
Factors Involved in the Apoptotic Cell Death Mechanism in Yellow Fever Hepatitis

 ,
,  ,
,  , , , , ,
, , , , ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Method
2.1. Ethics Statement
2.2. Histopathology and Immunohistochemistry
2.3. Quantitative Analysis and Photodocumentation
2.4. Statistical Analysis
3. Results
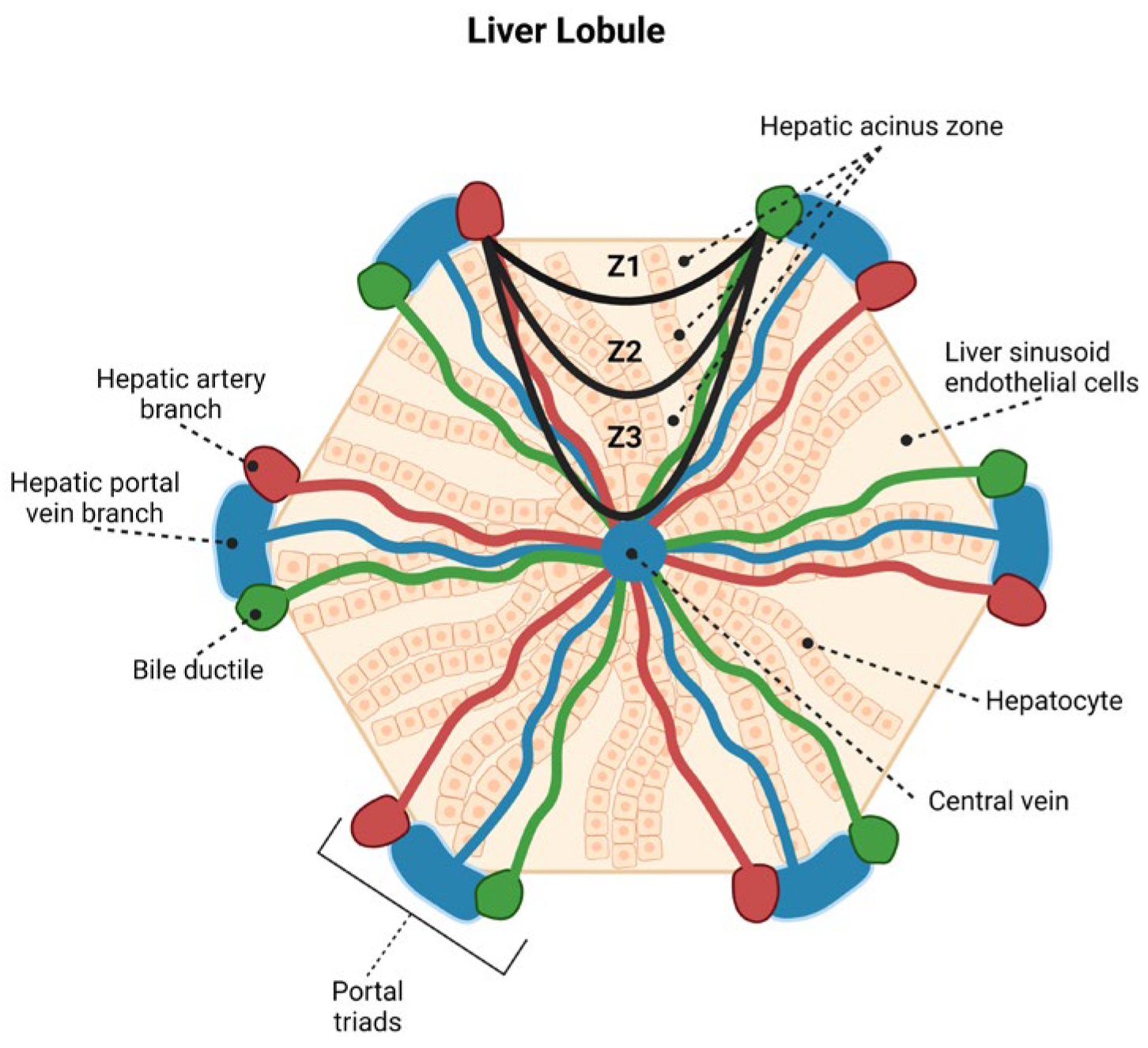
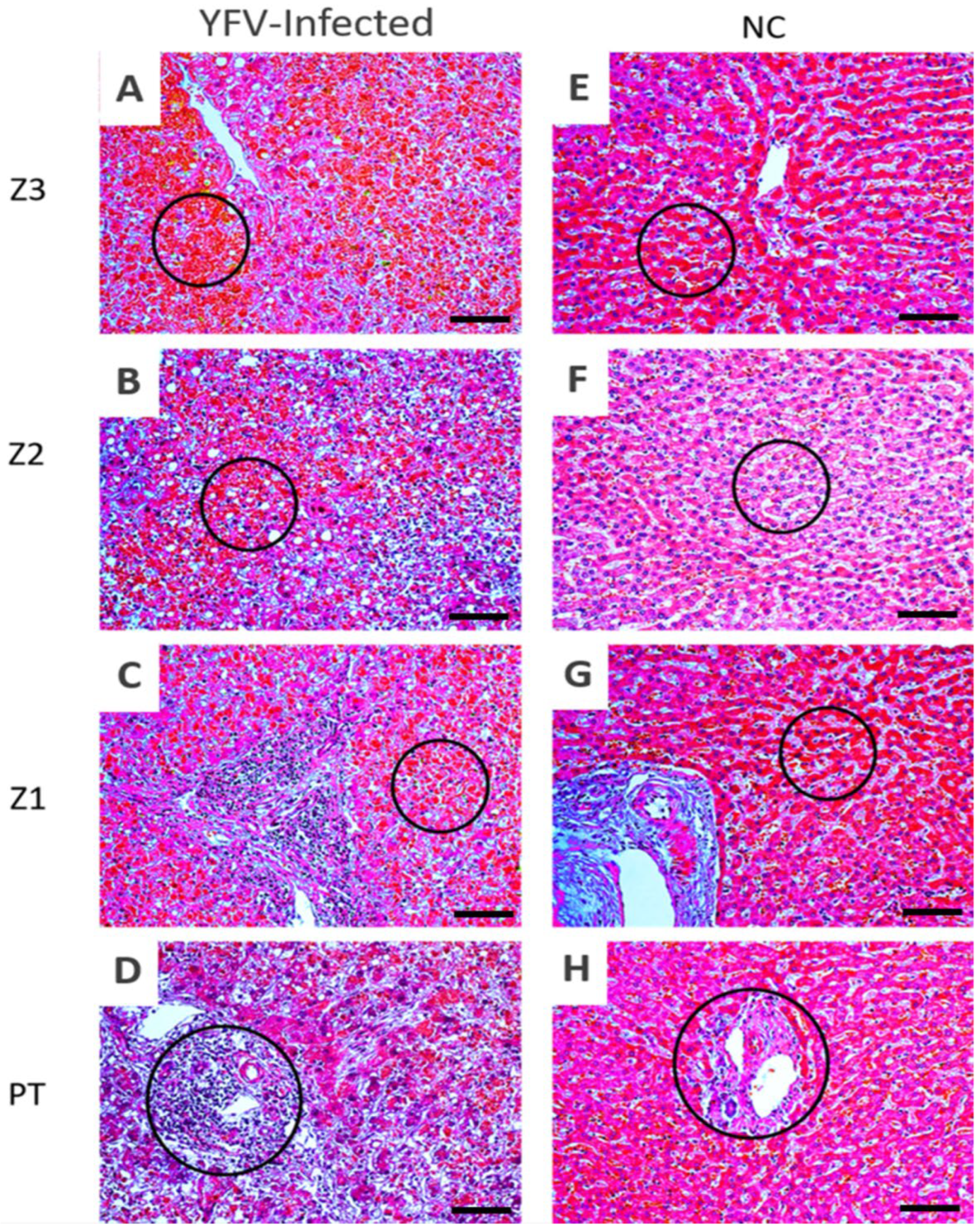
3.1. Histopathology
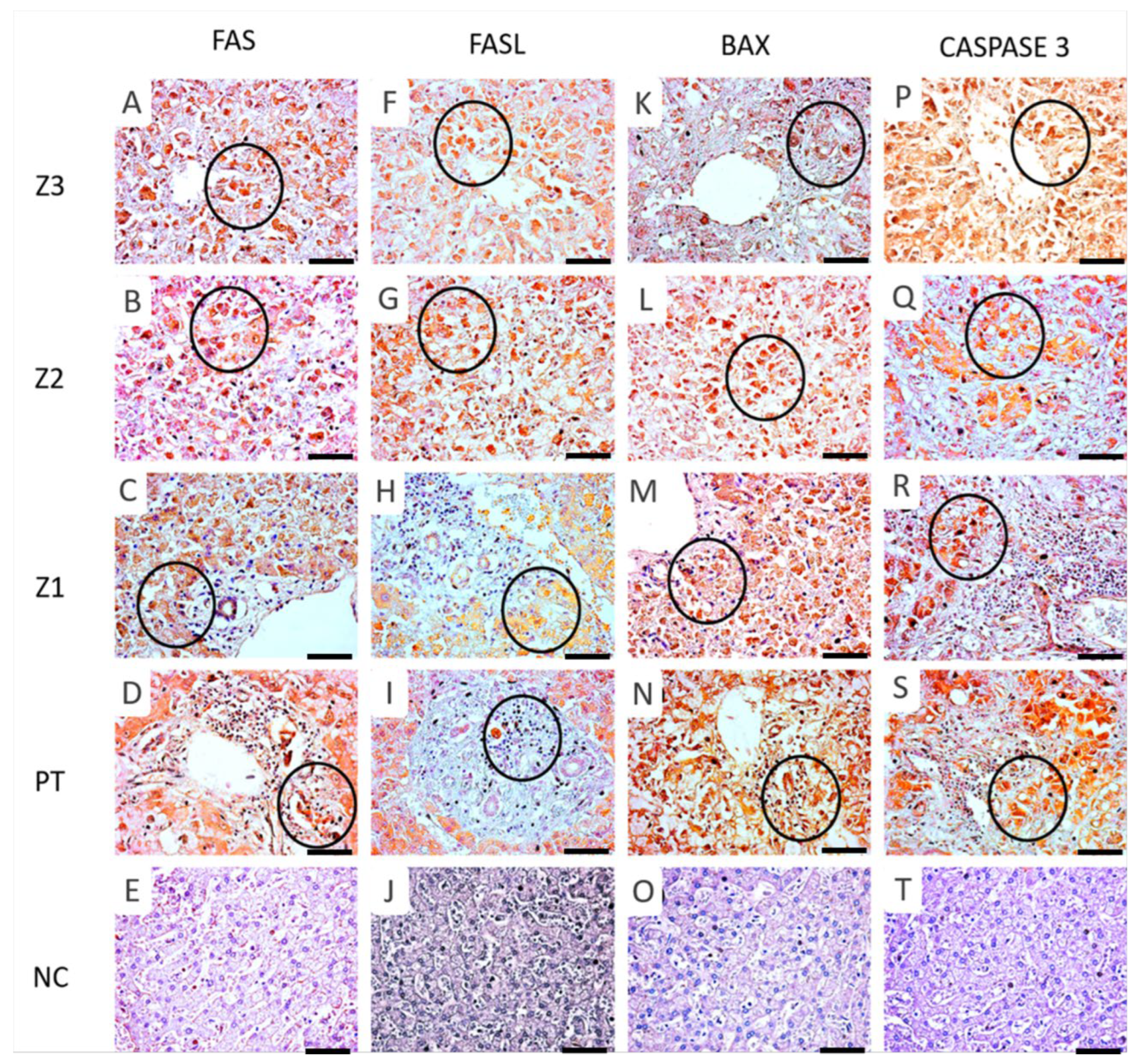
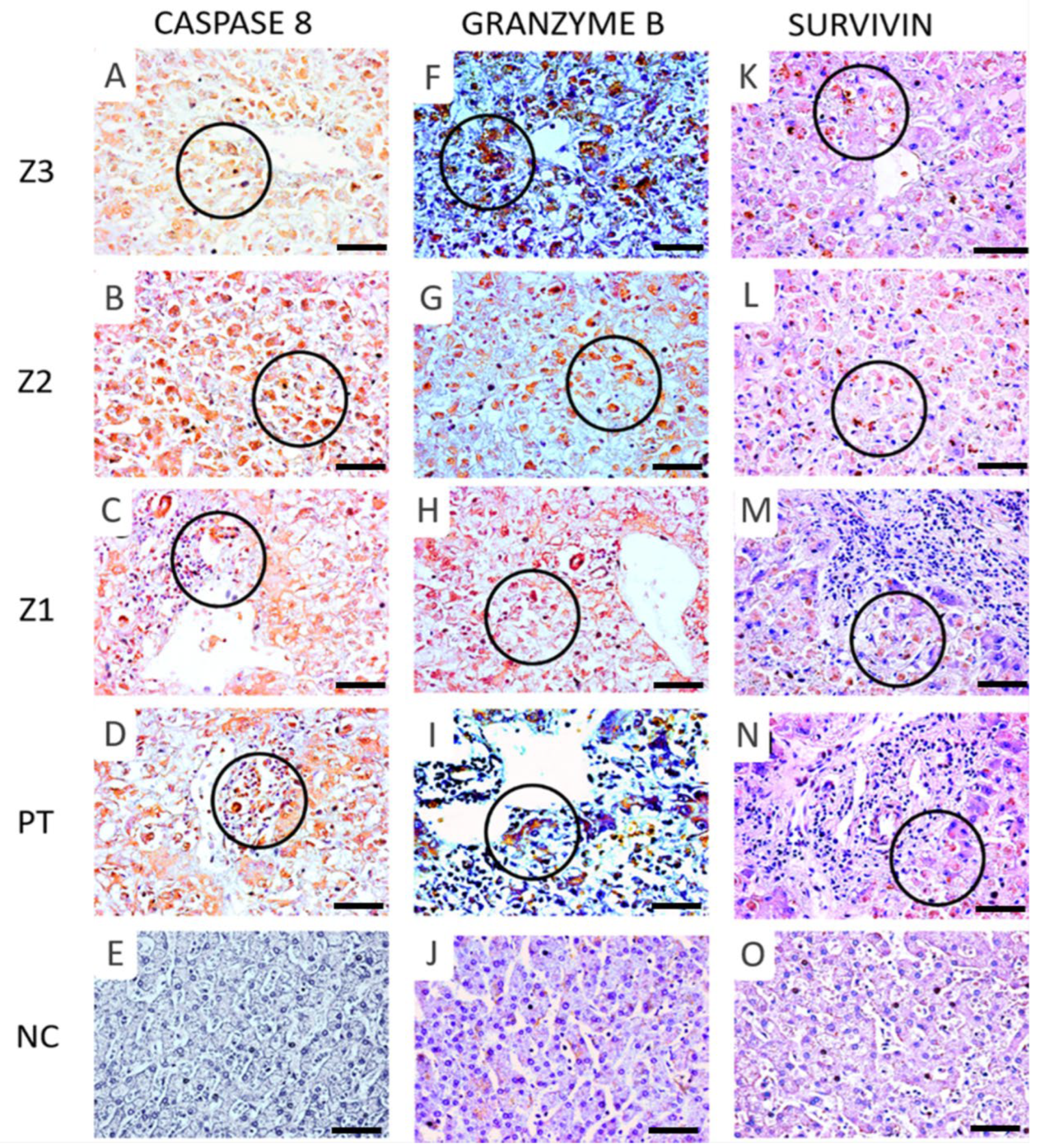
3.2. Immunoexpression of Apoptotic Markers
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vasconcelos, P.F.; Bryant, J.E.; Da Rosa, A.P.T.; Tesh, R.B.; Rodrigues, S.G.; Barrett, A.D. Genetic Divergence and Dispersal of Yellow Fever Virus, Brazil. Emerg. Infect. Dis. 2004, 10, 1578–1584. [Google Scholar] [CrossRef] [Green Version]
- Reed, W.; Carroll, J. Yellow Fever: 100 Years of Discovery the Etiology of Yellow Fever: An Additional Note. JAMA Class. 2008, 36, 1–3. [Google Scholar]
- Mutebi, J.-P.; Wang, H.; Li, L.; Bryant, J.; Barrett, A.D.T. Phylogenetic and Evolutionary Relationships among Yellow Fever Virus Isolates in Africa. J. Virol. 2001, 75, 6999–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto-Lima, D.; Madec, Y.; Bersot, M.I.; Campos, S.S.; Motta, M.D.A.; Santos, F.; Vazeille, M.; Vasconcelos, P.F.D.C.; Lourenço-De-Oliveira, R.; Failloux, A.-B. Potential risk of re-emergence of urban transmission of Yellow Fever virus in Brazil facilitated by competent Aedes populations. Sci. Rep. 2017, 7, 4848. [Google Scholar] [CrossRef] [PubMed]
