
Palladium-Catalyzed Direct (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole and 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)




Abstract
:1. Introduction
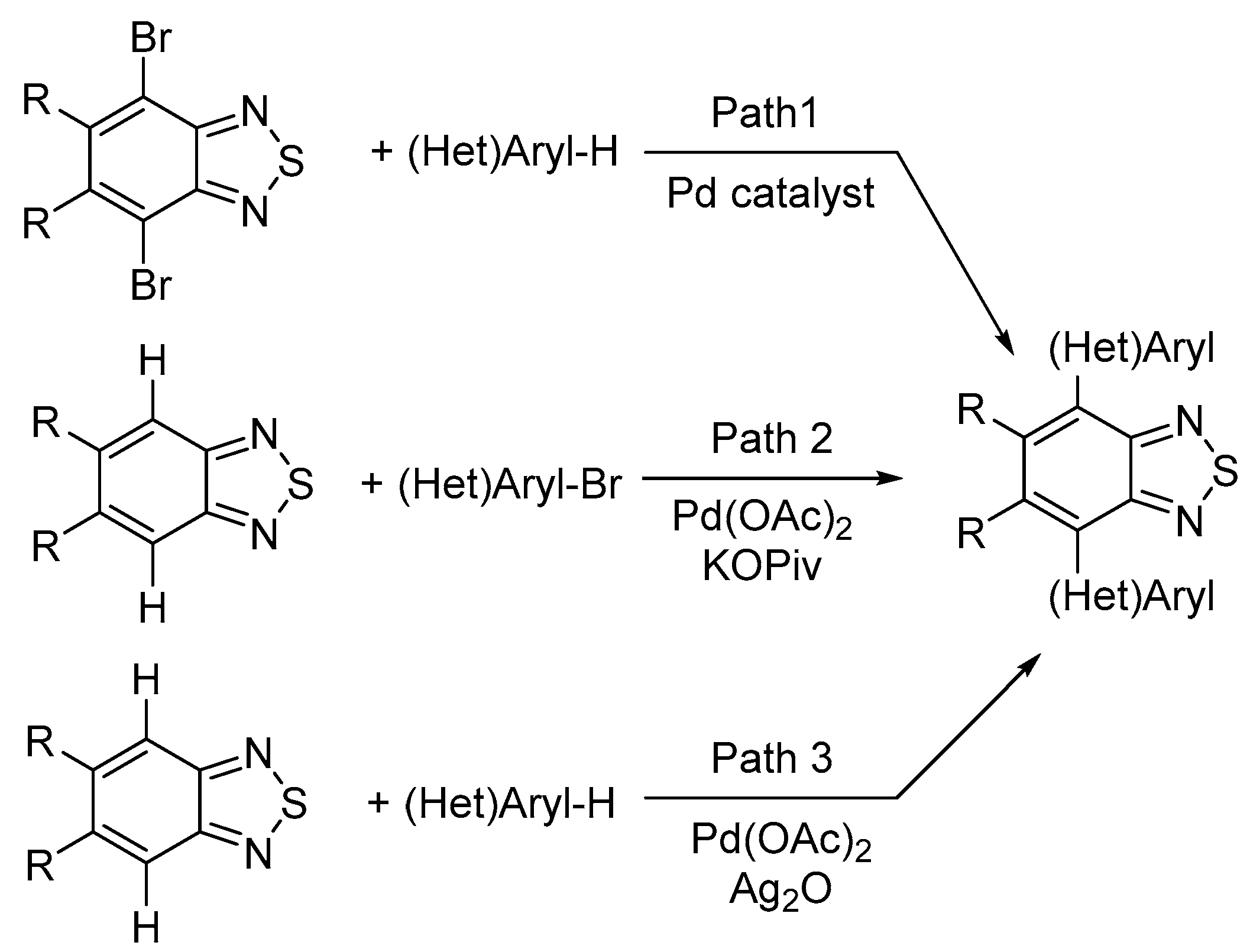
2. Results and Discussion
2.1. Palladium-Catalyzed (Het)arylation Reactions of 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2
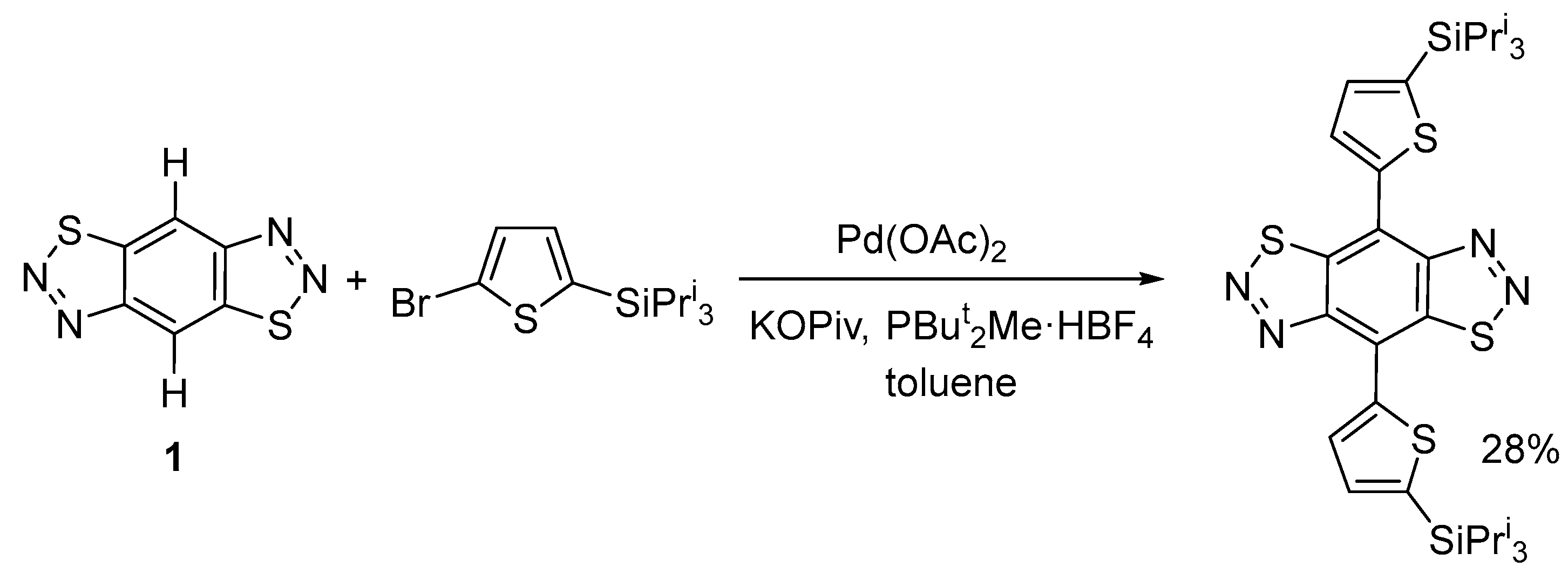
2.2. Palladium-Catalyzed (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1
2.3. Palladium-Catalyzed Oxidative (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1
2.4. Comparison of Suzuki and Stille Cross-Coupling Reactions with Direct (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1 and 4,8-dibromo Derivative 2
3. Experimental Section
3.1. Materials and Reagents
3.2. Analytical Instruments
3.3. General Procedure for the Synthesis of Mono-Substituted Products 4 from 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (Procedure A)
3.4. General Procedure for the Synthesis of Bis-Substituted Products 5 from 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (Procedure B)
3.5. General Procedure for the Preparation of Mono-Substituted Products 7 from Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1 (Procedure C)
3.6. General Procedure for the Preparation of Bis-Substituted Products 5 from Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1 (Procedure D)
3.7. General Procedure for the Preparation of Bis-Substituted Products 5 under C-H Oxidative Coupling Conditions (Procedure E)
3.8. Preparation of Preparation of 4-(5-(2-Ethylhexyl)thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 7a under C-H Oxidative Coupling Conditions (Procedure F)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chauhan, A.K.; Jha, P.; Aswal, D.K.; Yakhmi, J.V. Organic Devices: Fabrication, Applications, and Challenges. J. Electron. Mater. 2022, 51, 447–485. [Google Scholar] [CrossRef]
- Yan, J.; Saunders, B.R. Third-generation solar cells: A review and comparison of polymer: Fullerene, hybrid polymer and perovskite solar cells. RSC Adv. 2014, 4, 43286–43314. [Google Scholar] [CrossRef]
- Takimiya, K.; Osaka, I.; Nakano, M. π-Building Blocks for Organic Electronics: Revaluation of “Inductive” and “Resonance” Effects of π-Electron Deficient Units. Chem. Mater. 2014, 26, 587–593. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, J.; Qu, J.; Qian, P.-C.; Wong, W.-Y. Recent progress of electronic materials based on 2,1,3-benzothiadiazole and its derivatives: Synthesis and their application in organic light-emitting diodes. Sci. China Chem. 2021, 64, 341–357. [Google Scholar] [CrossRef]
