Introduction
Traditional treatment of bacterial related diseases is based upon chemicals that ultimately kill or inhibit the growth of the bacteria. These drugs work against microorganisms by breaching their cell walls, disrupting metabolism, or inhibiting macromolecule synthesis. With the re-emergence of many infectious diseases and the emergence of drug-resistant strains of bacteria, it is increasingly important to identify new therapeutic compounds with novel mechanisms of action to supplement existing anti-microbials.
The various regulatory systems in bacteria may represent novel targets. For example, it is now well understood that some pathogenic bacteria rely on the secretion and detection of small diffusible cues/signals, to control the expression of virulence [
1,
2,
3]. One such quorum sensing (QS) system, found in Gram-negative bacteria, is the
N-acylated homoserine lactone (AHL) system, which has been shown to control phenotypes such as virulence factor production and biofilm formation [
4,
5].
The essential features of this system include the signal molecule (AHLs), synthesized by the synthase LuxI and the signal receptor, LuxR, which also activates gene expression. It has been suggested that understanding the key interactions of the signal with its receptor could lead to the development of novel inhibitors of these systems [
6,
7] and hence the development of novel anti-microbial compounds.
Over 50 different Gram-negative species have been identified as using quorum sensing (QS) to control gene expression [
8,
9,
10,
11]. This display of QS, or cell population density-dependant control of gene expression plays an important role in many bacterial transcriptional profile [
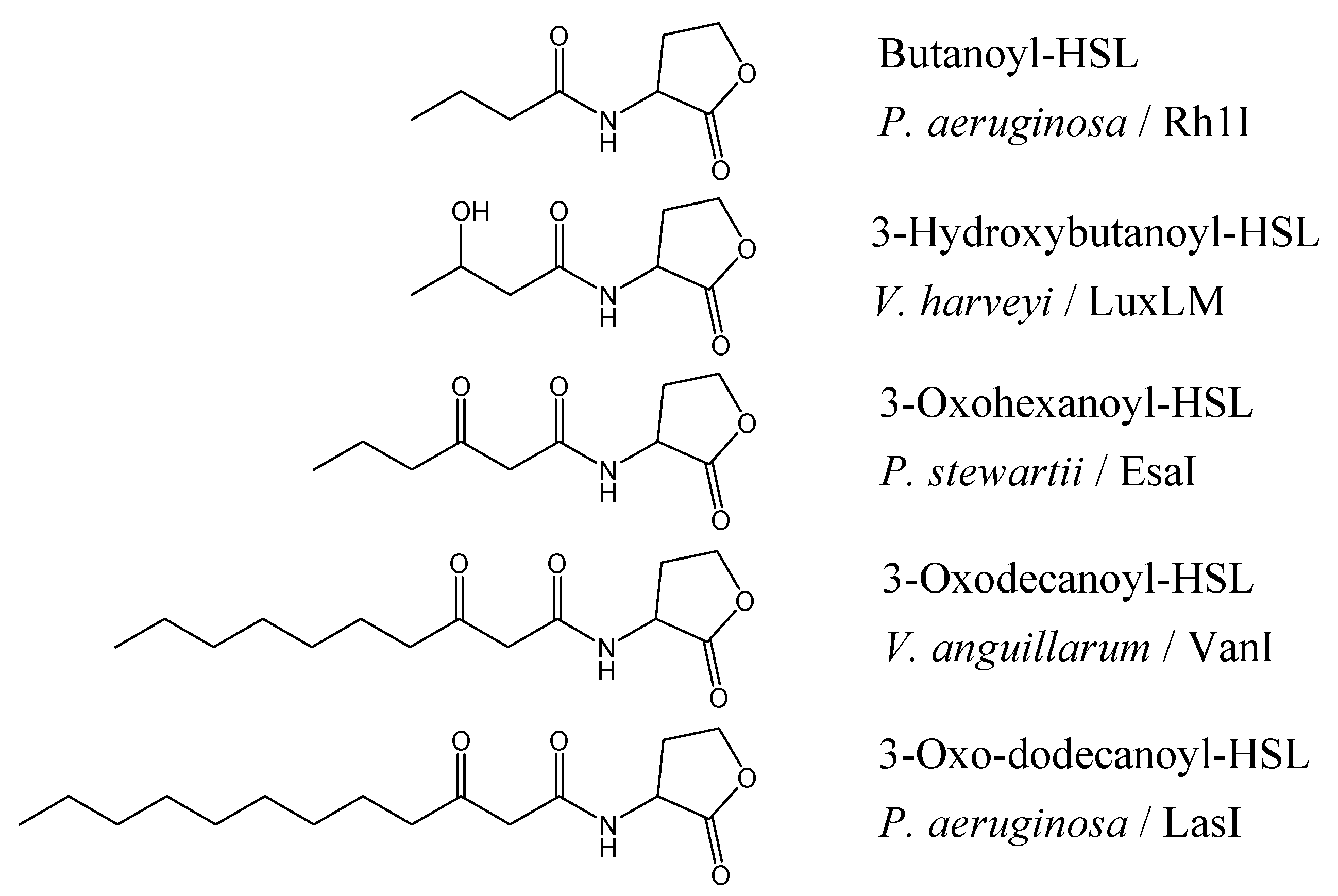
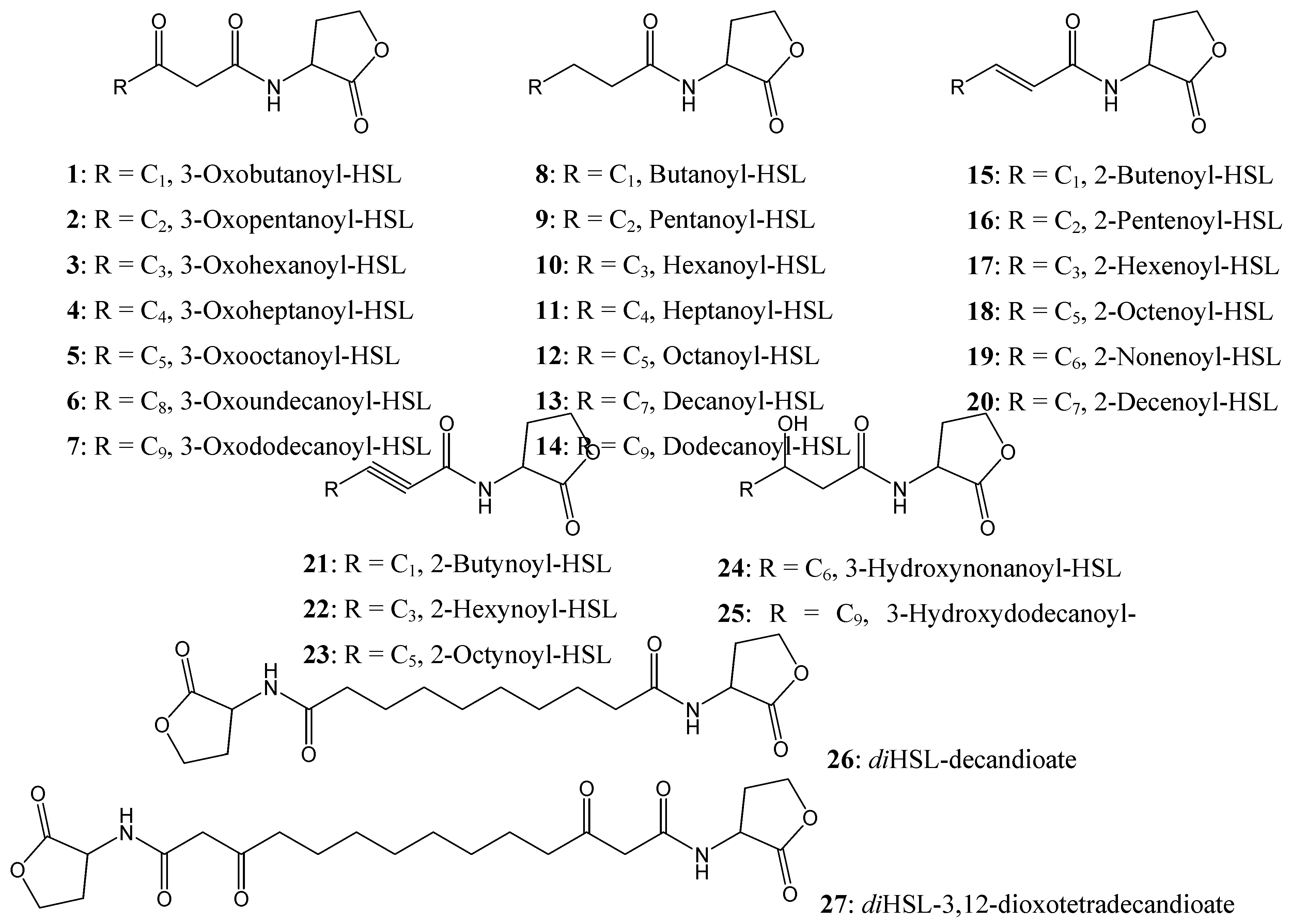
12]. The AHLs utilized by these bacteria all share a common lactone group (‘the head’), but differ in the length and the substitution of the acyl chain (‘the tail’), depending on the individual synthase gene (‘I’).
