
Escalation with Overdose Control is More Efficient and Safer than Accelerated Titration for Dose Finding

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
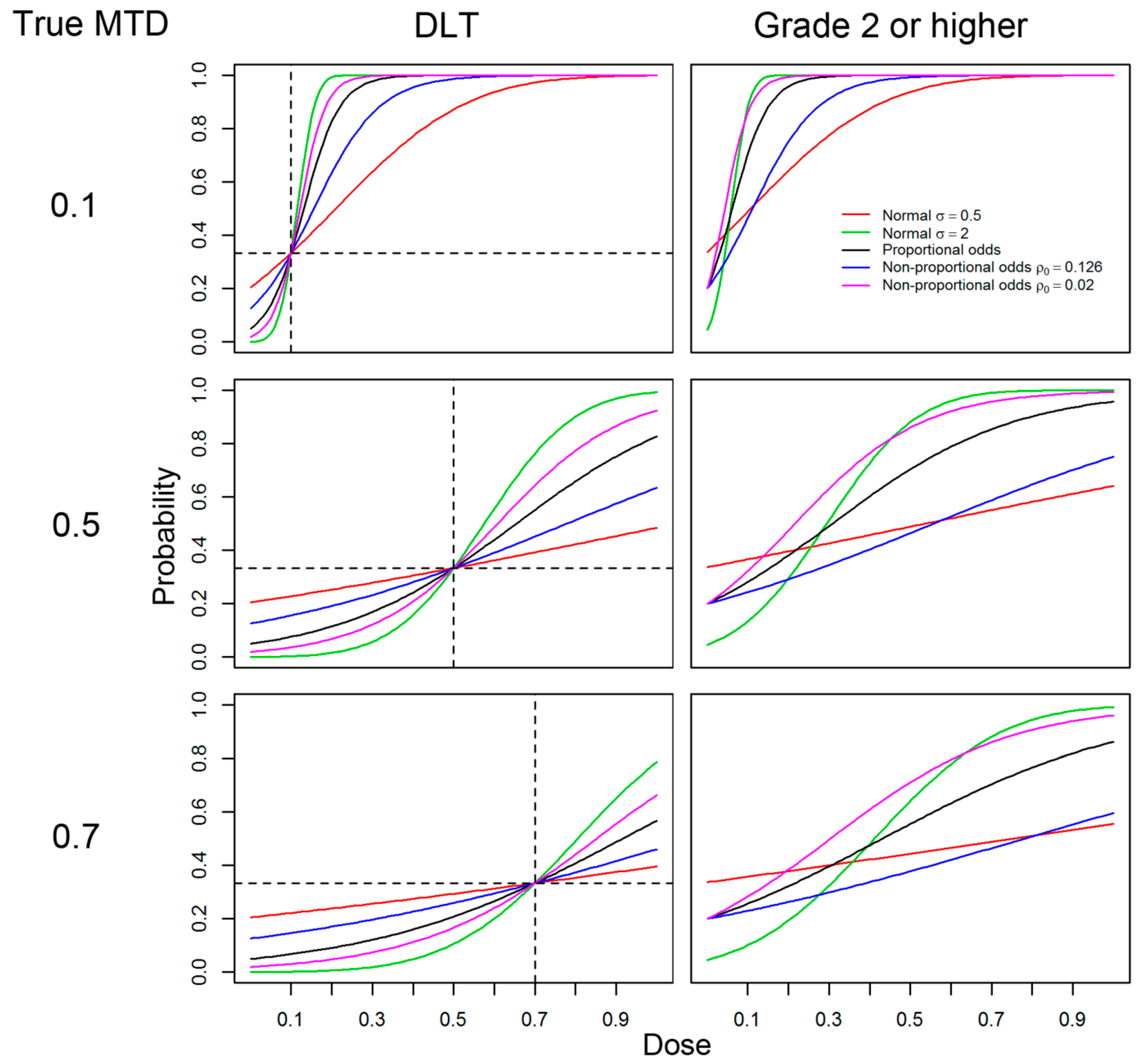
2.1. Models, Responses and General Settings
- (A)
- Logistic with link function F(w) = 1/(1+e−w),
- (B)
- Normal link function with shape parameter σ = 0.5,
- (C)
- Normal link function with shape parameter with σ = 2,
- (D)
- Non-proportional odds model (β1 ≠ β2) with logistic link function used on (A) with ρ0 = 0.126, and
- (E)
- (E)Non-proportional odds model with logistic link function used on (A) with ρ0 = 0.02.