- Servadio, J.L.; Muñoz-Zanzi, C.; Convertino, M. Estimating case fatality risk of severe Yellow Fever cases: Systematic literature review and meta-analysis. BMC Infect. Dis. 2021, 21, 89. [Google Scholar] [CrossRef]
- Ferreira, M.S.; Bezerra Júnior, P.S.; Cerqueira, V.D.; Rivero, G.R.C.; Oliveira Júnior, C.A.; Castro, P.H.G.; da Silva, G.A.; da Silva, W.B.; Imbeloni, A.A.; Sousa, J.R.; et al. Experimental Yellow Fever Virus Infection in the Squirrel Monkey (Saimiri Spp.) I: Gross Anatomical and Histopathological Findings in Organs at Necropsy. Mem. Inst. Oswaldo Cruz 2020, 115, 1–8. [Google Scholar] [CrossRef]
- de Oliveira, G.M.M.; Ferreira, R.M. Yellow Fever and Cardiovascular Disease: An Intersection of Epidemics. Arq. Bras. Cardiol. 2018, 110, 207–210. [Google Scholar] [CrossRef]
- Quaresma, J.A.; Barros, V.L.; Pagliari, C.; Fernandes, E.R.; Guedes, F.; Takakura, C.F.; Andrade, H.F., Jr.; Vasconcelos, P.F.; Duarte, M.I. Revisiting the liver in human yellow fever: Virus-induced apoptosis in hepatocytes associated with TGF-β, TNF-α and NK cells activity. Virology 2006, 345, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Monath, T.P.; Vasconcelos, P.F. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Quaresma, J.A.; Duarte, M.I.; Vasconcelos, P.F. Midzonal lesions in yellow fever: A specific pattern of liver injury caused by direct virus action and in situ inflammatory response. Med. Hypotheses 2006, 67, 618–621. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Pagliari, C.; Medeiros, D.B.A.; Duarte, M.I.S.; Vasconcelos, P.F.C. Immunity and immune response, pathology and pathologic changes: Progress and challenges in the immunopathology of yellow fever. Rev. Med. Virol. 2013, 23, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J. Arboviruses and apoptosis: The role of cell death in determining vector competence. J. Gen. Virol. 2016, 97, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Holanda, G.M.; Casseb, S.M.M.; Quaresma, J.A.S.; Vasconcelos, P.F.C.; Cruz, A.C.R. Yellow fever virus modulates cytokine mRNA expression and induces activation of caspase 3/7 in the human hepatocarcinoma cell line HepG2. Arch. Virol. 2019, 164, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, A.; Contamin, H.; Decelle, T.; Fournier, C.; Lang, J.; Deubel, V.; Marianneau, P. Host-cell interaction of attenuated and wild-type strains of yellow fever virus can be differentiated at early stages of hepatocyte infection. Microbes Infect. 2006, 8, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Brüning, J.; Weigel, B.; Pietschmann, T. Protein Interactions during the Flavivirus and Hepacivirus Life Cycle. Mol. Cell. Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Fernandes, E.R.; Pagliari, C.; Guedes, F.; Vasconcelos, P.F.D.C.; Junior, H.F.D.A.; Duarte, M.I.S. Immunohistochemical examination of the role of Fas ligand and lymphocytes in the pathogenesis of human liver yellow fever. Virus Res. 2006, 116, 91–97. [Google