The chain lengths vary from four to seventeen carbons, maybe unsubstituted, or 3-oxo- or 3-hydroxy substituted (
Figure 1) where the differences may exist to help the bacteria to differentiate signals from other QS bacterium within their environment. Not surprisingly, the diversity of AHLs is also mirrored in the diversity of the receptor protein (‘R’) which have a conserved DNA binding domain (C-terminal) [
13] and the N-terminal signal binding domain [
14].
Figure 1.
Structure of AHLs found in different Gram-negative bacteria and their signal synthases [
16].
Figure 1.
Structure of AHLs found in different Gram-negative bacteria and their signal synthases [
16].
Since the first reported observation of QS signaling in bacteria, many attempts have been directed towards producing synthetic analogues of these autoinducers to mimic natural AHL signaling molecules in an attempt to control the virulence gene expressions of the pathogenic bacteria and also to understand the requirements for signal binding inhibition [
6,
15,
17]. Most of these synthetic analogues contain ‘the head’, a homoserine lactone (HSL) moiety, and ‘the tail’, a fatty acyl group, with various chain lengths, saturation levels, and oxidation states. The biological activity of these compounds has been investigated and some compounds were found to possess significant antagonist behaviour. These compounds were synthesized to closely mirror the cognate autoinducer and the ability of the analogue to bind effectively to the receptor subsequently determines the extent of gene expression.
The binding of the autoinducer to the sensor can be compared to those of a ligand-binding to an enzyme active site. Thus, it is imperative to perform an analysis of the binding receptor site to establish molecular determinants involved in the binding of signal antagonists to the receptor protein. Recently, the crystal structure of TraR, the AHL receptor protein of
Agrobacterium tumefaciens complexed with its cognate autoinducer (3-oxooctanoyl-HSL) and DNA was reported by Vannini
et al. [
18] and Zang
et al. [
19].
Herein, we modeled the signaling molecules,
N-acylated homoserine lactone (AHLs) with the receptor protein (TraR –
A. tumefaciens) to determine structural constraints to ligand-binding. Based on the ligand docking experiments, we compared the docking data to the antagonist activity of known analogues to determine whether a correlation existed between the ligand-binding affinity with known inhibitory activity mediated by signal analogues using previously reported biological data for analogues synthesized by Zhu
et al. [
15].
Materials and Methods
Docking software used: Discovery Studio Modeling 1.2 – LigandFit (Accelrys).
TraR Protein Crystal Structure (PDB ID: 1h0m)
Protocol for ligand-receptor docking: The three-dimensional structure of TraR protein was taken from the pdb file (1h0m). The native autoinducer and all water molecules were removed. Hydrogen were added using the templates for the protein residues. The three-dimensional structures of the analogues were constructed by overlapping analogue conformers onto the reference ligand (5) in the template and then forming new complexes by exchanging atoms or sketching them. The ligands were then energy-minimized in the in-built Modeler available in Discovery Studio.
