The Pharmacokinetics of Vitamin C



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pharmacokinetics of Vitamin C
2.1. Oral Route of Administration
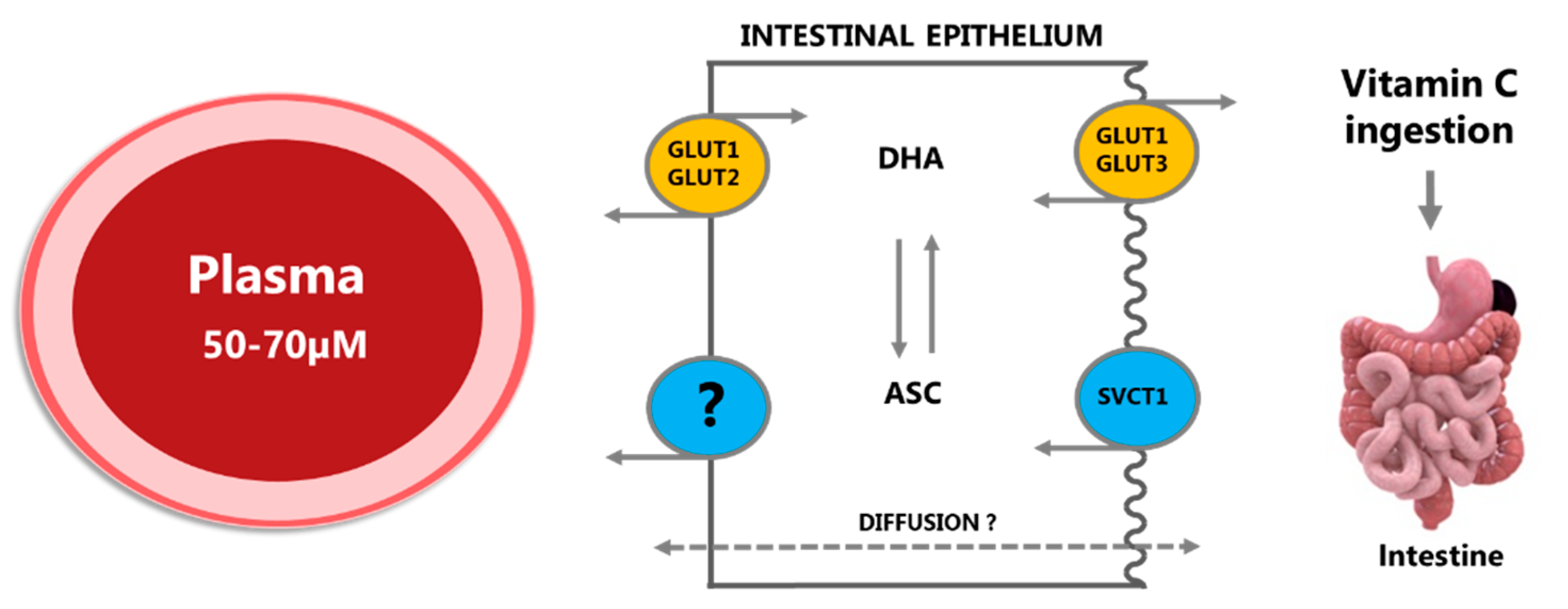
2.1.1. Absorption
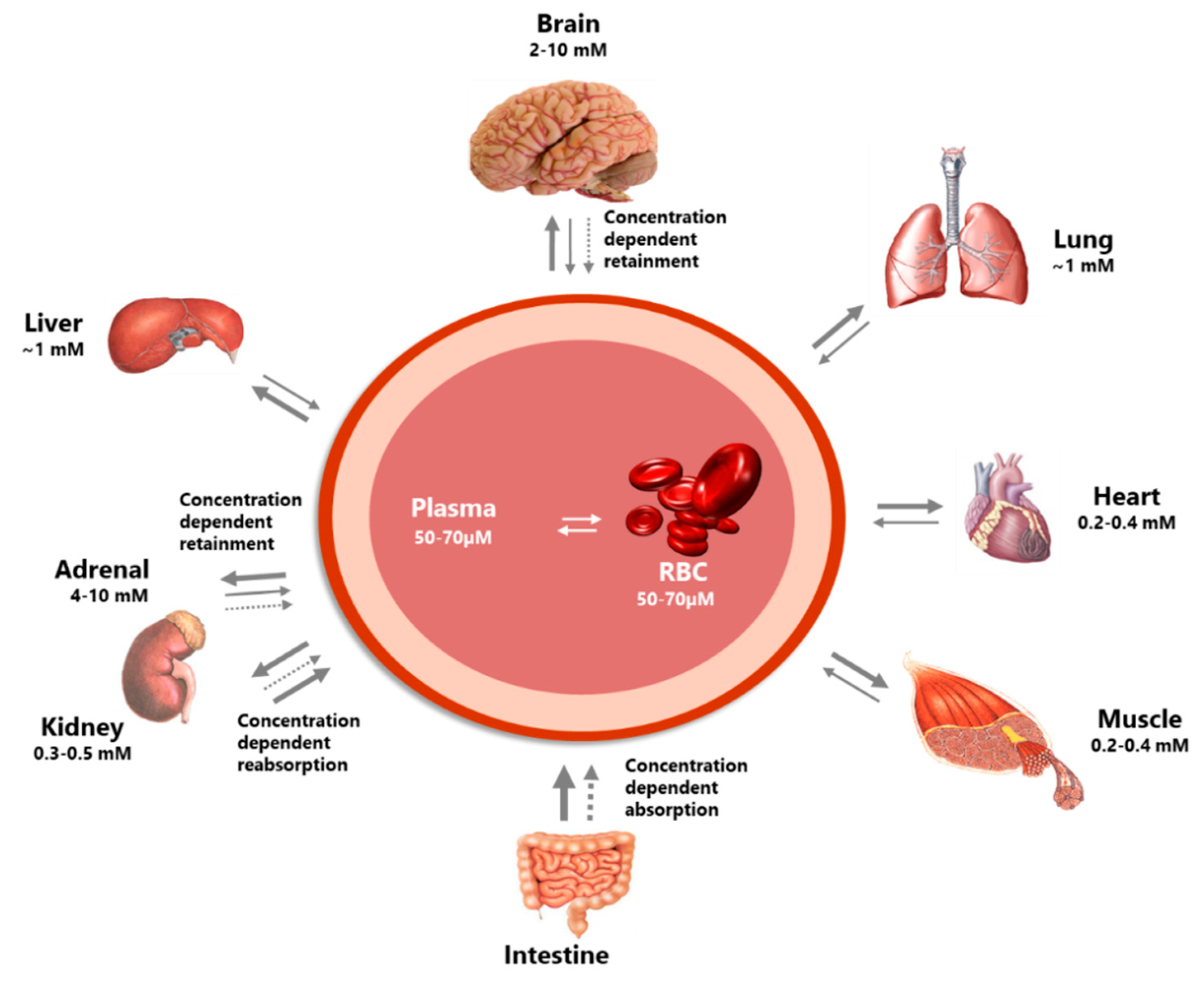
2.1.2. Distribution
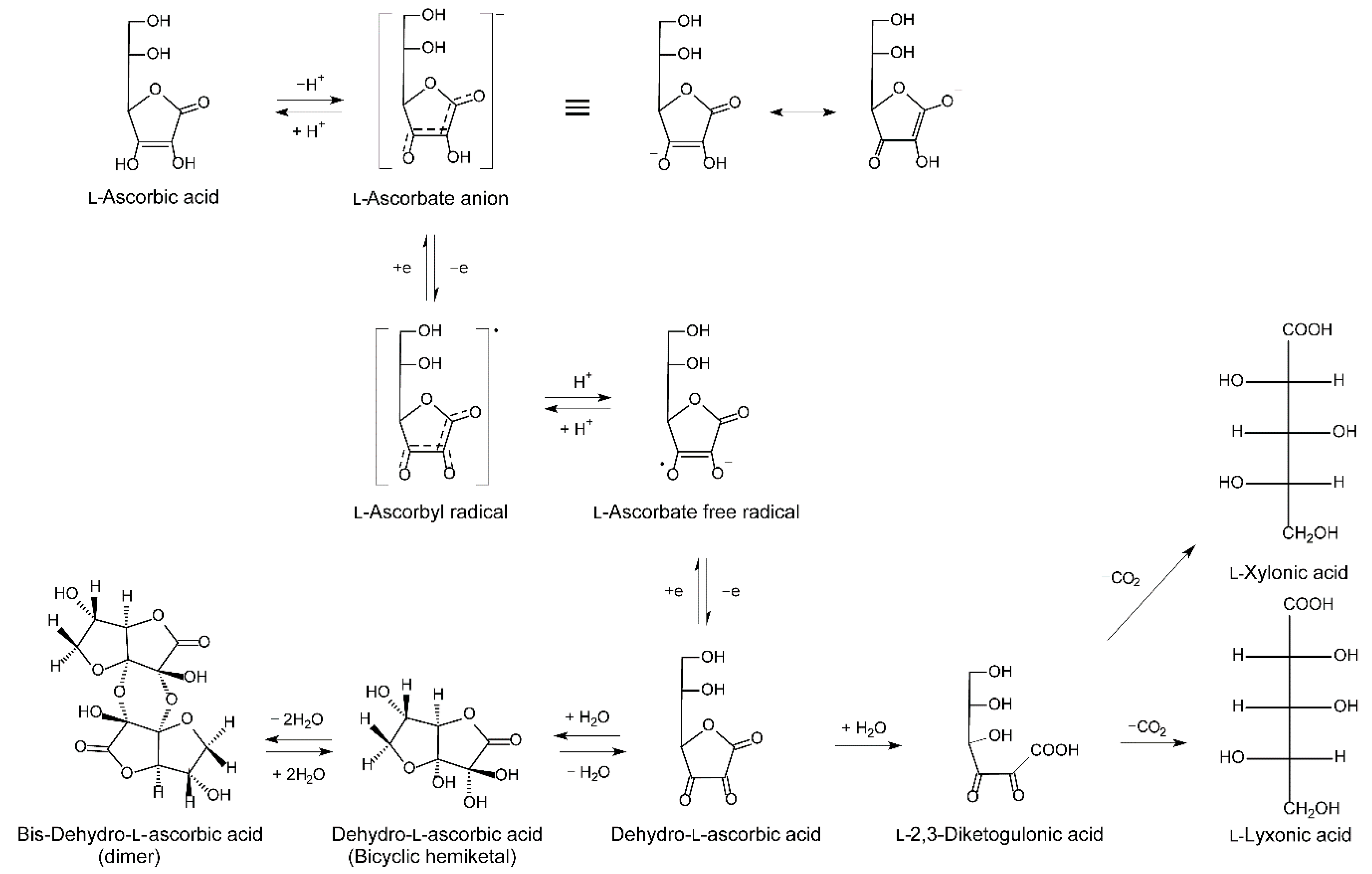
2.1.3. Metabolism
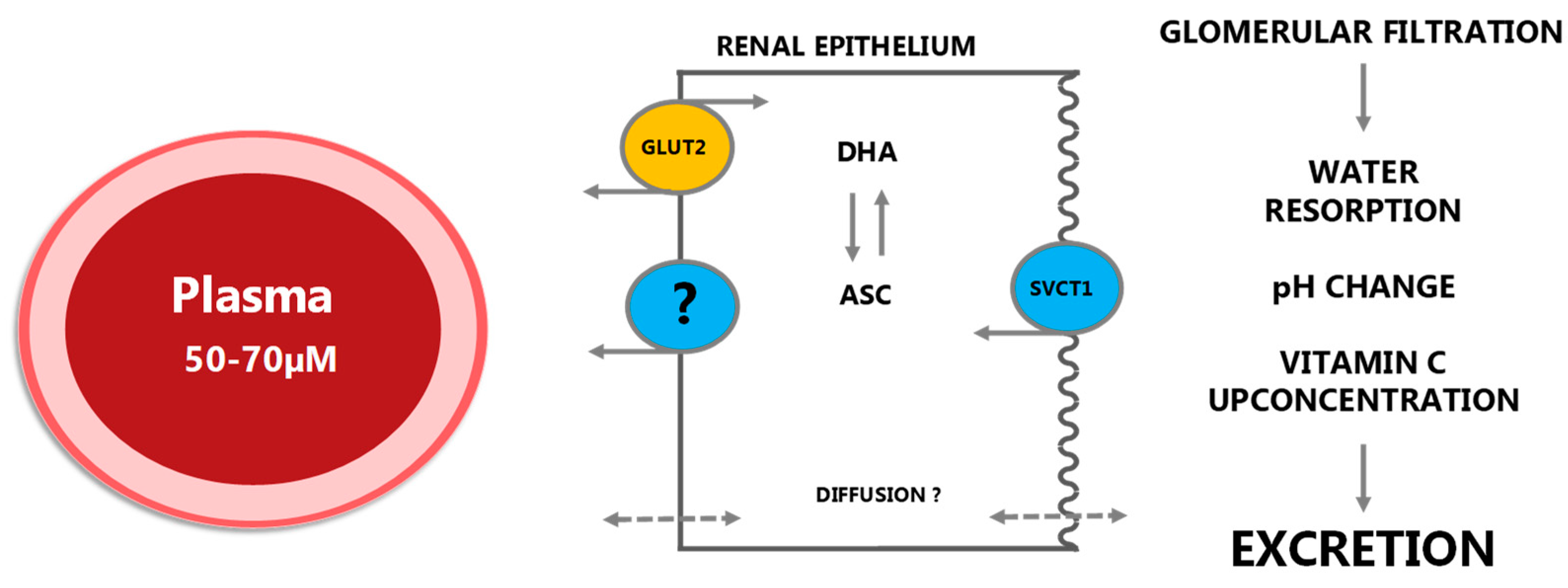
2.1.4. Excretion and Reuptake
2.1.5. Steady State Homeostasis of Vitamin C Following Oral Administration/Intake
2.1.6. Effect of Dosing Forms and Formulations
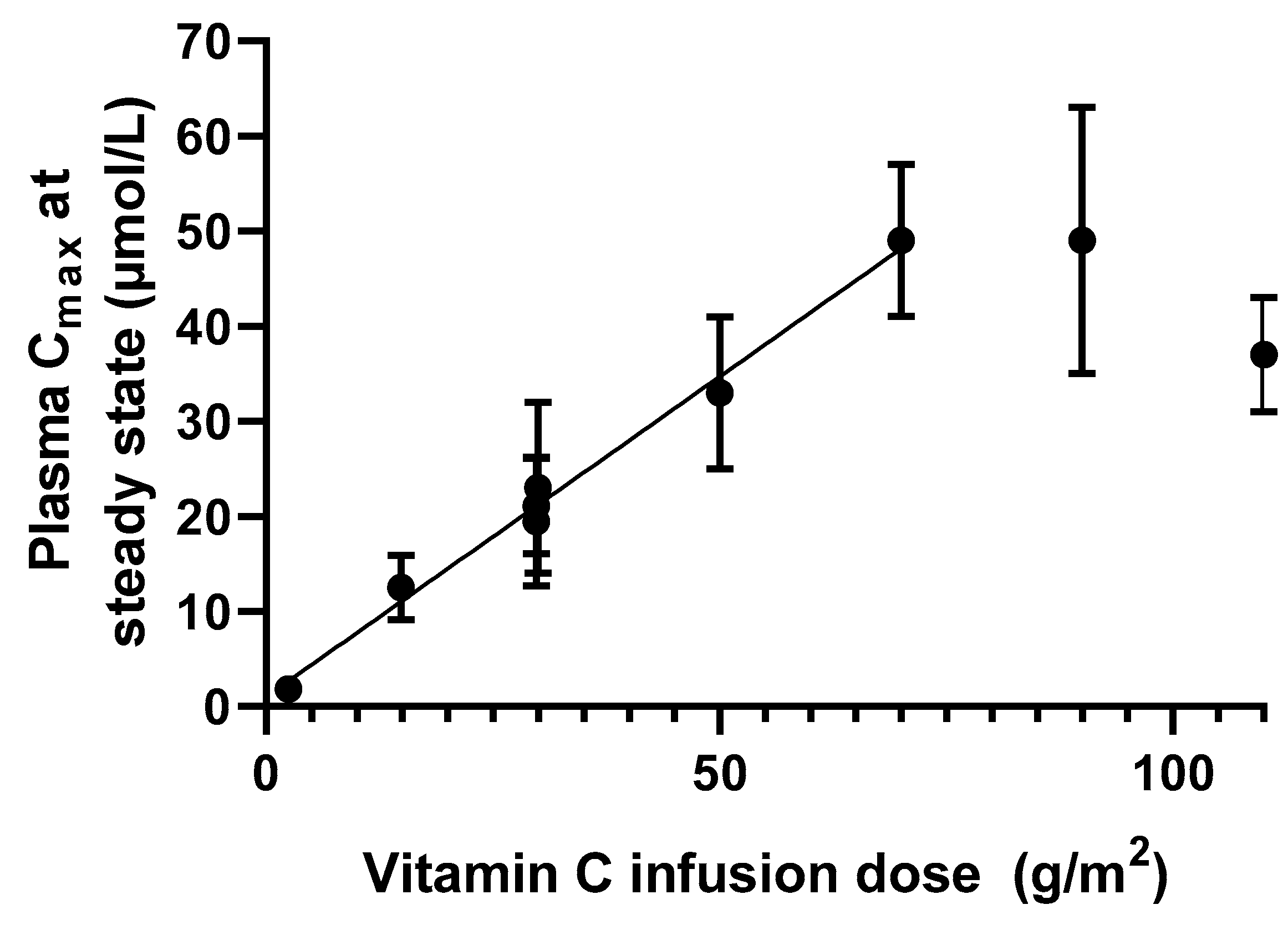
2.2. Intravenous Route of Administration
2.2.1. Distribution
2.2.2. Metabolism and Excretion
3. Factors Affecting Vitamin C Homeostasis and Requirements
3.1. Influence of Polymorphisms
3.2. Smoking
3.3. Pregnancy
3.4. Disease
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Yang, H. Conserved or lost: Molecular evolution of the key gene GULO in vertebrate vitamin C biosynthesis. Biochem. Genet. 2013, 51, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Lykkesfeldt, J. L-dehydroascorbic acid can substitute l-ascorbic acid as dietary vitamin C source in guinea pigs. Redox Biol. 2016, 7, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of vitamin C homeostasis during deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of vitamin C is tissue specific with early saturation of the brain and adrenal glands following differential oral dose regimens in guinea pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef]
- Smith, J.L.; Hodges, R.E. Serum levels of vitamin C in relation to dietary and supplemental intake of vitamin C in smokers and nonsmokers. Ann. N. Y. Acad. Sci. 1987, 498, 144–152. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.K.; Hojgaard, M.; Andersen, J.T.; Poulsen, H.E.; Lykkesfeldt, J.; Mikines, K.J. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: A pharmacokinetic evaluation. Basic Clin. Pharmacol. Toxicol. 2015, 116, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Cunningham, L.; Rose, R.C. Spontaneous decay of oxidized ascorbic acid (dehydro-L-ascorbic acid) evaluated by high-pressure liquid chromatography. Clin. Chem. 1990, 36, 1807–1809. [Google Scholar] [PubMed]
- Lykkesfeldt, J.; Loft, S.; Nielsen, J.B.; Poulsen, H.E. Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am. J. Clin. Nutr. 1997, 65, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Viscovich, M.; Poulsen, H.E. Ascorbic acid recycling in human erythrocytes is induced by smoking in vivo. Free Radic. Biol. Med. 2003, 35, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency increase lifestyle-associated vascular disease progression? Evidence based on experimental and clinical studies. Antiox. Redox Signal. 2013, 19, 2084–2104. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Vitamin C in human nutrition. In Vitamins in the Prevention of Human Disease; Herrmann, W., Obeid, R., Eds.; De Gruyter: Berlin, Germany, 2011; pp. 323–347. [Google Scholar]