- Rakitin, O.A. Recent Developments in the Synthesis of 1,2,5-Thiadiazoles and 2,1,3-Benzothiadiazoles. Synthesis 2019, 51, 4338–4347. [Google Scholar] [CrossRef]
- Rakitin, O.A. Fused 1,2,5-thia- and 1,2,5-selenadiazoles: Synthesis and application in materials chemistry. Tetrahedron Lett. 2020, 61, 152230. [Google Scholar] [CrossRef]
- Roncali, J. Molecular Engineering of the Band Gap of π-Conjugated Systems: Facing Technological Applications. Macromol. Rapid Commun. 2007, 28, 1761–1775. [Google Scholar] [CrossRef]
- Chmovzh, T.N.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Amelichev, S.A.; Rakitin, O.A. Synthesis of the 4,7-Dibromo Derivative of Highly Electron-Deficient [1,2,5]Thiadiazolo[3,4-d]pyridazine and Its Cross-Coupling Reactions. Eur. J. Org. Chem. 2018, 2018, 5668–5677. [Google Scholar] [CrossRef]
- Chmovzh, T.; Knyazeva, E.; Lyssenko, K.; Popov, V.; Rakitin, O. Safe Synthesis of 4,7-Dibromo[1,2,5]thiadiazolo[3,4-d]pyridazine and Its SNAr Reactions. Molecules 2018, 23, 2576. [Google Scholar] [CrossRef]
- Yamashita, Y.; Ono, K.; Tomura, M.; Tanaka, S. Synthesis and Properties of Benzobis(thiadiazole)s with Nonclassical π-Electron Ring Systems. Tetrahedron 1997, 53, 10169–10178. [Google Scholar] [CrossRef]
- Bianchi, L.; Zhang, X.; Chen, Z.; Chen, P.; Zhou, X.; Tang, Y.; Liu, B.; Guo, X.; Facchetti, A. New Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (iso-BBT)-Based Polymers for Application in Transistors and Solar Cells. Chem. Mater. 2019, 31, 6519–6529. [Google Scholar] [CrossRef]
- Chmovzh, T.N.; Alekhina, D.A.; Kudryashev, T.A.; Rakitin, O.A. Efficient Synthesis of 4,8-Dibromo Derivative of Strong Electron-Deficient Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) and Its SNAr and Cross-Coupling Reactions. Molecules 2022, 27, 7372. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C–H Bond Cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef] [PubMed]
- Bohra, H.; Wang, M. Direct C–H arylation: A “Greener” approach towards facile synthesis of organic semiconducting molecules and polymers. J. Mater. Chem. A 2017, 5, 11550–11571. [Google Scholar] [CrossRef]
- Mainville, M.; Leclerc, M. Direct (Hetero)arylation: A Tool for Low-Cost and Eco-Friendly Organic Photovoltaics. ACS Appl. Polym. Mater. 2021, 3, 2–13. [Google Scholar] [CrossRef]
- Albano, G.; Punzi, A.; Capozzi, M.A.M.; Farinola, G.M. Sustainable protocols for direct C–H bond arylation of (hetero)arenes. Green Chem. 2022, 24, 1809–1894. [Google Scholar] [CrossRef]