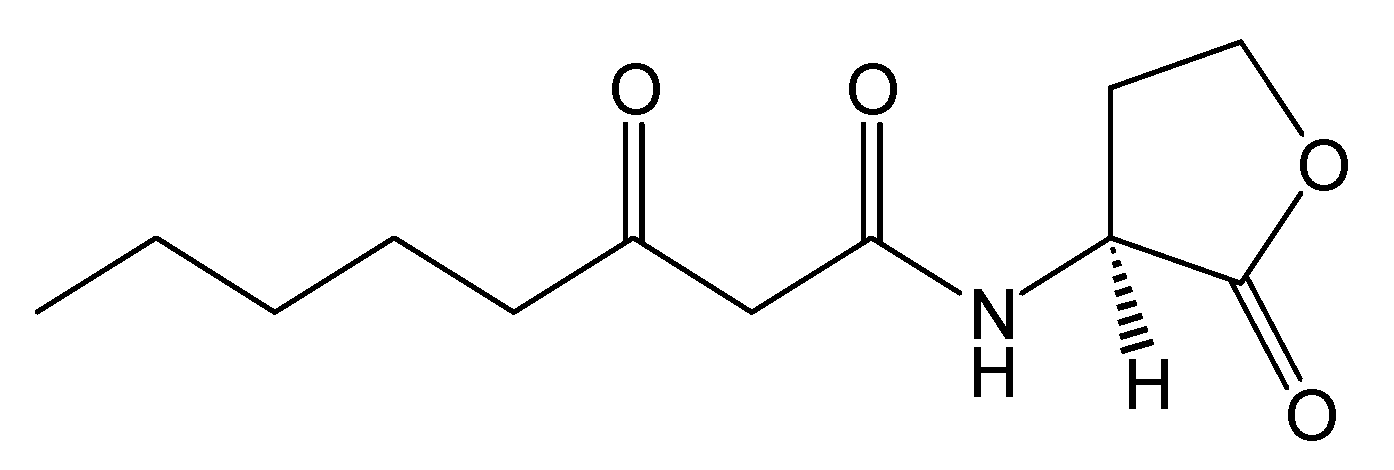
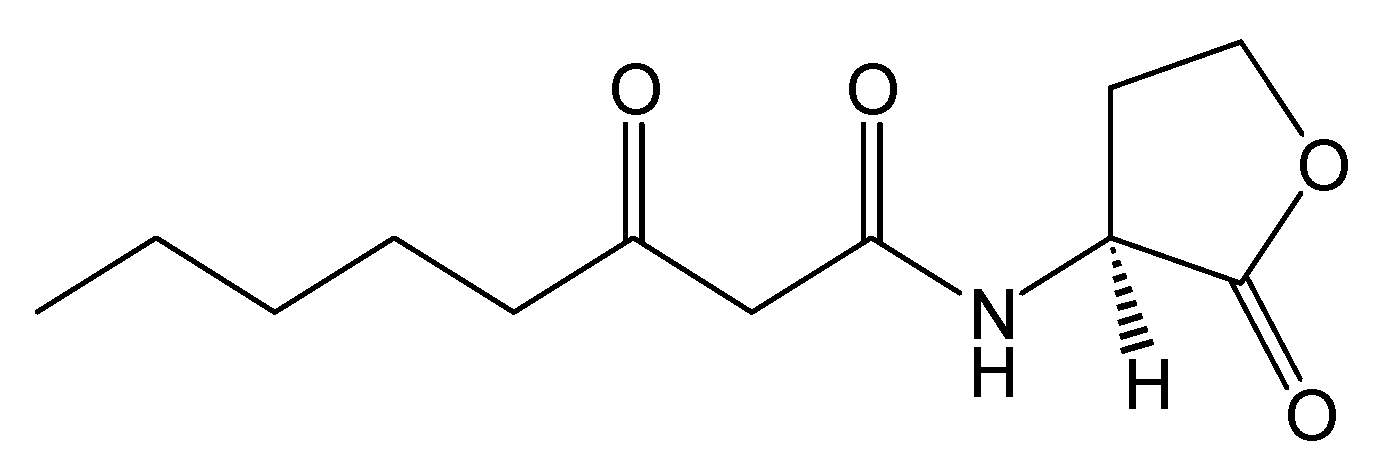
Figure 2.