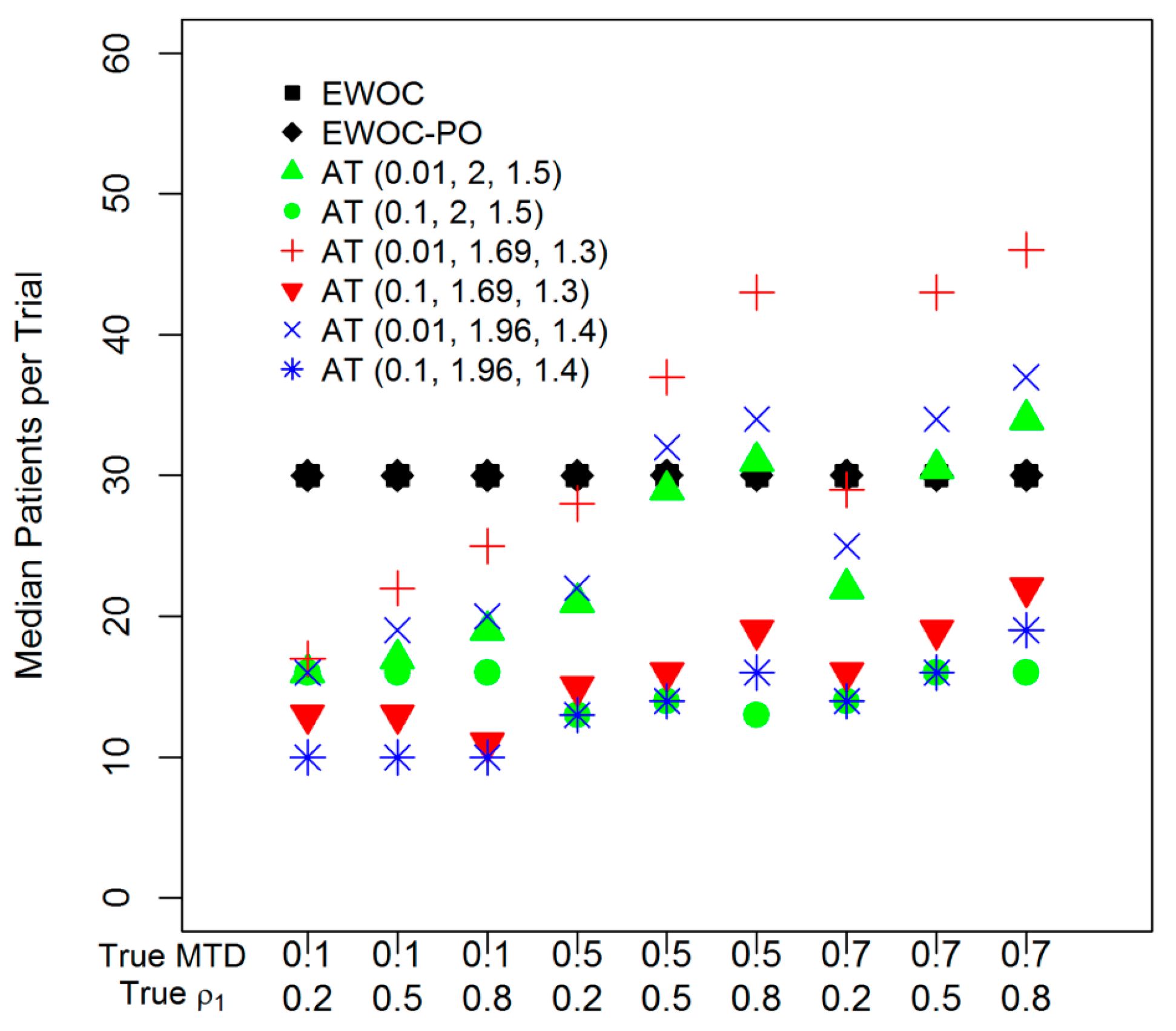
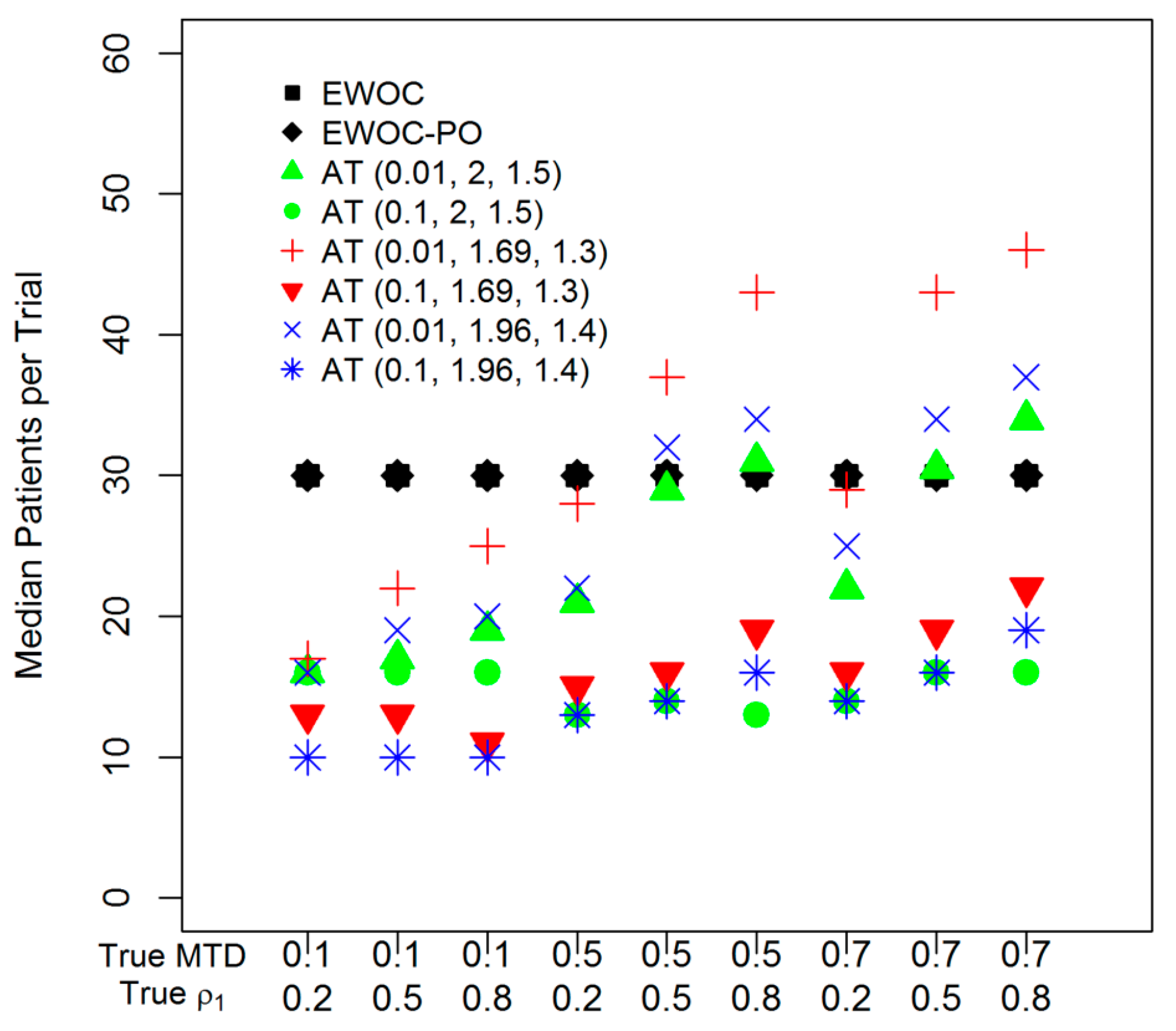
2.2. AT Designs
- (1)
- Double the dose in the accelerated phase and increase by 50% in the MF-UD phase (recommended by FDA);
- (2)
- Increase by 69% in the accelerated phase and 30% in the MF-UD phase; and
- (3)
- Increase by 96% in the accelerated phase and 40% in the MF-UD phase.
- Start trial in the accelerated phase by treating patients at the initial dose. Denote the dose level being used to treat patients as the current dose level.
- Accrue and treat one patient at the current dose level.
- Check the toxicity in the one patient at the current dose level.
- 3a.
- If there is a grade 2 toxicity or higher, end the accelerated phase and go to 5.
- 3b.
- If there is no toxicity, escalate the dose and go to 4.
- Escalate if possible.
- 4a.
- If the current dose level is the highest dose level; stop the trial and declare that the MTD is higher than the highest dose level.
- 4b.
- Otherwise, escalate to the next-higher dose level; go to 2. The next higher dose level is the current dose level multiplied by the increment used in the accelerated phase (e.g., 1.96).
- Accrue and treat patients in the MF-UD design phase so that there are three to be treated at the current dose level.
- 5a.