- Chen, C.; Maldonado, D.H.; Le Borgne, D.; Alary, F.; Lonetti, B.; Heinrich, B.; Donnio, B.; Moineau-Chane Ching, K.I. Synthesis of benzothiadiazole-based molecules via direct arylation: An eco-friendly way of obtaining small semi-conducting organic molecules. New J. Chem. 2016, 40, 7326–7337. [Google Scholar] [CrossRef]
- Dall′Agnese, C.; Hernández Maldonado, D.; Le Borgne, D.; Moineau-Chane Ching, K.I. Dissymmetrization of Benzothiadiazole by Direct C-H Arylation: A Way to Symmetrical and Unsymmetrical Elongated π-Conjugated Molecules. Eur. J. Org. Chem. 2017, 2017, 6872–6877. [Google Scholar] [CrossRef]
- Giannopoulos, P.; Raptis, D.; Theodosiou, K.; Andreopoulou, A.K.; Anastasopoulos, C.; Dokouzis, A.; Leftheriotis, G.; Lianos, P.; Kallitsis, J.K. Organic dyes end-capped with perfluorophenyl anchors: Synthesis, electrochemical properties and assessment of sensitization capacity of titania photoanodes. Dye. Pigment. 2018, 148, 167–179. [Google Scholar] [CrossRef]
- Chang, S.-W.; Waters, H.; Kettle, J.; Horie, M. Cyclopentadithiophene–benzothiadiazole oligomers: Synthesis via direct arylation, X-ray crystallography, optical properties, solution casted field-effect transistor and photovoltaic characteristics. Org. Electron. 2012, 13, 2967–2974. [Google Scholar] [CrossRef]
- Chang, S.-W.; Kettle, J.; Waters, H.; Horie, M. Cyclopentadithiophene–benzothiadiazole copolymers with permutations of repeating unit length and ratios; synthesis, optical and electrochemical properties and photovoltaic characteristics. RSC Adv. 2015, 5, 107276–107284. [Google Scholar] [CrossRef]
- Sharma, B.; Alam, F.; Dutta, V.; Jacob, J. Synthesis and Photovoltaic Studies on Novel Fluorene Based Cross-Conjugated Donor-Acceptor Type Polymers. Org. Electron. 2017, 40, 42–50. [Google Scholar] [CrossRef]
- Nitti, A.; Osw, P.; Abdullah, M.; Galbiati, A.; Pasini, D. Scalable Synthesis of Naphthothiophene-based D-π-D Extended Oligomers through Cascade Direct Arylation Processes. Synlett 2018, 29, 2577–2581. [Google Scholar] [CrossRef]
- Chaudhry, S.; Ryno, S.M.; Zeller, M.; McMillin, D.R.; Risko, C.; Mei, J. Oxidation Pathways Involving a Sulfide-Endcapped Donor–Acceptor–Donor π-Conjugated Molecule and Antimony(V) Chloride. J. Phys. Chem. B 2019, 123, 3866–3874. [Google Scholar] [CrossRef]
- Nitti, A.; Osw, P.; Calcagno, G.; Botta, C.; Etkind, S.I.; Bianchi, G.; Po, R.; Swager, T.M.; Pasini, D. One-Pot Regiodirected Annulations for the Rapid Synthesis of π-Extended Oligomers. Org. Lett. 2020, 22, 3263–3267. [Google Scholar] [CrossRef] [PubMed]
