Abstract
The G2P[4] genotype is among the rotavirus strains that circulate commonly in humans. Several countries have reported its immediate upsurge after the introduction of rotavirus vaccination, raising concern about sub-optimal vaccine effectiveness against this genotype in the long term. This study aimed to gain insight into the evolution of post-vaccine Zambian G2P[4] group A rotavirus (RVA) strains and their overall genetic make-up by analysis of sequence alignments at the amino acid (AA) level. Twenty-nine Zambian G2P[4] rotavirus strains were subjected to whole-genome sequencing using the Illumina MiSeq® platform. All the strains exhibited the typical DS-1-like genotype constellation, and the nucleotide sequences of the 11 genome segments showed high nucleotide similarities (>97%). Phylogenetic analyses together with representative global G2P[4] RVA showed that Zambian strains clustered into human lineages IV (for VP2, VP4, VP7, NSP1, and NSP5), V (for VP1, VP3, VP6, NSP2, and NSP3), and XXIII (for NSP4). The AA differences between the lineages where the study strains clustered and lineages of global reference strains were identified and analyzed. Selection pressure analysis revealed that AA site seven in the Viral Protein 3 (VP3) genome segment was under positive selection. This site occurs in the region of intrinsic disorder in the VP3 protein, and Zambian G2P[4] strains could potentially be utilizing this intrinsically disordered region to survive immune pressure. The Zambian G2P[4] strains from 2012 to 2016 comprised the G2P[4] strains that have been circulating globally since the early 2000s, highlighting the epidemiological fitness of these contemporary G2P[4] strains. Continuous whole-genome surveillance of G2P[4] strains remains imperative to understand their evolution during the post-vaccination period.
1. Introduction
Group A rotavirus (RVA)-induced acute gastroenteritis persists among the leading causes of mortality in children under five years, particularly in sub-Saharan Africa and southeast Asia [1]. To alleviate RVA disease burden, the World Health Organization (WHO) prequalified four rotavirus vaccines, namely Rotarix® (GlaxoSmithKline, Rixenstart, Belgium), RotaTeq® (Merck & Co., Whitehouse Station, NJ, USA), Rotavac® (Bharat Biotech, India), and Rotasiil® (Serum Institute, India), after extensive efficacy and safety studies (https://www.who.int/publications/i/item/WHO-IVB-2021.03 (accessed on 10 January 2023)). Vaccination and, to an extent, improvement in sanitation have substantially reduced RVA disease mortality cases from ~500,000 during the pre-vaccination era to ~128,000 during the post-vaccination period [1,2]. In Zambia, the Rotarix® vaccine was introduced in January 2012 in Lusaka as a pilot project and later rolled out countrywide in November 2013 [3,4], with vaccine coverage of 87% in 2021 (https://immunizationdata.who.int/pages/profiles/zmb.html (accessed on 17 January 2023)). Rotavirus vaccination has been shown to have a significant impact on rotavirus hospitalizations and deaths in Africa [5] although the widespread use of RVA vaccines has raised concerns about vaccine-induced selective pressure on circulating RVA strains, which may result in putative vaccine-escape mutants that may affect the effectiveness of RVA vaccines in the long term [6].
Rotavirus belongs to the Sedoreoviridae family of the order Reovirales and comprises 11 genome segments [7]. The ~18 kb rotavirus genome is organized into six structural proteins, namely viral proteins (VP1–4, 6, and 7) and five non-structural proteins (NSP1–5) [7]. The capsid proteins, VP7 (denoted G for its glycoprotein nature) and VP4 (denoted P due to its protease sensitivity), act as neutralizing antigens and are traditionally used to classify rotavirus strains into a G/P binomial scheme [8]. The G1P[8], G2P[4], G3P[8], G4P[8], G9P[8], and G12P[8] are the most commonly identified RVA genotype combinations in humans [9]. To fully describe rotavirus strains, a whole-genome-based classification scheme based on the genotype of each of the 11 genome segments is preferred and has revealed two major genotype constellations: a Wa-like constellation called genogroup one and a DS-1-like constellation called genogroup two [10]. The whole-genome classification nomenclature of RVA is Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx, representing the VP7 (Glycoprotein, G), VP4 (Protease sensitive, P), VP6 (Intermediate, I), VP1 (RNA dependent RNA polymerase, R), VP2 (Core, C), VP3 (Methyltransferase, M), NSP1 (Antagonist, A), NSP2 (NTpase, N), NSP3 (Translation enhancer, T), NSP4 (Enterotoxin, E), and NSP5 (pHosphoprotein, H) genome segments, respectively, with the letters describing the properties of the 11 genome segments and x indicating the numbers of the corresponding genotypes [10].
The G1P[8], G3P[8], G9P[8], and G12P[8] typically belong to the Wa-like genogroup [9]. However, atypical DS-1-like G1P[8] [11,12], G3P[8] [13,14], and G9P[8] strains [15] have been reported. On the other hand, the G2P[4] strains usually belong to the DS-1-like genogroup [16]. The rotavirus genome has a segmented nature that allows for reassortment events, leading to the emergence of reassortant RVA strains [17,18,19,20]. Additionally, the rotavirus RNA-dependent RNA polymerase, due to its error-prone nature, causes the buildup of sequential point mutations [7]. The varying mutation rates among different RVA genome segments reflect the different selective pressures exerted on different RVA genome segments [12,21,22]. The vast diversity of rotavirus strains is demonstrated by the 42 G, 58 P, 32 I, 28 R, 24 C, 24 M, 39 A, 28 N, 28 T, 32 E, and 28 H genotypes reported at the time of writing this work (https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg (accessed on 17 January 2023)).
The G2P[4] genotype has been observed to increase or maintain dominance after the introduction of the monovalent G1P[8] rotavirus vaccine in several countries such as Australia, Belgium, Brazil, Botswana, Japan, Kenya, Malawi, Saudi Arabia, South Africa, and Venezuela [6,23,24,25,26,27,28,29,30,31]. Although rotavirus vaccines offer cross-protection against different rotavirus genotypes, there is potential for differential vaccine effectiveness against strains from different genogroups such as G2P[4] [32]. As a result, it is imperative to elucidate the diversity and genetic evolution of G2P[4] rotaviruses, especially at the whole-genome level. Whole-genome studies of G2P[4] strains conducted in Italy and Japan reported a genetic shift in the global G2P[4] strains in the early 2000s [33,34]. A long-scale study looking into G2P[4] strains collected seven years before and seven years after rotavirus vaccine introduction in South Africa reported pre- and post-vaccination sub-lineages defined by AA substitutions observed outside known antigenic regions [35].