- If the maximum number of patients has been accrued, stop the trial. (The simulated trials were capped at 62 patients.)
- Check the number of patients at the current dose level.
- 6a.
- If there are three patients, go to 7.
- 6b.
- If there are more than three patients, go to 8.
- Check the number of toxicities (among three patients) at the current dose level.
- 7a.
- If there are zero toxicities, escalate and go to 10.
- 7b.
- If there is one toxicity, stay at the current dose and go to 5.
- 7c.
- If there are two or three toxicities, declare that the MTD has been exceeded and go to 9.
- Check the number of toxicities (among more than three patients) at the current dose level.
- 8a.
- If there are zero toxicities, stop the trial and declare that the MTD is the current dose.
- 8b.
- If there is one toxicity, escalate and go to 10 unless the MTD has been exceeded, then stop the trial and declare that the MTD is the current dose.
- 8c.
- If there are two toxicities, stop the trial; the MTD is the current dose.
- 8d.
- If there are three or four toxicities, declare that the MTD has been exceeded and go to 9 (it is impossible to have five or six toxicities among six patients at the same dose level under this MF-UD design).
- The MTD has been exceeded.
- 9a.
- If the current dose is the lowest dose, stop the trial; declare that the MTD is lower than the lowest dose level.
- 9b.
- If the next-lower dose level has more than three patients, stop the trial and declare that the MTD is the next lower dose level; otherwise, set the current dose level to be the next-lower dose level and go to 5. The next lower dose level is the current dose level divided by the increment used in the MF-UD phase (e.g., 1.4)
- Escalate if possible.
- 10a.
- If the current dose level is the highest dose level; stop the trial and declare that the MTD is higher than the highest dose level.
- 10b.
- Otherwise, escalate to the next-higher dose level; go to 5. The next higher dose level is the current dose level multiplied by the increment used in the MF-UD phase (e.g. 1.4)
2.3. EWOC Designs
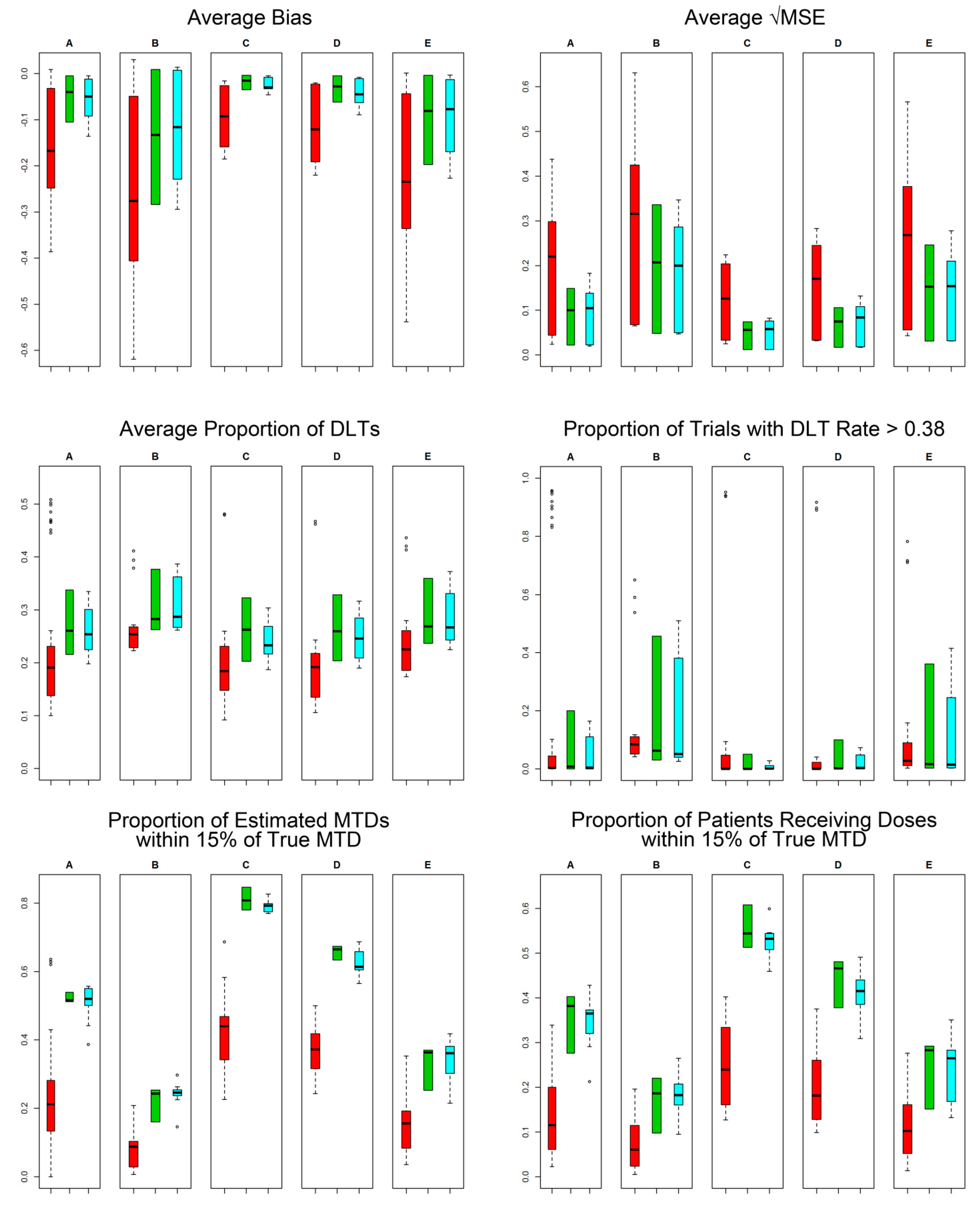
2.4. Efficiency and Safety Comparisons
3. Results


4. Concluding Remarks
“… the most effective and fastest way to improve the present situation is a change in the attitude of the gatekeeper—regulatory agencies. The US Food and Drug Administration should proactively encourage the adoption of statistical designs that would allow more patients to be treated at near-optimal doses while controlling for excessive toxicity. After all, the real justification for using the standard modified Fibonacci up-and-down design is tradition (i.e., “This is how we always have done it”) and comfort level, not scientific reasoning or clinical evidence.”