- Lombeck, F.; Komber, H.; Sepe, A.; Friend, R.H.; Sommer, M. Enhancing Phase Separation and Photovoltaic Performance of All-Conjugated Donor–Acceptor Block Copolymers with Semifluorinated Alkyl Side Chains. Macromolecules 2015, 48, 7851–7860. [Google Scholar] [CrossRef]
- Calascibetta, A.M.; Mattiello, S.; Sanzone, A.; Facchinetti, I.; Sassi, M.; Beverina, L. Sustainable Access to π-Conjugated Molecular Materials via Direct (Hetero)Arylation Reactions in Water and under Air. Molecules 2020, 25, 3717. [Google Scholar] [CrossRef]
- Efrem, A.; Wang, K.; Jia, T.; Wang, M. Direct Arylation Polymerization toward a Narrow Bandgap Donor-Acceptor Conjugated Polymer of Alternating 5,6-Difluoro-2,1,3-Benzothiadiazole and Alkyl-Quarternarythiophene: From Synthesis, Optoelectronic Properties to Devices. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1869–1879. [Google Scholar] [CrossRef]
- Miyake, H.; Tajima, T.; Takaguchi, Y. Synthesis and Light-absorption Characteristics of Thiophene Derivatives Bearing Ferrocenylthiocarbonyl Groups. Chem. Lett. 2017, 46, 48–50. [Google Scholar] [CrossRef]
- Matsidik, R.; Takimiya, K. Synthesis of Thiophene-annulated Naphthalene Diimide-based Small-Molecular Acceptors via Two-step C−H Activation. Chem. Asian J. 2019, 14, 1651–1656. [Google Scholar] [CrossRef]
- Takaguchi, Y.; Miyake, H.; Izawa, T.; Miyamoto, D.; Sagawa, R.; Tajima, T. Molecular Design of Benzothiadiazole-Based Dyes for Working with Carbon Nanotube Photocatalysts. Phosphorus. Sulfur. Silicon Relat. Elem. 2019, 194, 707–711. [Google Scholar] [CrossRef]
- Chávez, P.; Ngov, C.; de Frémont, P.; Lévêque, P.; Leclerc, N. Synthesis by Direct Arylation of Thiazole–Derivatives: Regioisomer Configurations–Optical Properties Relationship Investigation. J. Org. Chem. 2014, 79, 10179–10188. [Google Scholar] [CrossRef] [PubMed]
- Idris, I.; Tannoux, T.; Derridj, F.; Dorcet, V.; Boixel, J.; Guerchais, V.; Soulé, J.-F.; Doucet, H. Effective Modulation of the Photoluminescence Properties of 2,1,3-Benzothiadiazoles and 2,1,3-Benzoselenadiazoles by Pd-Catalyzed C–H Bond Arylations. J. Mater. Chem. C 2018, 6, 1731–1737. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, W.; Rojas, A.J.; Jucov, E.V.; Timofeeva, T.V.; Parker, T.C.; Barlow, S.; Marder, S.R. Controllable Direct Arylation: Fast Route to Symmetrical and Unsymmetrical 4,7-Diaryl-5,6-Difluoro-2,1,3-Benzothiadiazole Derivatives for Organic Optoelectronic Materials. J. Am. Chem. Soc. 2013, 135, 16376–16379. [Google Scholar] [CrossRef]