In Zambia, a four-year G2P[4] prevalence period post Rotarix® introduction was reported by surveillance based on conventional VP7 and VP4 genotyping [36]. Apart from the whole-genome sequencing report of a human-porcine reassortant G5P[6] strain and four intergenogroup reassortant strains [17,19], there is generally a paucity of published Zambian whole-genome G2P[4] RVA sequence data. The purpose of the present study was to perform whole-genome characterization of Zambian G2P[4] strains collected during routine surveillance to gain insight into their overall genetic makeup and evolution.
2. Materials and Methods
2.1. Ethics Statement
The Health Sciences Research Ethics Committee (HSREC) at the University of the Free State in Bloemfontein, South Africa, granted ethical permission for this study under ethics number (UFS-HSD2016/1082).
2.2. Stool Specimen and Strain Description
RVA-positive fecal specimens (n = 133) were obtained from children under the age of five years presenting with acute gastroenteritis as part of the ongoing World Health Organization Regional Office for Africa (WHO-AFRO) RVA surveillance program in Zambia. The stool samples were collected between 2011–2016 and were characterized into G and P types by conventional genotyping methods at the Diarrheal Pathogens Research Unit (DPRU), a WHO Rotavirus Reference Laboratory in South Africa (WHO RRL SA). Briefly, viral dsRNA was extracted from 10% fecal suspension using the QIAmp viral RNA extraction method (Qiagen, Hilden, Germany). The extracted RNA was reverse transcribed and amplified using consensus primer pairs Con2/Con3 and sBeg/End9 as described previously [37,38]. The resulting cDNA template was used for G and P typing using semi-nested RT-PCR amplification of the genes targeting VP7 and VP4. Out of the 133 samples, 29 were G2P[4] strains that were analyzed in this study.
2.3. Data Collection from GenBank
Datasets from different global geographical regions retrieved from GenBank were included for analysis (The accession numbers for the reference sequences used in this study are included as Supplementary Data S1).
2.4. Double-Stranded RNA Extraction
Rotavirus dsRNA was extracted as previously described [20]. Briefly, ~100 mg of fecal specimen was added to 200 µL of phosphate-buffered saline (PBS) solution, 0.01 M, pH (Sigma-Aldrich®, St Louis, MO, USA) to generate a fecal suspension. The fecal suspension was vortexed for 10 s and then left to stand for ten minutes at room temperature, after which a 300 µL of fecal suspension was added to 900 µL of TRI-Reagent®-LS (Molecular Research Center, Cincinnati, OH, USA). The fecal-TRI-Reagent solution was vortexed for 10 s and left to stand at room temperature for 10 min. A 300 µL volume of chloroform (Sigma-Aldrich®, St Louis, MO, USA) was added to the fecal-Tri-Reagent solution. After vortexing the solution for 10 s and letting it stand for 5 min at room temperature, the solution was centrifuged for 18,000× g for 20 min at 4 °C. The aqueous supernatant was removed and precipitated with 700 µL of ice-cold isopropanol (Sigma-Aldrich®, St Louis, MO, USA). Extracted RNA was incubated in 8 M Lithium chloride (Sigma-Aldrich®, St Louis, MO, USA) for 16 h at 4 °C to enrich for rotavirus dsRNA and then subsequently purified using the MinElute PCR purification kit (Qiagen, Hilden, Germany). Electrophoresis was then performed on a 5 µL aliquot of dsRNA in 1% 0.5 TBE agarose (Bioline, UK) gel stained with Pronasafe (Condalab, UK) for 1 h at 95 volts to check the integrity of the extracted and purified rotavirus dsRNA.
2.5. cDNA Synthesis
The complementary DNA (cDNA) was synthesized using the Maxima H Minus Double-Stranded cDNA Synthesis Kit (Thermo Fischer Scientific, Waltham, MA, USA), with modifications. Briefly, 13 µL of extracted viral RNA was denatured at 95 °C for 5 min, and then, 1 µL of random hexamer primer was added. Annealing was performed at 65 °C for 5 min. A 5 µL volume of First-Strand Reaction Mix was added to the solution, followed by 1 µL of First-Strand Enzyme. The mixture was incubated for 10 min at 25 °C, followed by 2 h at 50 °C. Afterward, the following components were added: 55 µL of nuclease-free water, 20 µL of Second-Strand Reaction Mix, and 5 µL of Second-Strand Enzyme, and the solution was incubated at 16 °C for 60 min. A 6 µL volume of 0.5 M EDTA, pH 8.0 was added to stop the reaction, and residual RNA was removed by adding 10 µL of RNase I.
2.6. DNA Library Preparations and Whole-Genome Sequencing
DNA libraries were generated using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) as per the manufacturer’s instructions. Briefly, the genomic cDNA was tagmented, indexed, and then amplified. Afterward, the amplified, tagmented, and indexed DNA was cleaned up using Ampure XP beads (Beckman Coulter, Pasadena, CA, USA). The library quality and fragment sizes were then validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). The DNA libraries were then normalized to 4 nM, and then, 5 µL of normalized libraries were then pooled together into a single tube. A 5 µL pool of the normalized libraries was denatured using 5 µL of 0.2 N sodium hydroxide. Afterward, 990 µL of hybridization buffer was added to the 10 µL of the denatured 4 nM DNA library to dilute to 20 pM with a further dilution to achieve a final loading concentration of 8 pM with a 20% Phix control. The combined library and Phix were loaded into the Illumina V3 reagent cartridge and sequencing performed for 600 cycles (301 bp X 2 paired-end) on a MiSeq® Illumina platform (Illumina, San Diego, CA, USA).
2.7. Genome Assembly
Quality control analysis of the sequenced raw data was performed using FASTQC v.0.11.9 [39]. Adapter sequences were trimmed from the raw FASTQ sequence data using BBDuk trimmer (https://sourceforge.net/projects/bbmap/ (accessed on 27 December 2022)). Reference-based mapping using the prototype DS-1-like reference strain (accession numbers HQ650116-HQ650126) was performed using Geneious Read Mapper 6.0.3 [40]. The Geneious Consensus Tool was used for consensus calling [40]. The Annotate and Predict Tool in Geneious Prime® version 2020.1.1 was used to annotate low coverage (<200) regions, whereby a coverage-annotated track indicating the annotated regions was generated to aid in consensus calling.