- Food and Nutrition Board Staff; Panel on Dietary Antioxidants; Institute of Medicine Staff. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium and Carotenoids: A Report of the Panel on Dietary Antioxidants and Related Compounds, Subcommitties on Upper Reference Levels of Nutrients and of the Interpretation and Use of Dietary Reference Intakes, and the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, Food and Nutrition Board, Institute of Medicine; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Carr, A.C.; Bozonet, S.M.; Pullar, J.M.; Simcock, J.W.; Vissers, M.C. A randomized steady-state bioavailability study of synthetic versus natural (kiwifruit-derived) vitamin C. Nutrients 2013, 5, 3684–3695. [Google Scholar] [CrossRef] [PubMed]
- Dachs, G.U.; Munn, D.G.; Carr, A.C.; Vissers, M.C.; Robinson, B.A. Consumption of vitamin C is below recommended daily intake in many cancer patients and healthy volunteers in Christchurch. N. Z. Med. J. 2014, 127, 73–76. [Google Scholar] [PubMed]
- Lykkesfeldt, J.; Christen, S.; Wallock, L.M.; Chang, H.H.; Jacob, R.A.; Ames, B.N. Ascorbate is depleted by smoking and repleted by moderate supplementation: A study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am. J. Clin. Nutr. 2000, 71, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- May, J.; Asard, H. Ascorbate recycling. In Vitamin C; Asard, H., May, J.M., Smirnoff, N., Eds.; BIOS Scientific Publishers Ltd.: Oxford, UK, 2004; pp. 189–202. [Google Scholar]
- Lykkesfeldt, J.; Bolbjerg, M.L.; Poulsen, H.E. Effect of smoking on erythorbic acid pharmacokinetics. Br. J. Nutr. 2003, 89, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Viscovich, M.; Lykkesfeldt, J.; Poulsen, H.E. Vitamin C pharmacokinetics of plain and slow release formulations in smokers. Clin. Nutr. 2004, 23, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J. Ascorbate and dehydroascorbic acid as reliable biomarkers of oxidative stress: Analytical reproducibility and long-term stability of plasma samples subjected to acidic deproteinization. Cancer Epidemiol. Prev. Biomark. 2007, 16, 2513–2516. [Google Scholar] [CrossRef]
- Todhunter, E.N.; Mc, M.T.; Ehmke, D.A. Utilization of dehydroascorbic acid by human subjects. J. Nutr. 1950, 42, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Linkswiler, H. The effect of the Ingestion of ascorbic acid and dehydroascorbic acid upon the blood levels of these two components in human subjects. J. Nutr. 1958, 64, 43–54. [Google Scholar] [CrossRef]
- Sabry, J.H.; Fisher, K.H.; Dodds, M.L. Human utilization of dehydroascorbic acid. J. Nutr. 1958, 64, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Malo, C.; Wilson, J.X. Glucose modulates vitamin C transport in adult human small intestinal brush border membrane vesicles. J. Nutr. 2000, 130, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kubler, W.; Gehler, J. Kinetics of intestinal absorption of ascorbic acid. Calculation of non-dosage-dependent absorption processes. Int. Z. Vitaminforsch. 1970, 40, 442–453. [Google Scholar] [PubMed]