- Zhang, J.; Parker, T.C.; Chen, W.; Williams, L.; Khrustalev, V.N.; Jucov, E.V.; Barlow, S.; Timofeeva, T.V.; Marder, S.R. C–H-Activated Direct Arylation of Strong Benzothiadiazole and Quinoxaline-Based Electron Acceptors. J. Org. Chem. 2016, 81, 360–370. [Google Scholar] [CrossRef]
- He, C.-Y.; Wu, C.-Z.; Zhu, Y.-L.; Zhang, X. Selective Thienylation of Fluorinated Benzothiadiazoles and Benzotriazoles for Organic Photovoltaics. Chem. Sci. 2014, 5, 1317–1321. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, K.; Yang, G.; Liu, J.; Li, Z.; Lin, H.; Liu, Y.; Zhao, J.; Zhang, J.; Huang, F.; et al. Terthiophene-Based D–A Polymer with an Asymmetric Arrangement of Alkyl Chains That Enables Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2015, 137, 14149–14157. [Google Scholar] [CrossRef]
- Facchetti, A.; Chen, Z.; Brown, J.E. Semiconducting Compounds and Related Devices. U.S. Patent 9,708,346, 19 October 2016. [Google Scholar]
- Marin, L.; Lutsen, L.; Vanderzande, D.; Maes, W. Quinoxaline derivatives with broadened absorption patterns. Org. Biomol. Chem. 2013, 11, 5866. [Google Scholar] [CrossRef]
- Zaitsev, K.V.; Lam, K.; Poleshchuk, O.K.; Kuz’mina, L.G.; Churakov, A.V. Oligothienyl catenated germanes and silanes: Synthesis, structure, and properties. Dalt. Trans. 2018, 47, 5431–5444. [Google Scholar] [CrossRef]
- Skorotetcky, M.S.; Krivtsova, E.D.; Borshchev, O.V.; Surin, N.M.; Svidchenko, E.A.; Fedorov, Y.V.; Pisarev, S.A.; Ponomarenko, S.A. Influence of the structure of electron-donating aromatic units in organosilicon luminophores based on 2,1,3-benzothiadiazole electron-withdrawing core on their absorption-luminescent properties. Dye. Pigment. 2018, 155, 284–291. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ar-H (Equiv) | Catalyst a | Base (Equiv) | Ligand b | Solvent | Conditions | Yields (%) | |
|---|---|---|---|---|---|---|---|---|
| 4 | 5 | |||||||
| 1 | 3a (2) | Pd(OAc)2 | PivOK (2) | - | DMA | 110 °C, 30 h | 0 | 0 |
| 2 | 3a (1) | Pd(OAc)2 | PivOK (1) | - | toluene | 110 °C, 30 h | 43 | 0 |
| 3 | 3a (2) | Pd(OAc)2 | PivOK (2) | - | xylene | 130 °C, 36 h | 0 | 40 |
| 4 | 3a (1) | Pd(OAc)2 | PivOK (1) | Xphos | toluene | 110 °C, 12 h | 0 | 0 |
| 5 | 3a (1) | Pd(OAc)2 | PivOK (1) | But3P | toluene | 110 °C, 12 h | 0 | 0 |
| 6 | 3a (1) | Pd(OAc)2 | PivOK (1) | dppf | toluene | 110 °C, 12 h | 0 | 0 |
| 7 | 3a (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | toluene | 110 °C, 12 h | 0 | 0 |
| 8 | 3a (2) | Pd(OAc)2 | PivOK (2) | Ph3P | DMA | 110 °C, 12 h | 0 | 0 |
| 9 | 3a (2) | Pd(OAc)2 | PivOK (2) | PBut2Me HBF4 | DMA | 110 °C, 12 h | 0 | 0 |
| 10 | 3a (2) | Pd(PPh3)4 | PivOCs (2) | - | xylene | 110 °C, 18 h | 38 | 0 |