Scholar] [CrossRef]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral Control of Mitochondrial Apoptosis. PLOS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Gores, G.J. Cellular and Molecular Mechanisms of Liver Injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte Death: A Clear and Present Danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. The Dual Regulation of Apoptosis by Flavivirus. Front. Microbiol. 2021, 12, 574. [Google Scholar] [CrossRef]
- Okamoto, T.; Suzuki, T.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Matsuura, Y. Regulation of Apoptosis during Flavivirus Infection. Viruses 2017, 9, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and Necrosis in the Liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [CrossRef] [Green Version]
- Garber, C.; Soung, A.; Vollmer, L.L.; Kanmogne, M.; Last, A.; Brown, J.; Klein, R.S. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 2019, 22, 1276–1288. [Google Scholar] [CrossRef]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sousa, J.R.; Azevedo, R.D.S.D.S.; Quaresma, J.A.S.; Vasconcelos, P.F.D.C. Cell Death And Zika Virus: An Integrated Network Of The Mechanisms Of Cell Injury. Infect. Drug Resist. 2019, 12, 2917–2921. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, e00887. [Google Scholar] [CrossRef] [Green Version]
- Suwanmanee, S.; Luplertlop, N. Immunopathogenesis of Dengue Virus-Induced Redundant Cell Death: Apoptosis and Pyroptosis. Viral Immunol. 2017, 30, 13–19. [Google Scholar] [CrossRef]