- Mayersohn, M. Ascorbic acid absorption in man—Pharmacokinetic implications. Eur. J. Pharmacol. 1972, 19, 140–142. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.C. Ascorbic acid efflux from human brain microvascular pericytes: Role of re-uptake. Biofactors 2015, 41, 330–338. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef]
- Mardones, L.; Ormazabal, V.; Romo, X.; Jana, C.; Binder, P.; Pena, E.; Vergara, M.; Zuniga, F.A. The glucose transporter-2 (GLUT2) is a low affinity dehydroascorbic acid transporter. Biochem. Biophys. Res. Commun. 2011, 410, 7–12. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.; Morrow, J.D. Mechanisms of ascorbic acid recycling in human erythrocytes. Biochim. Biophys. Acta 2001, 1528, 159–166. [Google Scholar] [CrossRef]
- Mendiratta, S.; Qu, Z.C.; May, J.M. Enzyme-dependent ascorbate recycling in human erythrocytes: Role of thioredoxin reductase. Free Radic. Biol. Med. 1998, 25, 221–228. [Google Scholar] [CrossRef]
- Mendiratta, S.; Qu, Z.C.; May, J.M. Erythrocyte ascorbate recycling: Antioxidant effects in blood. Free Radic. Biol. Med. 1998, 24, 789–797. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Increased oxidative damage in vitamin C deficiency is accompanied by induction of ascorbic acid recycling capacity in young but not mature guinea pigs. Free Radic. Res. 2002, 36, 567–574. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.C.; Whitesell, R.R. Ascorbic acid recycling enhances the antioxidant reserve of human erythrocytes. Biochemistry 1995, 34, 12721–12728. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Li, H.; Wang, Y.; Niyyati, M.; Wang, Y.; Leshin, J.; Levine, M. Low Red Blood Cell Vitamin C Concentrations Induce Red Blood Cell Fragility: A Link to Diabetes Via Glucose, Glucose Transporters, and Dehydroascorbic Acid. EBioMedicine 2015, 2, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Wang, Y.; Li, H.; Brinster, L.R.; Levine, M. Chemical Transport Knockout for Oxidized Vitamin C, Dehydroascorbic Acid, Reveals Its Functions in vivo. EBioMedicine 2017, 23, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2008, 34, 347–355. [Google Scholar] [CrossRef]
- Eck, P.; Kwon, O.; Chen, S.; Mian, O.; Levine, M. The human sodium-dependent ascorbic acid transporters SLC23A1 and SLC23A2 do not mediate ascorbic acid release in the proximal renal epithelial cell. Physiol. Rep. 2013, 1, e00136. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.E.; Harrison, F.E.; May, J.M. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem. Biophys. Res. Commun. 