| 11 | 3a (2) | Pd(PPh3)4 | PivOCs (2) | - | xylene | 130 °C, 16 h | 36 | |
| 12 | 3b (1) | Pd(OAc)2 | PivOK (1) | - | toluene | 110 °C, 30 h | 33 | 0 |
| 13 | 3b (2) | Pd(OAc)2 | PivOK (2) | - | xylene | 130 °C, 24 h | 0 | 36 |
| 14 | 3c (1) | Pd(OAc)2 | PivOK (1) | - | toluene | 110 °C, 30 h | 35 | 0 |
| 15 | 3c (2) | Pd(OAc)2 | PivOK (2) | - | xylene | 130 °C, 24 h | 0 | 29 |
| 16 | 3d (1) | Pd(OAc)2 | PivOK (1) | - | toluene | 110 °C, 30 h | 31 | 0 |
| 17 | 3d (2) | Pd(OAc)2 | PivOK (2) | - | xylene | 130 °C, 24 h | 0 | 30 |

| Entry | Ar-X (Equiv) | Catalyst a | Base(Equiv) | Ligand b | Solvent | Conditions | Yields (%) | |
|---|---|---|---|---|---|---|---|---|
| 7a | 5a | |||||||
| 1 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | - | toluene | 110 °C, 12 h | 0 | 0 |
| 2 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | Xphos | toluene | 110 °C, 12 h | 0 | 0 |
| 3 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | But3P | toluene | 110 °C, 12 h | 0 | 0 |
| 4 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | dppf | toluene | 110 °C, 12 h | 0 | 0 |
| 5 | 6a(Br) (1) | Pd(PPh3)4 | PivOK (1) | - | toluene | 110 °C, 12 h | 0 | 0 |
| 6 | 6a(Br) (1) | Pd2(dba)3 | PivOK (1) | - | toluene | 110 °C, 12 h | 0 | 0 |
| 7 | 6a(Br) (1) | Pd(OAc)2 | PivOCs (1) | CsF/TBAB | toluene | 110 °C, 12 h | 0 | 0 |
| 8 | 6a(Br) (1) | PdCl2(PPh3)2 | PivOK (1) | - | toluene | 110 °C, 12 h | 0 | 0 |
| 9 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | toluene | 110 °C, 12 h | 20 | 0 |
| 10 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | toluene | 110 °C, 36 h | 45 | 0 |
| 11 | 6a(Br) (2) | Pd(OAc)2 | PivOK (2) | PBut2Me HBF4 | xylene | 130 °C, 36 h | 0 | 55 |
| 12 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | DMF | 110 °C, 30 h | 0 | 0 |
| 13 | 6a(Br) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | DMA | 120 °C, 24 h | 0 | 0 |
| 14 | 6a(I) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | toluene | 110 °C, 24 h | 40 | 20 |
| 15 | 6a(I) (2) | Pd(OAc)2 | PivOK (2) | PBut2Me HBF4 | xylene | 130 °C, 36 h | 0 | 54 |
| 16 | 6a(Cl) (1) | Pd(OAc)2 | PivOK (1) | PBut2Me HBF4 | toluene | 110 °C, 24 h | 2 | 0 |

| Entry | Ar-X (Equiv) | Solvent | Conditions | Yields (%) | |
|---|---|---|---|---|---|
| 7 | 5 | ||||
| 1 | 6a(Br) (1) | toluene | 110 °C, 36 h | 45 | 0 |
| 2 | 6a(Br) (2) | xylene | 130 °C, 36 h | 0 | 55 |
| 3 | 6b(Br) (1) | toluene | 110 °C, 36 h | 30 | 3 |
| 4 | 6b(Br) (2) | xylene | 130 °C, 36 h | 0 | 35 |
| 5 | 6c(Br) (1) | toluene | 110 °C, 36 h | 38 | 4 |
| 6 | 6c(Br) (2) | xylene | 130 °C, 36 h | 0 | 34 |