- Roy, S.G.; Sadigh, B.; Datan, E.; Lockshin, A.R.; Zakeri, Z. Regulation of cell survival and death during Flavivirus infections. World J. Biol. Chem. 2014, 5, 93–105. [Google Scholar] [CrossRef]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.-M.; Lee, C.-K.; Wu-Hsieh, B.A.-Y. Intrahepatic Infiltrating NK and CD8 T Cells Cause Liver Cell Death in Different Phases of Dengue Virus Infection. PLoS ONE 2012, 7, e46292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, R.S.S.; De Sousa, J.R.; Araujo, M.T.F.; Filho, A.J.M.; De Alcantara, B.N.; Araujo, F.M.C.; Queiroz, M.G.L.; Cruz, A.C.R.; Vasconcelos, B.H.B.; Chiang, J.O.; et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Tian, Y.; Sette, A.; Weiskopf, D. Transcriptomic immune profiles of human flavivirus-specific T-cell responses. Immunology 2019, 160, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.E.; Howerth, E.W. Survivin: A Bifunctional Inhibitor of Apoptosis Protein. Veter. Pathol. 2004, 41, 599–607. [Google Scholar] [CrossRef]
- Nassar, A.; Lawson, D.; Cotsonis, G.; Cohen, C. Survivin and Caspase-3 Expression in Breast Cancer: Correlation with Prognostic Parameters, Proliferation, Angiogenesis, and Outcome. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 113–120. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Reference | Dilution |
|---|---|---|
| BAX | Abcam/ab32503 | 1:100 |
| SURVIVIN | Novus/NB500-201 | 1:100 |
| FAS | Dako/m3555 | 1:100 |
| FASL | Abcam/ab15285 | 1:100 |
| GRANZYME B | Dako/m7235 | 1:100 |
| CASPASE 3 | Abcam/ab4051 | 1:100 |