2011, 414, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, D.; Lindblad, M.M.; Paidi, M.D.; Hasselholt, S.; Lykkesfeldt, J.; Tveden-Nyborg, P. In vivo vitamin C deficiency in guinea pigs increases ascorbate transporters in liver but not kidney and brain. Nutr. Res. 2014, 34, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Birck, M.M.; Lykkesfeldt, J. High dietary fat and cholesterol exacerbates chronic vitamin C deficiency in guinea pigs. Br. J. Nutr. 2011, 105, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Prenatal vitamin C deficiency results in differential levels of oxidative stress during late gestation in foetal guinea pig brains. Redox Biol. 2014, 2, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, J.G.; Paidi, M.D.; Lindblad, M.M.; Birck, M.M.; Kjaergaard, A.B.; Dantzer, V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Maternal vitamin C deficiency during pregnancy results in transient fetal and placental growth retardation in guinea pigs. Eur. J. Nutr. 2015, 54, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, J.G.; Tveden-Nyborg, P.; Lykkesfeldt, J. Prolonged maternal vitamin C deficiency overrides preferential fetal ascorbate transport but does not influence perinatal survival in guinea pigs. Br. J. Nutr. 2013, 110, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic vitamin C deficiency does not accelerate oxidative stress in ageing brains of guinea pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency result in impaired brain development in infants? Redox Rep. 2009, 14, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- May, J.M. Vitamin C transport and its role in the central nervous system. In Water Soluble Vitamins; Springer: Dordrecht, The Netherlands, 2012; pp. 85–103. [Google Scholar]
- Smirnoff, N. Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radic. Biol. Med. 2018, 122, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Schafer, F.Q. Ascorbate as an antioxidant. In Vitamin C: Its Functions and Biochemistry in Animals and Plants; Asard, H., May, J.M., Smirnoff, N., Eds.; BIOS Scientific Publishers Limited: Oxford, UK, 2004; pp. 173–188. [Google Scholar]
- Banhegyi, G.; Braun, L.; Csala, M.; Puskas, F.; Mandl, J. Ascorbate metabolism and its regulation in animals. Free Radic. Biol. Med. 1997, 23, 793–803. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Paris, H.L.; Beals, J.W.; Binns, S.E.; Giordano, G.R.; Scalzo, R.L.; Schweder, M.M.; Blair, E.; Bell, C. Liposomal-encapsulated Ascorbic Acid: Influence on Vitamin C Bioavailability and Capacity to Protect Against Ischemia-Reperfusion Injury. Nutr. Metab. Insights 2016, 9, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Fuchigami, Y.; Hagimori, M.; Sato, H.; Ogawa, K.; Munakata, C.; Wada, M.; Maruyama, K.; Kawakami, S. Evaluation of the targeted delivery of 5-fluorouracil and ascorbic acid into the brain with ultrasound-responsive nanobubbles. J. Drug Target. 2018, 26, 684–691. [Google Scholar] [CrossRef]