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Gatsonis, C.; Greenhouse, J.B. Bayesian Methods for Phase I Clinical Trials. Stat. Med. 1992, 11, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, E.; McCabe, M.S.; Grochow, L.; Yamamoto, S.; Rubinstein, L.; Budd, T.; Shoemaker, D.; Emanuel, E.J.; Grady, C. Risks and Benefits of Phase 1 Oncology Trials, 1991 through 2002. New Engl. J. Med. 2005, 352, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Babb, J.; Rogatko, A.; Zacks, S. Cancer Phase I Clinical Trials: Efficient Dose Escalation with Overdose Control. Stat. Med. 1998, 17, 1103–1120. [Google Scholar] [CrossRef]
- Zacks, S.; Rogatko, A.; Babb, J. Optimal Bayesian-Feasible Dose Escalation for Cancer Phase I Trials. Stat. Probab. Lett. 1998, 38, 215–220. [Google Scholar] [CrossRef]
- Tighiouart, M.; Rogatko, A. Dose Finding with Escalation with Overdose Control (EWOC) in Cancer Clinical Trials. Stat. Sci. 2010, 25, 217–226. [Google Scholar] [CrossRef]
- Cheung, Y.K. Coherence Principles in Dose-Finding Studies. Biometrika 2005, 92, 863–873. [Google Scholar] [CrossRef]
- Babb, J.S.; Rogatko, A. Patient Specific Dosing in a Cancer Phase I Clinical Trial. Stat. Med. 2001, 20, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Babb, J.S.; Rogatko, A. Bayesian Methods for Cancer Phase I Clinical Trials; Marcel Dekker: New York, NY, USA, 2004; pp. 1–40. [Google Scholar]
- Tighiouart, M.; Cook-Wiens, G.; Rogatko, A. Incorporating a Patient Dichotomous Characteristic in Cancer Phase I Clinical Trials Using Escalation with Overdose Control. J. Probab. Stat. 2012, 2012, 10. [Google Scholar] [CrossRef]
- Xu, Z.; Tighiouart, M.; Rogatko, A. EWOC 2.0: Interactive Software for Dose Escalation in Cancer Phase I Clinical Trials. Drug Inf. J. 2007, 41, 221–228. [Google Scholar]
- Wang, H.; Tighiouart, M.; Huang, S.C.; Berel, D.; Cook-Wiens, G.; Bresee, C.; Li, Q.; Rogatko, A. The Integrated Web Portal for Escalation with Overdose Control (EWOC). Open Med. Inf. J. 2013, 7, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Lunn, D.J.; Thomas, A.; Best, N.; Spiegelhalter, D. Winbugs—A Bayesian Modelling Framework: Concepts, Structure, and Extensibility. Stat. Comput. 2000, 10, 325–337. [Google Scholar] [CrossRef]
- Tighiouart, M.; Rogatko, A.; Babb, J.S. Flexible Bayesian Methods for Cancer Phase I Clinical Trials. Dose Escalation with Overdose Control. Stat. Med. 2005, 24, 2183–2196. [Google Scholar] [CrossRef] [PubMed]
- Tighiouart, M.; Cook-Wiens, G.; Rogatko, A. Escalation with Overdose Control Using Ordinal Toxicity Grades for Cancer Phase I Clinical Trials. J. Probab. Stat. 2012, 2012. [Google Scholar] [CrossRef]
- Cheng, J.D.; Babb, J.S.; Langer, C.; Aamdal, S.; Robert, F.; Engelhardt, L.R.; Fernberg, O.; Schiller, J.; Forsberg, G.; Alpaugh, R.K.; et al. Individualized Patient Dosing in Phase I Clinical Trials: The Role of Escalation with Overdose Control in PNU-214936. J. Clin. Oncol. 