2.8. Whole-Genome Genotyping
The genome segments were genotyped using the web-based Virus Pathogen Database and Analysis Resource (ViPR) to generate full-genome constellations [41].
2.9. Phylogenetic Analysis
Basic Local Alignment Search Tool (BLAST) and the National Center for Biotechnology Information’s (NCBI) Virus Variation Resource were used to compile reference sequences [42,43]. The phylogenetic trees for the genome segments encoding VP4 and VP7 were created using previously described lineage designations [35,44]. A previously proposed lineage framework was used to assign lineages to non-G and non-P genome segments [45].
The open reading frame (ORF) sequences for each genome segment were aligned using the Multiple Sequence Comparison by Log-Expectation (MUSCLE) tool [46] in Molecular Evolutionary Genetic Analysis (MEGA) version X [47]. The Model Test in MEGA X was used to estimate the best evolutionary model. The evolutionary models used were Generalized Time Reversible + Gamma + Inversions (GTR + G + I) for VP1, VP2, and VP3; Tamura-3-parameter + Gamma + Inversions (T92 + G + I) for NSP2, NSP3, and NSP4; Tamura-3-parameter + Gamma (T92 + G) for VP4 and VP6; and Tamura-3 parameter + Inversions (T92 + I) for VP7, NSP1, and NSP5. For model selection analysis, maximum likelihood was chosen as the statistical method, and gaps/missing data were handled using partial deletion with an 80% site coverage cut-off and moderate-branch swap filtering. The MEGA X software was used to generate maximum likelihood phylogenetic trees for each genome segment with a 1000 bootstrap support.
2.10. Inference of Selective Pressures
In order to enhance the estimation of sites in the genome of G2P[4] RVA strains undergoing negative and positive selection, we employed three evolutionary analyses method available from the DataMonkey webserver [48]. These are fixed-effects likelihood (FEL) [49], Fast Unbiased Bayesian AppRoximation (FUBAR) [50], and Mixed Effects Model of Evolution (MEME) [51]. The basis of these three selective pressure analyses methods are the estimation of non-synonymous (dN) and synonymous (dS) substitution rates on a per-site basis for the coding alignment of the G2P[4] RVA genome segments. The FEL and MEME methods utilize a maximum likelihood approach, while FUBAR employs a Bayesian approach [48]. The FEL and MEME analysis was performed with p-value threshold of 0.1, while FUBAR was performed with a posterior probability of 0.9.
3. Results
3.1. Whole Genotype Analysis
All the 29 study strains had a pure DS-1-like genotype constellation (G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2). The genome segments from the 29 G2P[4] strains showed nucleotide sequence identities of >97% amongst each other (Supplementary Data S2).
3.2. Phylogenetic and Sequence Analysis
Phylogenetic analyses together with representative global G2P[4] RVAs showed that Zambian strains clustered into human lineages IV (for VP4, VP7, VP2, NSP1, and NSP5), V (for VP1, VP3, VP6, NSP2, and NSP3), and XXIII (for NSP4) (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11).
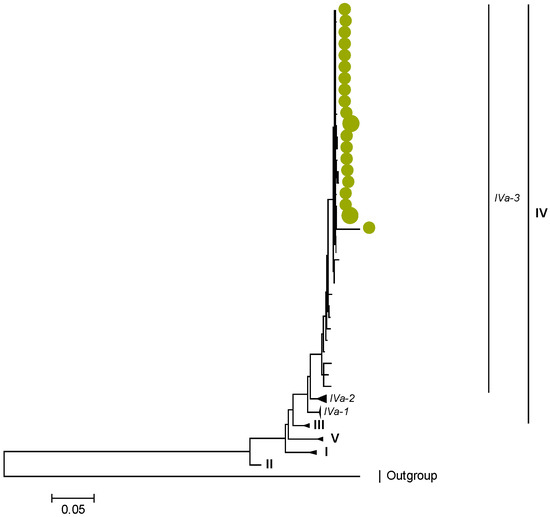
Figure 1.
VP7. Maximum Likelihood phylogenetic tree of the genome segment encoding VP7 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals, while sub-lineages are in italic. The scale number indicates the number of nucleotide substitutions per site.
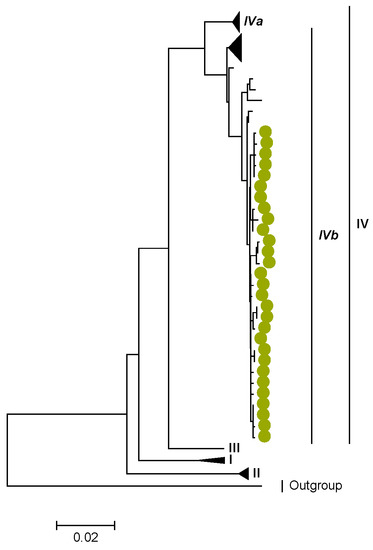
Figure 2.
VP4. Maximum Likelihood phylogenetic tree of the genome segment encoding VP4 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals, while sub-lineages are in italic. The scale number indicates the number of nucleotide substitutions per site.
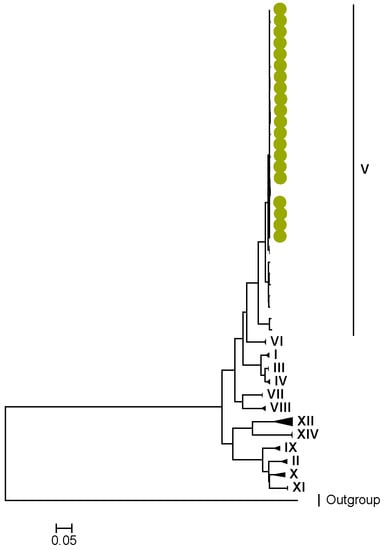
Figure 3.