| CASPASE 8 | Abcam/ab25901 | 1:100 |
| Markers (Cells/mm2) | Z3 | Tukey (p ≤ 0.05) | Z2 | Tukey (p ≤ 0.05) | Z1 | Tukey (p ≤ 0.05) | TP | Tukey (p ≤ 0.05) | ANOVA (p ≤ 0.05) |
|---|---|---|---|---|---|---|---|---|---|
| BAX | 68.53 ± 8.36 | *** | 89.9 ± 4.9 | *** | 59.64 ± 4.37 | *** | 54.24 ± 5.54 | *** | *** |
| Control | 17.28 ± 4.5 | 17.9 ± 9.0 | 18.56 ± 6.36 | 16.0 ± 3.9 | |||||
| GRANZYME B | 50.92 ± 6.9 | *** | 61.64 ± 4.25 | *** | 49.22 ± 5.35 | *** | 43.7 ± 5.7 | *** | *** |
| Control | 17.92 ± 3.2 | 16.96 ± 3.3 | 15.36 ± 3.85 | 15.68 ± 3.64 | |||||
| FAS | 45.56 ± 4.99 | *** | 55.38 ± 3.3 | *** | 41.9 ± 4.3 | *** | 40.97 ± 4.4 | *** | *** |
| Control | 14.4 ± 4.0 | 15.36 ± 8.2 | 15.36 ± 3.8 | 12.16 ± 3.8 | |||||
| FASL | 61.56 ± 3.9 | *** | 84.13 ± 7.5 | *** | 60.34 ± 5.3 | *** | 57.37 ± 4.0 | *** | *** |
| Control | 17.28 ± 3.8 | 16.64 ± 6.3 | 18.8 ± 3.6 | 17.2 ± 2.8 | |||||
| SURVIVIN | 46.1 ± 7.2 | *** | 54.32 ± 5.2 | *** | 44.8 ± 6.0 | *** | 44.11 ± 3.7 | *** | *** |
| Control | 16.28 ± 3.4 | 16.96 ± 3.3 | 15.68 ± 2.6 | 13.44 ± 3.1 | |||||
| CASPASE 3 | 60.65 ± 7.6 | *** | 75.57 ± 9.9 | *** | 49.45 ± 6.1 | *** | 48.06 ± 4.7 | *** | *** |
| Control | 15.0 ± 4.0 | 19.8 ± 3.8 | 14.4 ± 2.2 | 14.76 ± 2.8 | |||||
| CASPASE 8 | 61.94 ± 6.0 | *** | 80.91 ± 7.9 | *** | 60.95 ± 9.9 | *** | 54.78 ± 4.6 | *** | *** |
