axis gene expression patterns are prognostic of survival in epithelial ovarian cancer. Endocr.-Relat. Cancer 2007, 14, 781–790. [Google Scholar] [CrossRef]
- Dale, O.T.; Aleksic, T.; Shah, K.A.; Han, C.; Mehanna, H.; Rapozo, D.C.; Sheard, J.D.H.; Goodyear, P.; Upile, N.S.; Robinson, M.; et al. IGF-1R expression is associated with HPV-negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis 2015, 36, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-S.; Kim, E.S.; Liu, D.; Lee, J.J.; Solis, L.; Behrens, C.; Lippman, S.M.; Hong, W.K.; Wistuba, I.I.; Lee, H.-Y. Prognostic implications of tumoral expression of insulin like growth factors 1 and 2 in patients with non–small-cell lung cancer. Clin. Lung Cancer 2014, 15, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.A.; Varewijck, A.J. IGF-IR targeted therapy: Past, present and future. Front. Endocrinol. 2014, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.-S.; Mulvihill, M.J.; Rosenfeld-Franklin, M.; Cooke, A.; Feng, L.; Mak, G.; O’Connor, M.; Yao, Y.; Pirritt, C.; Buck, E.; et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor–dependent tumor growth in vivo. Mol. Cancer Ther. 2007, 6, 2158–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Kim, E.S.; Nava-Parada, P.; Alam, S.; Johnson, F.M.; Stephens, A.W.; Simantov, R.; Poondru, S.; Gedrich, R.; Lippman, S.M.; et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzanov, I.; Lindsay, C.R.; Goff, L.; Sosman, J.; Gilbert, J.; Berlin, J.; Poondru, S.; Simantov, R.; Gedrich, R.; Stephens, A.; et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Fassnacht, M.; Berruti, A.; Baudin, E.; Demeure, M.J.; Gilbert, J.; Haak, H.; Kroiss, M.; Quinn, D.I.; Hesseltine, E.; Ronchi, C.L.; et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol. 2015, 16, 426–435. [Google Scholar] [CrossRef] [Green Version]
- van der Veeken, J.; Oliveira, S.; Schiffelers, R.M.; Storm, G.; van Bergen En Henegouwen, P.M.; Roovers, R.C. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: Implications for cancer therapy. Curr. Cancer Drug Targets 2009, 9, 748–760. [Google Scholar] [CrossRef]
- Qi, H.W.; Shen, Z.; Fan, L.H. Combined inhibition of insulin-like growth factor-1 receptor enhances the effects of gefitinib in a human non-small cell lung cancer resistant cell line. Exp. Ther. Med. 2011, 2, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Mizuuchi, H.; Sato, K.; Takemoto, T.; Iwasaki, T.; Mitsudomi, T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int. J. Cancer 2014, 135, 1002–1006. [Google Scholar] [CrossRef]
- Camirand, A.; Zakikhani, M.; Young, F.; Pollak, M. Inhibition of insulin-like growth factor-1 receptor signaling enhances growth-inhibitory and proapoptotic effects of gefitinib (Iressa) in human breast cancer cells. Breast Cancer Res. 2005, 7, R570. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.; Gee, J.M.W.; Barrow, D.; Tonge, D.; Holloway, B.; Nicholson, R. Inhibition of insulin receptor isoform-A signalling restores sensitivity to gefitinib in previously de novo resistant colon cancer cells. Br. J. Cancer 2006, 95, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Urtasun, N.; Vidal-Pla, A.; Pérez-Torras, S.; Mazo, A. Human pancreatic cancer stem cells are sensitive to dual inhibition of IGF-IR and ErbB receptors. BMC Cancer 2015, 15, 223. [Google Scholar] [CrossRef] [Green Version]
- Zanella, E.R.; Galimi, F.; Sassi, F.; Migliardi, G.; Cottino, F.; Leto, S.M.; Lupo, B.; Erriquez, J.; Isella, C.; Comoglio, P.M.; et al. IGF2 is an actionable target that identifies a distinct subpopulation of colorectal cancer patients with marginal response to anti-EGFR therapies. Sci. Transl. Med. 2015, 7, 272ra12. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A landscape of pharmacogenomic interactions in cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.D.; Bignell, G.; Butler, A.; Edkins, S.; Hinton, J.; Beare, D.; Swamy, S.; Santarius, T.; Chen, L.; Widaa, S.; et al. PICNIC: An algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics 2010, 11, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- The Cancer Cell Line Encyclopedia Consortium; The Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- The International HapMap Consortium. The international hapmap project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, E.S.; Olshen, A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, J.; Zheng, B.; Kong, Q.; Jiang, Y.; Liu, S.; Yang, Y.; Han, X.; Sheng, Y.; Zhang, Y.; Cheng, L.; et al. Prioritization of can-didate cancer drugs based on a drug functional similarity network constructed by integrating pathway activities and drug activities. Mol. Oncol. 