VP1. Maximum Likelihood phylogenetic tree of the genome segment encoding VP1 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
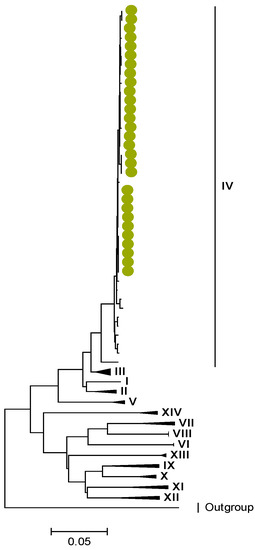
Figure 4.
VP2. Maximum Likelihood phylogenetic tree of the genome segment encoding VP2 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
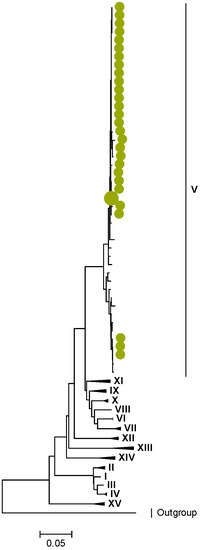
Figure 5.
VP3. Maximum Likelihood phylogenetic tree of the genome segment encoding VP3 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
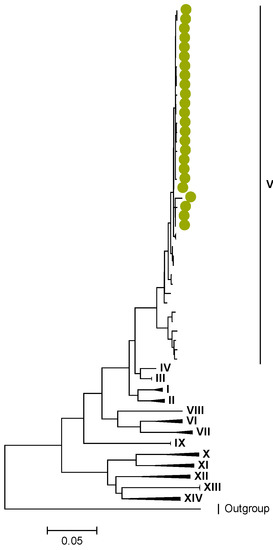
Figure 6.
VP6. Maximum Likelihood phylogenetic tree of the genome segment encoding VP6 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
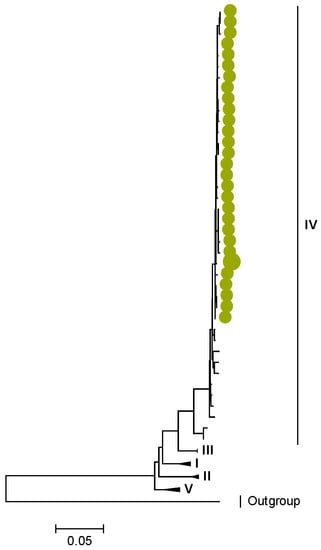
Figure 7.
NSP1. Maximum Likelihood phylogenetic tree of the genome segment encoding NSP1 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
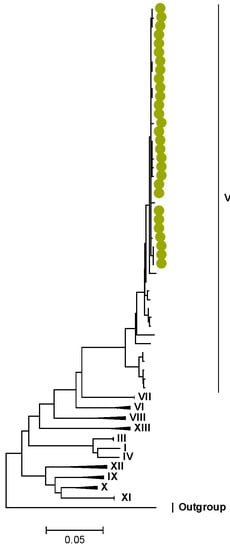
Figure 8.
NSP2. Maximum Likelihood phylogenetic tree of the genome segment encoding NSP2 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
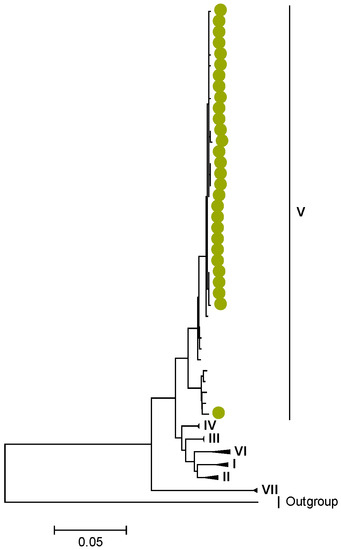
Figure 9.
NSP3. Maximum Likelihood phylogenetic tree of the genome segment encoding NSP3 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
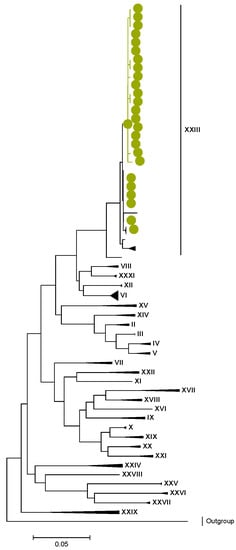
Figure 10.
NSP4. Maximum Likelihood phylogenetic tree of the genome segment encoding NSP4 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
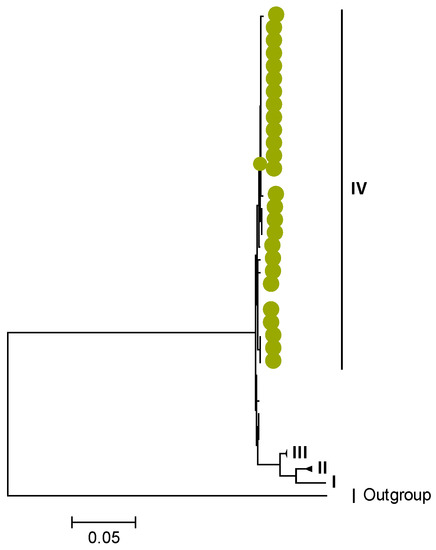
Figure 11.
NSP5. Maximum Likelihood phylogenetic tree of the genome segment encoding NSP5 of Zambian G2P[4] sequences. The study strains are indicated with light-green circular symbols. Lineages are indicated in Roman numerals. The scale number indicates the number of nucleotide substitutions per site.
Zambian G2 sequences clustered further into sub-lineage IVa_3 (Figure 1). The AA substitutions (I44M, D96N, S178N, and I287V) were observed between G2 strains in lineages I, II, III, and V in respect to the G2 strains in lineage IV (Table 1 and Table 2).

Table 1.
Amino acid (AA) differences observed across the G2 lineages.
Table 2.
The AA properties of observed AA substitutions across G2 lineages and region of occurrence in VP7.
We identified AA substitutions (I20V, S598L, and M630I) between P[4] strains in lineages I, II, and III in respect to the P[4] strains in lineage IV, where the study P[4] sequences clustered (Table 3 and Table 4). Within lineage IV, Zambian P[4] sequences further clustered into sub-lineage IVb (Figure 2).
Table 3.
Amino acid (AA) differences observed across the P[4] lineages.
Table 4.
The AA properties of observed AA substitutions across P[4] lineages and region of occurrence in VP4.