- Peng, Y.; Zhao, Y.; Chen, Y.; Yang, Z.; Zhang, L.; Xiao, W.; Yang, J.; Guo, L.; Wu, Y. Dual-targeting for brain-specific liposomes drug delivery system: Synthesis and preliminary evaluation. Bioorg. Med. Chem. 2018, 26, 4677–4686. [Google Scholar] [CrossRef]
- Stephenson, C.M.; Levin, R.D.; Spector, T.; Lis, C.G. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother. Pharmacol. 2013, 72, 139–146. [Google Scholar] [CrossRef]
- Chen, P.; Yu, J.; Chalmers, B.; Drisko, J.; Yang, J.; Li, B.; Chen, Q. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs 2012, 23, 437–444. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- Campbell, E.J.; Vissers, M.C.M.; Wohlrab, C.; Hicks, K.O.; Strother, R.M.; Bozonet, S.M.; Robinson, B.A.; Dachs, G.U. Pharmacokinetic and anti-cancer properties of high dose ascorbate in solid tumours of ascorbate-dependent mice. Free Radic. Biol. Med. 2016, 99, 451–462. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C.; Hicks, K.O. Pharmacokinetic modeling of ascorbate diffusion through normal and tumor tissue. Free Radic. Biol. Med. 2014, 77, 340–352. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Alexander, M.S.; Waldron, T.J.; Sibenaller, Z.A.; Spitz, D.R.; Buettner, G.R.; Allen, B.G.; Cullen, J.J. Pharmacological Ascorbate as a Means of Sensitizing Cancer Cells to Radio-Chemotherapy While Protecting Normal Tissue. Semin. Radiat. Oncol. 2019, 29, 25–32. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Ou, J.; Zhu, X.; Lu, Y.; Zhao, C.; Zhang, H.; Wang, X.; Gui, X.; Wang, J.; Zhang, X.; Zhang, T.; et al. The safety and pharmacokinetics of high dose intravenous ascorbic acid synergy with modulated electrohyperthermia in Chinese patients with stage III-IV non-small cell lung cancer. Eur. J. Pharm. Sci. 2017, 109, 412–418. [Google Scholar] [CrossRef]
- Kobylecki, C.J.; Afzal, S.; Davey Smith, G.; Nordestgaard, B.G. Genetically high plasma vitamin C, intake of fruit and vegetables, and risk of ischemic heart disease and all-cause mortality: A Mendelian randomization study. Am. J. Clin. Nutr. 2015, 101, 1135–1143. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Smoking depletes vitamin C: Should smokers be recommended to take supplements? In Cigarette Smoke and Oxidative Stress; Halliwell, B., Poulsen, H.E., Eds.; Springer: Heidelberg, Berlin, 2006; pp. 237–260. [Google Scholar]
- Preston, A.M.; Rodriguez, C.; Rivera, C.E. Plasma ascorbate in a population of children: Influence of age, gender, vitamin C intake, BMI and smoke exposure. P. R. Health Sci. J. 2006, 25, 137–142. [Google Scholar]
- Preston, A.M.; Rodriguez, C.; Rivera, C.E.; Sahai, H. Influence of environmental tobacco smoke on vitamin C status in children. Am. J. Clin. Nutr. 2003, 77, 167–172. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Prieme, H.; Loft, S.; Poulsen, H.E. Effect of smoking cessation on plasma ascorbic acid concentration. BMJ 1996, 313, 91. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Viscovich, M.; Poulsen, H.E. Plasma malondialdehyde is induced by smoking: A study with balanced antioxidant profiles. Br. J. Nutr. 2004, 92, 203–206. [Google Scholar] [CrossRef]