| Control | 14.08 ± 2.3 | 22.72 ± 4.2 | 16.0 ± 2.9 | 14.4 ± 2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Costa Lopes, J.; Falcão, L.F.M.; Martins Filho, A.J.; Carvalho, M.L.G.; Mendes, C.C.H.; Olímpio, F.A.; do Socorro Cabral Miranda, V.; dos Santos, L.C.; Chiang, J.O.; Cruz, A.C.R.; et al. Factors Involved in the Apoptotic Cell Death Mechanism in Yellow Fever Hepatitis. Viruses 2022, 14, 1204. https://doi.org/10.3390/v14061204
da Costa Lopes J, Falcão LFM, Martins Filho AJ, Carvalho MLG, Mendes CCH, Olímpio FA, do Socorro Cabral Miranda V, dos Santos LC, Chiang JO, Cruz ACR, et al. Factors Involved in the Apoptotic Cell Death Mechanism in Yellow Fever Hepatitis. Viruses. 2022; 14(6):1204. https://doi.org/10.3390/v14061204
Chicago/Turabian Styleda Costa Lopes, Jeferson, Luiz Fábio Magno Falcão, Arnaldo Jorge Martins Filho, Marcos Luiz Gaia Carvalho, Caio Cesar Henriques Mendes, Fábio Alves Olímpio, Vanessa do Socorro Cabral Miranda, Lais Carneiro dos Santos, Jannifer Oliveira Chiang, Ana Cecilia Ribeiro Cruz, and et al. 2022. "Factors Involved in the Apoptotic Cell Death Mechanism in Yellow Fever Hepatitis" Viruses 14, no. 6: 1204. https://doi.org/10.3390/v14061204
APA Styleda Costa Lopes, J., Falcão, L. F. M., Martins Filho, A. J., Carvalho, M. L. G., Mendes, C. C. H., Olímpio, F. A., do Socorro Cabral Miranda, V., dos Santos, L. C., Chiang, J. O., Cruz, A. C. R., Galúcio, V. C. A., do Socorro da Silva Azevedo, R., Martins, L. C., Duarte, M. I. S., de Sousa, J. R., da Costa Vasconcelos, P. F., & Quaresma, J. A. S. (2022). Factors Involved in the Apoptotic Cell Death Mechanism in Yellow Fever Hepatitis. Viruses, 14(6), 1204. https://doi.org/10.3390/v14061204