Amino acid position 159 of lineage V of R2 sequences where Zambian strains clustered featured an arginine, while other R2 lineages had a lysine (Tables S1 and S2). Three AA differences (N/D28I, I70V, and V538I) were observed between C2 sequences in lineages I–III and V–XIV in respect to lineage IV, where the study strains clustered (Tables S3 and S4). We observed three AA differences (S49N, V209I, and M348V) for the VP3 sequences between lineages I–V and VI–XIV in respect to the lineage V, where Zambian sequences grouped (Tables S5 and S6).
In the genome segment encoding NSP1, three AA differences were observed between the lineages I–II, V, and the lineage IV of the study strains. These differences are represented by l273F, H319R, and K429R, as shown in Tables S7 and S8. Similarly, in NSP2, NSP3, NSP4, and NSP5, there were conservative AA differences between the reference lineages and the lineage of the study strains. These differences are represented by L273F in NSP2, M61I in NSP3, R137Q in NSP4, and V122M in NSP5, as shown in Tables S9–S16.
3.3. Selection Pressure Analysis
The AA sites in the genome segments of Zambian G2P[4] strains were undergoing purifying selection with the exception of the AA site seven in the VP3 gene, which was under diversifying/positive selection (Figure 12 and Table S17).
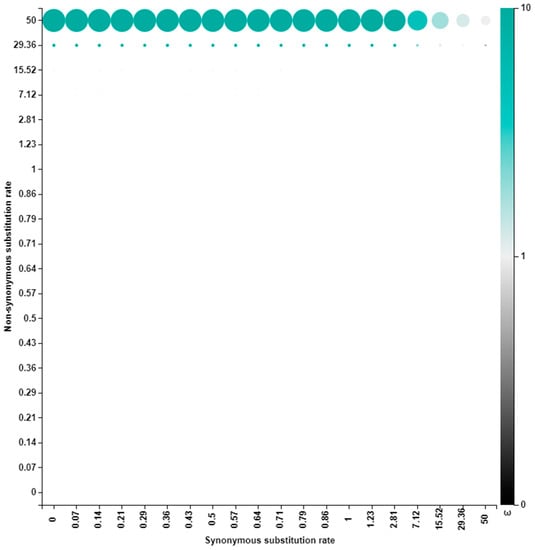
Figure 12.
Posterior distributions of VP3 site seven over the discretized rate grid. The size of a dot is proportional to the posterior weight allocated to that grid point, and the color shows the intensity of selection.
This site was found to have two AA: isoleucine occurring in ~40% of the strains and arginine in ~60% of the strains (Table S18).
4. Discussion
The study reports on whole-genome analysis of 29 G2P[4] RVA strains collected from Zambia from 2011 to 2016. All the Zambian G2P[4] gene sequences exhibited a pure DS-1-like constellation, high nucleotide similarities (>97%), and clustered alongside contemporary human G2P[4] lineages. In essence, Zambian G2P[4] strains emerged independent of interspecies-transmission events, suggesting they were of human origin. These findings are consistent with whole-genome studies of G2P[4] studies in several countries, whereby the reported G2P[4] strains evolved devoid of reassortment with animal genes [23,44,52,53].
The Zambian G2 sequences, which were all post-vaccine, clustered into sub-lineage IVa_3, which is relatable to a South African study wherein post-vaccine G2 strains clustered in sub-lineage IVa_3, while pre-vaccine strains clustered in sub-lineage IVa_1 [35]. In that study, G2 sequences in sub-lineage IVa_3 were found to have a serine at position 15, while sequences in sub-lineage IVa_1 featured phenyalanine, indicating that the AA substitution could be of epidemiological relevance. The aspartate to asparagine substitution at escape mutation site 96, which hallmarks the G2 sequences from the year 2000 [44], could be a selective advantage to enable antibody-dependent enhancement, potentially favoring the production of non-neutralizing antibodies to this protein region [54]. Therefore, this AA site possibly plays a significant role in epidemiological fitness and consequent transmissibility of lineage IV G2 strains. Methionine identification at the Cytotoxic T lymphocyte (CTL) region [55] of lineage IV G2 sequences could have far-reaching biological implications. Apart from protein initiation, methionine’s unbranched side chain provides energetic stability, leading to different conformations [56]. Additionally, the presence of the sulfur atom makes methionine an optimal AA for transient protein–protein interactions [57] that could interfere with the clearance of rotavirus infection.
According to the literature, the isoleucine substitution for valine residue is suggested to shift the equilibrium to a more compact conformation [58]. Therefore, the occurrence of this AA substitution at the hemagglutination site 120 [59] of Zambian P[4] lineage IV strains could be posing possible functional changes such as improving interaction with the sialic-acid-containing structure during cell attachment. Notably, the P[4] study sequences were found to cluster in sub-lineage IVb. This is similar to what was observed in a South African study of post-vaccine P[4] strains [35]. The South African study reported that post-vaccine P[4] sequences in sub-lineage IVb had an arginine at position 162, while pre-vaccine P[4] sequences had a glycine. The epidemiological relevance of this radical AA substitution warrants more exploration as to whether it contributes to epidemiological fitness of the contemporary circulating G2P[4] strains. The radical AA substitution of serine for leucine from polar to nonpolar residue [60] at position 598 of the VP5 portion could be enhancing the permeabilization effect of membranes to enable VP4 entry into the host cell [61], as the presence of the bulkier hydrophobic leucine side chain induces local rearrangements that may likely induce small but significant differences in function [60]. Although we noted that most of the AA differences between the lineages in the rest of the genome segments were conservative, we posit that the intrinsic link between the AA ambiguities and rotavirus protein multifunctionality requires in-depth exploration to comprehensively understand the epidemiological fitness of the lineages in the post-vaccination context era.
Most AA sites were undergoing purifying selection, probably as a strategy to purge deleterious polymorphisms that arise due to the error-prone nature of the RNA polymerase enzyme [7]. However, AA site seven in the VP3 was under positive selection. Arginine and isoleucine that were observed in this site have extreme physicochemical disparities, whereby arginine is a polar positively charged residue, while isoleucine is a non-polar neutrally charged residue [62]. The AA residues under strong selective pressure are suggested to have essential catalytic functions [63]. Therefore, this AA site is likely selected to enhance the methylase capping activity of the VP3. Additionally, apart from lying in the signal sequence domain region, site seven was also found among the disordered protein regions of VP3 [64]. Viruses utilize intrinsically disordered regions to survive in harsh environments to evade the host immune system and to hijack and manipulate host cellular proteins [65]. Therefore, selection for this effect could be at play in this region.