- Canoy, D.; Wareham, N.; Welch, A.; Bingham, S.; Luben, R.; Day, N.; Khaw, K.T. Plasma ascorbic acid concentrations and fat distribution in 19,068 British men and women in the European Prospective Investigation into Cancer and Nutrition Norfolk cohort study. Am. J. Clin. Nutr. 2005, 82, 1203–1209. [Google Scholar] [CrossRef]
- Schectman, G. Estimating ascorbic acid requirements for cigarette smokers. Ann. N. Y. Acad. Sci. 1993, 686, 335–345. [Google Scholar] [CrossRef]
- Schectman, G.; Byrd, J.C.; Gruchow, H.W. The influence of smoking on vitamin C status in adults. Am. J. Public Health 1989, 79, 158–162. [Google Scholar] [CrossRef]
- Schectman, G.; Byrd, J.C.; Hoffmann, R. Ascorbic acid requirements for smokers: Analysis of a population survey. Am. J. Clin. Nutr. 1991, 53, 1466–1470. [Google Scholar] [CrossRef]
- Kallner, A.B.; Hartmann, D.; Hornig, D.H. On the requirements of ascorbic acid in man: Steady-state turnover and body pool in smokers. Am. J. Clin. Nutr. 1981, 34, 1347–1355. [Google Scholar] [CrossRef]
- Hansen, S.N.; Schou-Pedersen, A.M.V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Spatial Memory Dysfunction Induced by Vitamin C Deficiency Is Associated with Changes in Monoaminergic Neurotransmitters and Aberrant Synapse Formation. Antioxidants 2018, 7, 82. [Google Scholar] [CrossRef]
- Hansen, S.N.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency affect cognitive development and function? Nutrients 2014, 6, 3818–3846. [Google Scholar] [CrossRef]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo (-/-) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef]
- Jauniaux, E.; Poston, L.; Burton, G.J. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum. Reprod. Update 2006, 12, 747–755. [Google Scholar] [CrossRef]
- Juhl, B.; Lauszus, F.F.; Lykkesfeldt, J. Poor Vitamin C Status Late in Pregnancy Is Associated with Increased Risk of Complications in Type 1 Diabetic Women: A Cross-Sectional Study. Nutrients 2017, 9, 186. [Google Scholar] [CrossRef]
- Chappell, L.C.; Seed, P.T.; Kelly, F.J.; Briley, A.; Hunt, B.J.; Charnock-Jones, D.S.; Mallet, A.; Poston, L. Vitamin C and E supplementation in women at risk of preeclampsia is associated with changes in indices of oxidative stress and placental function. Am. J. Obstet. Gynecol. 2002, 187, 777–784. [Google Scholar] [CrossRef]
- Beazley, D.; Ahokas, R.; Livingston, J.; Griggs, M.; Sibai, B.M. Vitamin C and E supplementation in women at high risk for preeclampsia: A double-blind, placebo-controlled trial. Am. J. Obstet. Gynecol. 2005, 192, 520–521. [Google Scholar] [CrossRef]