| 7 | 6e(Br) (1) | toluene | 110 °C, 36 h | 29 | 3 |
| 8 | 6e(Br) (2) | xylene | 130 °C, 36 h | 0 | 39 |
| 9 | 6f(Br) (1) | toluene | 110 °C, 36 h | 15 | 0 |
| 10 | 6f(Br) (2) | xylene | 130 °C, 36 h | 0 | 20 |
| 11 | 6f(I) (1) | toluene | 110 °C, 36 h | 45 | 3 |
| 12 | 6f(I) (2) | xylene | 130 °C, 36 h | 0 | 50 |
| 13 | 6g(I) (1) | toluene | 110 °C, 36 h | 45 | 5 |
| 14 | 6g(I) (2) | xylene | 130 °C, 36 h | 0 | 50 |
| 15 | 6h(I) (1) | toluene | 110 °C, 36 h | 60 | 5 |
| 16 | 6h(I) (2) | xylene | 130 °C, 36 h | 0 | 55 |
| 17 | 6i(I) (1) | toluene | 110 °C, 36 h | 45 | 2 |
| 18 | 6i(I) (2) | xylene | 130 °C, 36 h | 0 | 49 |
| 19 | 6j(I) (1) | toluene | 110 °C, 36 h | 40 | 0 |
| 20 | 6j(I) (2) | xylene | 130 °C, 36 h | 0 | 25 |

| Entry | Ar-H (Eqv) | Catalyst a | Oxidizing Agent (Equiv) | Conditions | Yields (%) | |
|---|---|---|---|---|---|---|
| 7 | 5 | |||||
| 1 | 3a (2) | Pd(TFA)2 | Ag2O (2) | 110 °C, 24 h | 0 | 0 |
| 2 | 3a (2) | Pd(OAc)2 | Ag2O (2) | 110 °C, 36 h | 10 | 40 |
| 3 | 3a (2) | Pd(OAc)2 | AgOAc (2) | 110 °C, 48 h | 10 | 15 |
| 4 | 3a (2) | Pd(OAc)2 | AgBF4 (2) | 110 °C, 24 h | 0 | 0 |
| 5 | 3a (2) | Pd(OAc)2 | AgClO4 (2) | 110 °C, 24 h | 0 | 0 |
| 6 | 3a (2) | Pd(OAc)2 | AgNO3 (2) | 110 °C, 24 h | 4 | 0 |
| 7 | 3a (1) | Pd(OAc)2 | Ag2O (1) | 90 °C, 36 h | 30 | 2 |
| 8 | 3a (3) | Pd(OAc)2 | Ag2O (2) | 110 °C, 48 h | 0 | 55 |
| 9 | 3a (3) | Pd(OAc)2 | Ag2O (2) | 120 °C, 48 h | 0 | 50 |
| 10 | 3b (3) | Pd(OAc)2 | Ag2O (2) | 110 °C, 48 h | 0 | 29 |
| 11 | 3c (3) | Pd(OAc)2 | Ag2O (2) | 110 °C, 48 h | 0 | 35 |
| 12 | 3e (3) | Pd(OAc)2 | Ag2O (2) | 110 °C, 48 h | 0 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmovzh, T.N.; Kudryashev, T.A.; Alekhina, D.A.; Rakitin, O.A. Palladium-Catalyzed Direct (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole and 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole). Molecules 2023, 28, 3977. https://doi.org/10.3390/molecules28093977
Chmovzh TN, Kudryashev TA, Alekhina DA, Rakitin OA. Palladium-Catalyzed Direct (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole and 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole). Molecules. 2023; 28(9):3977. https://doi.org/10.3390/molecules28093977
Chicago/Turabian StyleChmovzh, Timofey N., Timofey A. Kudryashev, Daria A. Alekhina, and Oleg A. Rakitin. 2023. "Palladium-Catalyzed Direct (Het)arylation Reactions of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole and 4,8-Dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)" Molecules 28, no. 9: 3977. https://doi.org/10.3390/molecules28093977