Our analysis would have been more enriched with analysis performed in the context of pre-vaccination Zambian G2P[4] samples. However, natural fluctuations are known to impact the prevalence of circulating rotavirus strains [53], and sampling for the current study was performed based on availability. Regardless, our study findings provide valuable whole-genome insights regarding the evolution of G2P[4] strains in Zambia.
5. Conclusions
The G2P[4] strains circulating in Zambia from 2012 to 2016 belong to the lineages of G2P[4] strains that have been circulating globally since the early 2000s, highlighting the epidemiological fitness of these contemporary strains. The analysis of AA substitutions defining the lineages of the circulating G2P[4] strains provides an insightful perspective to understanding their evolution.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/v15020501/s1, Supplementary Data S1: Accession numbers for the reference sequences used in the construction of the maximum likelihood phylogenetic trees; Supplementary Data S2: Nucleotide similarity analysis amongst genome segments of Zambian G2P[4] strains 3; Supplementary Data S3: Tables S1–S17.
Author Contributions
M.M.N. and J.M.M. conceptualized the main project; M.M.N., J.S., E.M.M., J.M.M. and M.L.S. obtained the samples; P.N.M., M.T.M., N.M. and M.M.N. conducted the laboratory experiments; formal and bioinformatic analysis was performed by P.N.M., R.-L.P. and M.M.N.; original draft manuscript preparation was performed by P.N.M.; review of the drafts was performed by all co-authors; M.M.N. sourced the funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research was principally funded by a World Health Organization (WHO) grant awarded to M.M.N (Reference 2022/1252000-0). Additional grants from the Bill and Melinda Gates Foundation (BMGF-OPP1180423_2017), the South African Medical Research Foundation through the Self-Initiated Research grant, the National Research Foundation (NRF-120814), and the Poliomyelitis Research Foundation (PRF-19/16 awarded to M.M.N. and PRF 21/77 awarded to P.N.M.). The views and opinions expressed in this article are not necessarily those of the funding and affiliated institutions but solely of the authors of this publication.
Institutional Review Board Statement
The Health Sciences Research Ethics Committee (HSREC) at the University of the Free State in Bloemfontein, South Africa, granted ethical permission for this study under ethics number (UFS-HSD2016/1082).
Informed Consent Statement
The diarrheal stool samples were collected as a routine diagnostic clinical specimen when the parents brought their child to a health facility for clinical management, requiring no written informed consent. As part of the World Health Organization (WHO) coordinated rotavirus surveillance network, the archived rotavirus-positive specimens were anonymized and utilized for whole-genomic characterization under a Technical Service agreement to the University of the Free State Next-Generation Sequencing Unit, a WHO-Collaborating Centre for Vaccine-Preventable Diseases Surveillance and Pathogen Genomics based in Bloemfontein, South Africa. The WHO Research Ethics Review Committee granted an “exemption activity”, noting that the procedures involved in the study are part of routine hospital-based rotavirus surveillance.
Data Availability Statement
All the gene sequences in this study were submitted in the NCBI GenBank database under accession numbers OQ132993–OQ133311 and are included as supplementary data S4.
Acknowledgments
We would like to acknowledge Jeffrey Mphahlele in facilitating obtaining of the samples. Further, we acknowledge the assistance in retrieving the archival stool samples by Ina Peenze, Khutso Mothapo, and Dorah Makinita at the WHO-RRL in Pretoria, South Africa. We also acknowledge Sebotsana Rasebotsa, Lesedi Mosime, Gilmore Pambuka, Ayodeji Ogunbayo, and Teboho Mooko for assistance with the laboratory work. The provision of technical ICT support by Stephanus Riekert is duly acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Parashar, U.D.; Gibson, C.J.; Bresee, J.S.; Glass, R.I. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 2006, 12, 304–306. [Google Scholar] [CrossRef]
- Mpabalwani, E.M.; Simwaka, C.J.; Mwenda, J.M.; Mubanga, C.P.; Monze, M.; Matapo, B.; Parashar, U.D.; Tate, J.E. Impact of rotavirus vaccination on diarrhoeal hospitalisations in children aged <5 Years in Lusaka, Zambia. Clin. Infect. Dis. 2016, 62, S183–S187. [Google Scholar] [CrossRef]
- Chilengi, R.; Rudd, C.; Bolton, C.; Guffey, B.; Masumbu, P.K.; Stringer, J. Successes, challenges and lessons learned in accelerating introduction of rotavirus immunisation in Zambia. World J. Vaccines 2015, 5, 43–53. [Google Scholar] [CrossRef]
- Shah, M.P.; Mwenda, J.M.; Tate, J.E.; Steele, A.D.; Parashar, U.D. Estimated reductions in hospitalizations and deaths from childhood diarrhea following implementation of rotavirus vaccines in Africa. Expert Rev. Vaccines 2017, 16, 987–995. [Google Scholar] [CrossRef]
- Zeller, M.; Heylen, E.; Tamim, S.; McAllen, J.K.; Kirkness, E.F.; Akopov, A.; De Coster, S.; Van Ranst, M.; Matthijnssens, J. Comparative analysis of the Rotarix™ vaccine strain and G1P[8] rotaviruses detected before and after vaccine introduction in Belgium. PeerJ 2017, 5, e2733. [Google Scholar] [CrossRef]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Heath/Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef]
- Dóró, R.; László, B.; Martella, V.; Leshem, E.; Gentsch, J.; Parashar, U.; Bányai, K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect. Genet. Evol. 2014, 28, 446–461. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef]