2019, 13, 2259–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Beck, A.H.; Aerts, H.J.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2013, 504, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haverty, P.M.; Lin, E.; Tan, J.; Yu, Y.; Lam, B.; Lianoglou, S.; Neve, R.M.; Martin, S.; Settleman, J.; Yauch, R.L.; et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 2016, 533, 333–337. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1—Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Tanimoto, T. IBM Internal Report 17th Nov; DJ Rogers and TT Tanimoto. Science 1957, 132, 1115–1118. [Google Scholar]
- Giri, V.; Sivakumar, T.V.; Cho, K.M.; Kim, T.Y.; Bhaduri, A. RxnSim: A tool to compare biochemical reactions. Bioinformatics 2015, 31, 3712–3724. [Google Scholar] [CrossRef] [Green Version]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Perlman, L.; Gottlieb, A.; Atias, N.; Ruppin, E.; Sharan, R. Combining drug and gene similarity measures for drug-target elucidation. J. Comput. Biol. 2011, 18, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Zabłocka, A.; Kazana, W.; Sochocka, M.; Stańczykiewicz, B.; Janusz, M.; Leszek, J.; Orzechowska, B. Inverse correlation between Alzheimer’s disease and cancer: Short overview. Mol. Neurobiol. 2021, 58, 6335–6349. [Google Scholar] [CrossRef]
- Chen, D.; Hao, S.; Xu, J. Revisiting the relationship between Alzheimer’s disease and cancer with a circRNA perspective. Front. Cell Dev. Biol. 2021, 9, 647197. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| GDSC Multi-omics Pathway | GDSC mRNA-Pathway | GDSC mRNA Expression | CCLE Multi-omics Pathway | CCLE mRNA-Pathway | CCLE mRNA Expression | |
|---|---|---|---|---|---|---|
| NDCG | 0.815 | 0.816 | 0.381 | 0.976 | 0.978 | 0.798 |
| Sum of squared error | 1.176 | 1.173 | 3.540 | 0.728 | 0.662 | 2.633 |
| Database | Drug1 | Drug2 | Functional Sim | SMILES Sim | PPI Sim |
|---|---|---|---|---|---|
| GDSC | (5Z)-7-Oxozeaenol | GSK2126458 | 0.998365459 | 0.162790698 | 1.08E−70 |
| GDSC | MS-275 | OSI-930 | 0.998193161 | 0.362694301 | 3.30E−77 |
| GDSC | GW-2580 | VX-11e | 0.997189963 | 0.242063492 | 3.71E−84 |
| GDSC | ABT-869 | AC220 | 0.996300765 | 0.257462687 | 0.9 |
| GDSC | 681640 | Methotrexate | 0.996231842 | 0.236734694 | 6.46E−66 |
| GDSC | AG-014699 | PHA-793887 | 0.996134942 | 0.223529412 | 5.37E−72 |
| GDSC | JQ12 | Vinblastine | 0.995927571 | - | 1.54E−91 |
| GDSC | KIN001-055 | T0901317 | 0.99574713 | 0.161111111 | 4.90E−75 |
| GDSC | PFI-1 | Tamoxifen | 0.995377083 | 0.208092486 | 2.38E−66 |
| GDSC | PFI-1 | SB590885 | 0.995238003 | 0.194029851 | 2.74E−83 |
| CCLE | L-685458 | ZD-6474 | 0.994678 | 0.11349 | 1.18E−67 |
| CCLE | 17-AAG | Paclitaxel | 0.994097 | 0.272071 | 0.9 |
| CCLE | 17-AAG | Panobinostat | 0.99304 | 0.075472 | 3.97E−71 |
| CCLE | TKI258 | ZD-6474 | 0.992772 | 0.227439 | 0.9 |
| CCLE | Paclitaxel | Topotecan | 0.992513 | 0.291483 | 0.9 |
| CCLE | L-685458 | TKI258 | 0.989722 | 0.129946 | 2.38E−66 |
| CCLE | PHA-665752 | Sorafenib | 0.98863 | 0.157855 | 0.9 |
| CCLE | AZD6244 | Sorafenib | 0.98829 | 0.169047 | 0.9 |
| CCLE | Erlotinib | Sorafenib | 0.987973 | 0.256565 | 0.9 |
| CCLE | AZD6244 | Nutlin-3 | 0.987127 | 0.137107 | 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, M.; Jiang, L.; Meng, Z.; Xu, J. Computational Drug Repurposing Based on a Recommendation System and Drug–Drug Functional Pathway Similarity. Molecules 2022, 27, 1404. https://doi.org/10.3390/molecules27041404
Shao M, Jiang L, Meng Z, Xu J. Computational Drug Repurposing Based on a Recommendation System and Drug–Drug Functional Pathway Similarity. Molecules. 2022; 27(4):1404. https://doi.org/10.3390/molecules27041404
Chicago/Turabian StyleShao, Mengting, Leiming Jiang, Zhigang Meng, and Jianzhen Xu. 2022. "Computational Drug Repurposing Based on a Recommendation System and Drug–Drug Functional Pathway Similarity" Molecules 27, no. 4: 1404. https://doi.org/10.3390/molecules27041404
APA StyleShao, M., Jiang, L., Meng, Z., & Xu, J. (2022). Computational Drug Repurposing Based on a Recommendation System and Drug–Drug Functional Pathway Similarity. Molecules, 27(4), 1404. https://doi.org/10.3390/molecules27041404