- Villar, J.; Purwar, M.; Merialdi, M.; Zavaleta, N.; Thi Nhu Ngoc, N.; Anthony, J.; De Greeff, A.; Poston, L.; Shennan, A.; WHO Vitamin C and Vitamin E trial group. World Health Organisation multicentre randomised trial of supplementation with vitamins C and E among pregnant women at high risk for pre-eclampsia in populations of low nutritional status from developing countries. BJOG 2009, 116, 780–788. [Google Scholar] [CrossRef]
- Chappell, L.C.; Seed, P.T.; Briley, A.L.; Kelly, F.J.; Lee, R.; Hunt, B.J.; Parmar, K.; Bewley, S.J.; Shennan, A.H.; Steer, P.J.; et al. Effect of antioxidants on the occurrence of pre-eclampsia in women at increased risk: A randomised trial. Lancet 1999, 354, 810–816. [Google Scholar] [CrossRef]
- Juhl, B.; Lauszus, F.F.; Lykkesfeldt, J. Is Diabetes Associated with Lower Vitamin C Status in Pregnant Women? A Prospective Study. Int. J. Vitam. Nutr. Res. 2016, 86, 184–189. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Lykkesfeldt, J. Role of marginal vitamin C deficiency in atherogenesis: In vivo models and clinical studies. Basic Clin. Pharmacol. Toxicol. 2009, 104, 419–433. [Google Scholar] [CrossRef]
- Traber, M.G.; Buettner, G.R.; Bruno, R.S. The relationship between vitamin C status, the gut-liver axis, and metabolic syndrome. Redox Biol. 2019, 21, 101091. [Google Scholar] [CrossRef]
- Carr, A.C.; Rosengrave, P.C.; Bayer, S.; Chambers, S.; Mehrtens, J.; Shaw, G.M. Hypovitaminosis C and vitamin C deficiency in critically ill patients despite recommended enteral and parenteral intakes. Crit. Care 2017, 21, 300. [Google Scholar] [CrossRef]
- Marik, P.E. Vitamin C for the treatment of sepsis: The scientific rationale. Pharmacol. Ther. 2018, 189, 63–70. [Google Scholar] [CrossRef]
- Carr, A.C.; Shaw, G.M.; Fowler, A.A.; Natarajan, R. Ascorbate-dependent vasopressor synthesis: A rationale for vitamin C administration in severe sepsis and septic shock? Crit. Care 2015, 19, 418. [Google Scholar] [CrossRef]
- Fowler, A.A., 3rd; Syed, A.A.; Knowlson, S.; Sculthorpe, R.; Farthing, D.; DeWilde, C.; Farthing, C.A.; Larus, T.L.; Martin, E.; Brophy, D.F.; et al. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J. Transl. Med. 2014, 12, 32. [Google Scholar] [CrossRef]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, Vitamin C, and Thiamine for the Treatment of Severe Sepsis and Septic Shock: A Retrospective Before-After Study. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef]
- Hill, A.; Clasen, K.C.; Wendt, S.; Majoros, A.G.; Stoppe, C.; Adhikari, N.K.J.; Heyland, D.K.; Benstoem, C. Effects of Vitamin C on Organ Function in Cardiac Surgery Patients: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 2103. [Google Scholar] [CrossRef]
- Maxwell, S.R.; Thomason, H.; Sandler, D.; Leguen, C.; Baxter, M.A.; Thorpe, G.H.; Jones, A.F.; Barnett, A. HAntioxidant status in patients with uncomplicated insulin-dependent and non-insulin-dependent diabetes mellitus. Eur. J. Clin. Investig. 1997, 27, 484–490. [Google Scholar] [CrossRef]