- Mwangi, P.N.; Mogotsi, M.T.; Rasebotsa, S.P.; Seheri, M.L.; Mphahlele, M.J.; Ndze, V.N.; Dennis, F.E.; Jere, K.C.; Nyaga, M.M. Uncovering the first atypical ds-1-like G1P[8] rotavirus strains that circulated during pre-rotavirus vaccine introduction era in South Africa. Pathogens 2020, 9, 391. [Google Scholar] [CrossRef]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of double-and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-like backbone after rotavirus vaccine introduction in Malawi. J. Virol. 2018, 92, e01246-17. [Google Scholar] [CrossRef]
- Mhango, C.; Banda, A.; Chinyama, E.; Mandolo, J.J.; Kumwenda, O.; Malamba-Banda, C.; Barnes, K.G.; Kumwenda, B.; Jambo, K.; Donato, C.M.; et al. Comparative whole genome analysis reveals re-emergence of typical human Wa-like and DS-1-like G3 rotaviruses after Rotarix vaccine introduction in Malawi. medRxiv 2022. [Google Scholar] [CrossRef]
- Mwanga, M.J.; Verani, J.R.; Omore, R.; Tate, J.E.; Parashar, U.D.; Murunga, N.; Gicheru, E.; Breiman, R.F.; Nokes, D.J.; Agoti, C.N. Multiple introductions and predominance of rotavirus group A genotype G3P[8] in coastal Kenya in 2018, 4 years after nationwide vaccine introduction. Pathogens 2020, 9, 981. [Google Scholar] [CrossRef]
- Fukuda, S.; Tacharoenmuang, R.; Guntapong, R.; Upachai, S.; Singchai, P.; Ide, T.; Hatazawa, R.; Sutthiwarakom, K.; Kongjorn, S.; Onvimala, N.; et al. Full genome characterization of novel DS-1-like G9P[8] rotavirus strains that have emerged in Thailand. PLoS ONE 2020, 15, e0231099. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Maringa, W.M.; Simwaka, J.; Mwangi, P.N.; Mpabalwani, E.M.; Mwenda, J.M.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia. Viruses 2021, 13, 1872. [Google Scholar] [CrossRef]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic characterization of an African G4P[6] human rotavirus strain identified in a diarrheic child in Kenya: Evidence for porcine-to-human interspecies transmission and reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef]
- Maringa, W.M.; Mwangi, P.N.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Peenze, I.; Esona, M.D.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Molecular characterisation of a rare reassortant porcine-like G5P[6] rotavirus strain detected in an unvaccinated child in Kasama, Zambia. Pathogens 2020, 9, 663. [Google Scholar] [CrossRef]
- Rasebotsa, S.; Mwangi, P.N.; Mogotsi, M.T.; Sabiu, S.; Magagula, N.B.; Rakau, K.; Uwimana, J.; Mutesa, L.; Muganga, N.; Murenzi, D.; et al. Whole genome and in-silico analyses of G1P[8] rotavirus strains from pre-and post-vaccination periods in Rwanda. Sci. Rep. 2020, 10, 1–22. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Tan, Y.; Seheri, M.L.; Halpin, R.A.; Akopov, A.; Stucker, K.M.; Fedorova, N.B.; Shrivastava, S.; Steele, A.D.; Mwenda, J.M.; et al. Whole-genome sequencing and analyses identify high genetic heterogeneity, diversity and endemicity of rotavirus genotype P[6] strains circulating in Africa. Infect. Genet. Evol. 2018, 63, 79–88. [Google Scholar] [CrossRef]
- Donker, N.C.; Kirkwood, C.D. Selection and evolutionary analysis in the nonstructural protein NSP2 of rotavirus A. Infect. Genet. Evol. 2012, 12, 1355–1361. [Google Scholar] [CrossRef]
- Donato, C.M.; Zhang, Z.A.; Donker, N.C.; Kirkwood, C.D. Characterization of G2P[4] rotavirus strains associated with increased detection in Australian states using the RotaTeq® vaccine during the 2010-2011 surveillance period. Infect. Genet. Evol. 2014, 28, 398–412. [Google Scholar] [CrossRef]
- Gurgel, R.Q.; Cuevas, L.E.; Vieira, S.C.F.; Barros, V.C.F.; Fontes, P.B. Predominance of rotavirus P[4]G2 in a vaccinated population, Brazil. Emerg. Infect. Dis. 2007, 13, 1571–1573. [Google Scholar] [CrossRef]
- Mokomane, M.; Esona, M.D.; Bowen, M.D.; Tate, J.E.; Steenhoff, A.P.; Lechiile, K.; Gaseitsiwe, S.; Seheri, L.M.; Magagula, N.B.; Weldegebriel, G.; et al. Diversity of Rotavirus Strains Circulating in Botswana before and after introduction of the Monovalent Rotavirus Vaccine. Vaccine 2019, 37, 6324–6328. [Google Scholar] [CrossRef]
- Khandoker, N.; Thongprachum, A.; Takanashi, S.; Okitsu, S.; Nishimura, S. Molecular epidemiology of rotavirus gastroenteritis in Japan during 2014-2015: Characterization of re-emerging G2P[4] after rotavirus vaccine introduction. J. Med. Virol. 2018, 90, 1040–1046. [Google Scholar] [CrossRef]
- Wandera, E.A.; Mohammad, S.; Bundi, M.; Komoto, S.; Nyangao, J.; Kathiiko, C.; Odoyo, E.; Miring’u, G.; Taniguchi, K.; Ichinose, Y. Impact of rotavirus vaccination on rotavirus and all-cause gastroenteritis in peri-urban Kenyan children. Vaccine 2017, 35, 5217–5223. [Google Scholar] [CrossRef]
- Mhango, C.; Mandolo, J.J.; Wachepa, R.; Kanjerwa, O.; Malamba-Banda, C.; Matambo, P.B.; Barnes, K.G.; Chaguza, C.; Shawa, I.T.; Nyaga, M.M.; et al. Rotavirus Genotypes in Hospitalized Children with Acute Gastroenteritis Before and After Rotavirus Vaccine Introduction in Blantyre, Malawi, 1997–2019. J. Infect. Dis. 2020, 225, 2127–2136. [Google Scholar] [CrossRef]
- Al-Ayed, M.S.Z.; Asaad, A.M.; Qureshi, M.A.; Hawan, A.A. Epidemiology of group A rotavirus infection after the introduction of monovalent vaccine in the National Immunization Program of Saudi Arabia. J. Med. Virol. 2017, 89, 429–434. [Google Scholar] [CrossRef]
- Page, N.A.; Steele, A.D. Antigenic and genetic characterization of serotype G2 human rotavirus strains from South Africa from 1984 to 1998. J. Med. Virol. 2004, 72, 320–327. [Google Scholar] [CrossRef]