2004, 22, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Alpaugh, K.; Hedlund, G.; Forsberg, G.; Langer, C.; Rogatko, A.; Hawkins, R.; Dueland, S.; Lassen, U.; Cohen, R.B. Phase I Dose Escalation, Pharmacokinetic and Pharmacodynamic Study of Naptumomab Estafenatox Alone in Patients with Advanced Cancer and with Docetaxel in Patients with Advanced Non-small-cell Lung Cancer. J. Clin. Oncol. 2009, 27, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Rogatko, A.; Schoeneck, D.; Jonas, W.; Tighiouart, M.; Khuri, F.R.; Porter, A. Translation of Innovative Designs into Phase I Trials. J. Clin. Oncol. 2007, 25, 4982–4986. [Google Scholar] [CrossRef] [PubMed]
- Storer, B.E. Design and Analysis of Phase-I Clinical-Trials. Biometrics 1989, 45, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Freidlin, B.; Rubinstein, L.; Arbuck, S.G.; Collins, J.; Christian, M.C. Accelerated Titration Designs for Phase I Clinical Trials in Oncology. J. Natl. Cancer Inst. 1997, 89, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Shih, W.J. Statistical Properties of the Traditional Algorithm-Based Designs for Phase I Cancer Clinical Trials. Biostatistics 2001, 2, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Gerke, O.; Siedentop, H. Optimal Phase I Dose-Escalation Trial Designs in Oncology—A Simulation Study. Stat. Med. 2008, 27, 5329–5344. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equations of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo Sampling Methods Using Markov Chains and Their Applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Smith, B.J. Boa: An R Package for MCMC Output Convergence Assessment and Posterior Inference. J. Stat. Softw. 2007, 21, 1–37. [Google Scholar]
- Kang, S.H.; Ahn, C.W. An Investigation of the Traditional Algorithm-Based Designs for Phase 1 Cancer Clinical Trials. Drug Inf. J. 2002, 36, 865–873. [Google Scholar] [CrossRef]
- Rogatko, A.T.M.; Bresee, C.; Cook-Wiens, G.; Li, Q.; Huang, S.; Moyse, H.; Yatawara, M. Escalation with Overdose Control—Web Portal. Available online: https://biostatistics.csmc.edu/ewoc/index.php (accessed on 24 July 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogatko, A.; Cook-Wiens, G.; Tighiouart, M.; Piantadosi, S. Escalation with Overdose Control is More Efficient and Safer than Accelerated Titration for Dose Finding. Entropy 2015, 17, 5288-5303. https://doi.org/10.3390/e17085288
Rogatko A, Cook-Wiens G, Tighiouart M, Piantadosi S. Escalation with Overdose Control is More Efficient and Safer than Accelerated Titration for Dose Finding. Entropy. 2015; 17(8):5288-5303. https://doi.org/10.3390/e17085288
Chicago/Turabian StyleRogatko, André, Galen Cook-Wiens, Mourad Tighiouart, and Steven Piantadosi. 2015. "Escalation with Overdose Control is More Efficient and Safer than Accelerated Titration for Dose Finding" Entropy 17, no. 8: 5288-5303. https://doi.org/10.3390/e17085288
APA StyleRogatko, A., Cook-Wiens, G., Tighiouart, M., & Piantadosi, S. (2015). Escalation with Overdose Control is More Efficient and Safer than Accelerated Titration for Dose Finding. Entropy, 17(8), 5288-5303. https://doi.org/10.3390/e17085288