- Stankova, L.; Riddle, M.; Larned, J.; Burry, K.; Menashe, D.; Hart, J.; Bigley, R. Plasma ascorbate concentrations and blood cell dehydroascorbate transport in patients with diabetes mellitus. Metabolism 1984, 33, 347–353. [Google Scholar] [CrossRef]
- Feskens, E.J.; Virtanen, S.M.; Rasanen, L.; Tuomilehto, J.; Stengard, J.; Pekkanen, J.; Nissinen, A.; Kromhout, D. Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study. Diabetes Care 1995, 18, 1104–1112. [Google Scholar]
- Paolisso, G.; Balbi, V.; Volpe, C.; Varricchio, G.; Gambardella, A.; Saccomanno, F.; Ammendola, S.; Varricchio, M.; D’Onofrio, F. Metabolic benefits deriving from chronic vitamin C supplementation in aged non-insulin dependent diabetics. J. Am. Coll. Nutr. 1995, 14, 387–392. [Google Scholar] [CrossRef]
- Song, Y.; Xu, Q.; Park, Y.; Hollenbeck, A.; Schatzkin, A.; Chen, H. Multivitamins, individual vitamin and mineral supplements, and risk of diabetes among older U.S. adults. Diabetes Care 2011, 34, 108–114. [Google Scholar] [CrossRef]
- Mason, S.A.; Baptista, R.; Della Gatta, P.A.; Yousif, A.; Russell, A.P.; Wadley, G.D. High-dose vitamin C supplementation increases skeletal muscle vitamin C concentration and SVCT2 transporter expression but does not alter redox status in healthy males. Free Radic. Biol. Med. 2014, 77, 130–138. [Google Scholar] [CrossRef]
- Mason, S.A.; Rasmussen, B.; van Loon, L.J.C.; Salmon, J.; Wadley, G.D. Ascorbic acid supplementation improves postprandial glycaemic control and blood pressure in individuals with type 2 diabetes: Findings of a randomized cross-over trial. Diabetes Obes. Metab. 2019, 21, 674–682. [Google Scholar] [CrossRef]
- Odermarsky, M.; Lykkesfeldt, J.; Liuba, P. Poor vitamin C status is associated with increased carotid intima-media thickness, decreased microvascular function, and delayed myocardial repolarization in young patients with type 1 diabetes. Am. J. Clin. Nutr. 2009, 90, 447–452. [Google Scholar] [CrossRef]
- Tousoulis, D.; Antoniades, C.; Tountas, C.; Bosinakou, E.; Kotsopoulou, M.; Toutouzas, P.; Stefanadis, C. Vitamin C affects thrombosis/ fibrinolysis system and reactive hyperemia in patients with type 2 diabetes and coronary artery disease. Diabetes Care 2003, 26, 2749–2753. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. https://doi.org/10.3390/nu11102412
Lykkesfeldt J, Tveden-Nyborg P. The Pharmacokinetics of Vitamin C. Nutrients. 2019; 11(10):2412. https://doi.org/10.3390/nu11102412
Chicago/Turabian StyleLykkesfeldt, Jens, and Pernille Tveden-Nyborg. 2019. "The Pharmacokinetics of Vitamin C" Nutrients 11, no. 10: 2412. https://doi.org/10.3390/nu11102412
APA StyleLykkesfeldt, J., & Tveden-Nyborg, P. (2019). The Pharmacokinetics of Vitamin C. Nutrients, 11(10), 2412. https://doi.org/10.3390/nu11102412