- Vizzi, E.; Piñeros, O.A.; Oropeza, M.D.; Naranjo, L.; Suárez, J.A. Human rotavirus strains circulating in Venezuela after vaccine introduction: Predominance of G29[P4] and reemergence of G1P[8]. Virol. J. 2017, 14, 58. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Zeller, M.; Heylen, E.; De Coster, S.; Vercauteren, J.; Braeckman, T.; Van Herck, K.; Meyer, N.; Pirçon, J.Y.; Soriano-Gabarro, M.; et al. Higher proportion of G2P[4] rotaviruses in vaccinated hospitalized cases compared with unvaccinated hospitalized cases, despite high vaccine effectiveness against heterotypic G2P[4] rotaviruses. Clin. Microbiol. Infect. 2014, 20, O702–O710. [Google Scholar] [CrossRef]
- Giammanco, G.M.; Bonura, F.; Zeller, M.; Heylen, E.; Van Ranst, M.; Martella, V.; Banyai, K.; Matthijnssens, J.; De Grazia, S. Evolution of DS-1-like human G2P[4] rotaviruses assessed by complete genome analyses. J. Gen. Virol. 2014, 95, 91–109. [Google Scholar] [CrossRef]
- Doan, Y.H.; Nakagomi, T.; Agbemabiese, C.A.; Nakagomi, O. Changes in the distribution of lineage constellations of G2P[4] Rotavirus A strains detected in Japan over 32 years (1980–2011). Infect. Genet. Evol. 2015, 34, 423–433. [Google Scholar] [CrossRef]
- Mwangi, P.N.; Page, N.A.; Seheri, M.L.; Mphahlele, M.J.; Nadan, S.; Esona, M.D.; Kumwenda, B.; Kamng’ona, A.W.; Donato, C.M.; Steele, A.D.; et al. Evolutionary changes between pre-and post-vaccine South African group A G2P[4] rotavirus strains, 2003–2017. Microb. Genom. 2022, 8, 000809. [Google Scholar] [CrossRef]
- Simwaka, J.C.; Mpabalwani, E.M.; Seheri, M.; Peenze, I.; Monze, M.; Matapo, B.; Parashar, U.D.; Mufunda, J.; Mphahlele, J.M.; Tate, J.E.; et al. Diversity of rotavirus strains circulating in children under five years of age who presented with acute gastroenteritis before and after rotavirus vaccine introduction, University Teaching Hospital, Lusaka, Zambia, 2008–2015. Vaccine 2018, 36, 7243–7247. [Google Scholar] [CrossRef]
- Gentsch, J.R.; Glass, R.I.; Woods, P.; Gouvea, V.; Gorziglia, M.; Flores, J.; Das, B.K.; Bhan, M.K. Identification of group A rotavirus gene 4 types by polymerase chain reaction. J. Clin. Microbiol. 1992, 30, 1365–1373. [Google Scholar] [CrossRef]
- Gouvea, V.; Glass, R.I.; Woods, P.; Taniguchi, K.; Clark, H.F.; Forrester, B.; Fang, Z.Y. Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J. Clin. Microbiol. 1990, 28, 276–282. [Google Scholar] [CrossRef]
- Andrews, S.F. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 October 2022).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2019, 47, D23. [Google Scholar] [CrossRef]
- Doan, Y.H.; Nakagomi, T.; Cunliffe, N.A.; Pandey, B.D.; Sherchand, J.B. The occurrence of amino acid substitutions D96N and S242N in VP7 of emergent G2P[4] rotaviruses in Nepal in 2004–2005: A global and evolutionary perspective. Arch. Virol. 2011, 156, 1960–1978. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Damanka, S.A.; Dennis, F.E.; Lartey, B.L. Sub-genotype phylogeny of the non-G, non-P genes of genotype 2 Rotavirus A strains. PLoS ONE 2019, 14, e0217422. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Doan, Y.H.; Do, L.P.; Damanka, S. Genomic constellation and evolution of Ghanaian G2P[4] rotavirus strains from a global perspective. Infect. Genet. Evol. 2016, 45, 122–131. [Google Scholar] [CrossRef]
- Dennis, A.F.; McDonald, S.M.; Payne, D.C.; Mijatovic-Rustempasic, S.; Esona, M.D.; Edwards, K.M.; Chappell, J.D.; Patton, J.T. Molecular epidemiology of contemporary G2P[4] human rotaviruses cocirculating in a single US community: Footprints of a globally transitioning genotype. J. Virol. 2014, 88, 3789–3801. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. BioRxiv 2020. [Google Scholar]
- Franco, M.A.; Tin, C.; Greenberg, H.B. CD8+ T cells can mediate almost complete short-term and partial long-term immunity to rotavirus in mice. J. Virol. 1997, 71, 4165–4170. [Google Scholar] [CrossRef]
- Aledo, J.C. Methionine in proteins: The Cinderella of the proteinogenic amino acids. Protein Sci. 2019, 28, 1785–1796. [Google Scholar] [CrossRef]
- Manta, B.; Gladyshev, V.N. Regulated methionine oxidation by monooxygenases. Free. Radic. Biol. Med. 2017, 109, 141–155. [Google Scholar] [CrossRef]
- Keating, D.H.; Cronan, J.E. An isoleucine to valine substitution in Escherichia coli acyl carrier protein results in a functional protein of decreased molecular radius at elevated pH. J. Biol. Chem. 1996, 271, 15905–15910. [Google Scholar] [CrossRef]
- Fuentes-Pananá, E.M.; López, S.; Gorziglia, M.; Arias, C.F. Mapping the hemagglutination domain of rotaviruses. J. Virol. 1995, 69, 2629–2632. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. Bioinform. Genet. 2003, 317, 289. [Google Scholar]
- Dowling, W.; Denisova, E.; LaMonica, R.; Mackow, E.R. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J. Virol. 2000, 74, 6368–6376. [Google Scholar] [CrossRef] [PubMed]
- Creixell, P.; Schoof, E.M.; Tan, C.S.H.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.M.; Snyder, M.J.; Dennis, A.F.; Patton, J.T. Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J. Virol. 2014, 88, 9072–9085. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Singh, A.; Kumar, P.; Uversky, V.N.; Rao, C.D.; Giri, R. Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int. J. Biol. Macromol. 2020, 144, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.M.; Verma, N.C.; Rao, C.; Uversky, V.N.; Nandi, C.K. Intrinsically disordered proteins of viruses: Involvement in the mechanism of cell regulation and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2020, 174, 1–78. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).