Structure of N-(3-oxooctanoyl)-L-homoserine lactone.
Figure 2.
Structure of N-(3-oxooctanoyl)-L-homoserine lactone.
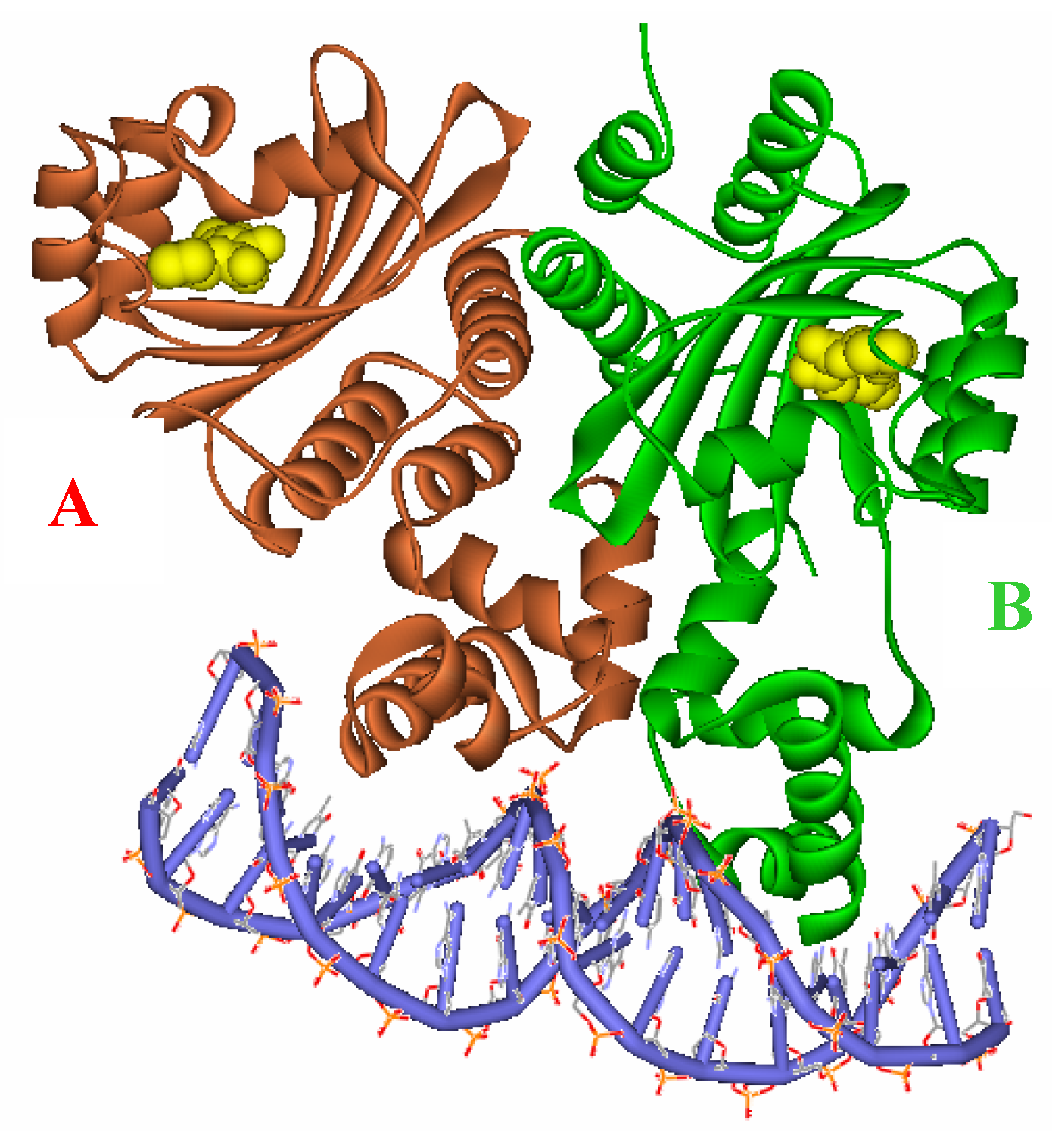
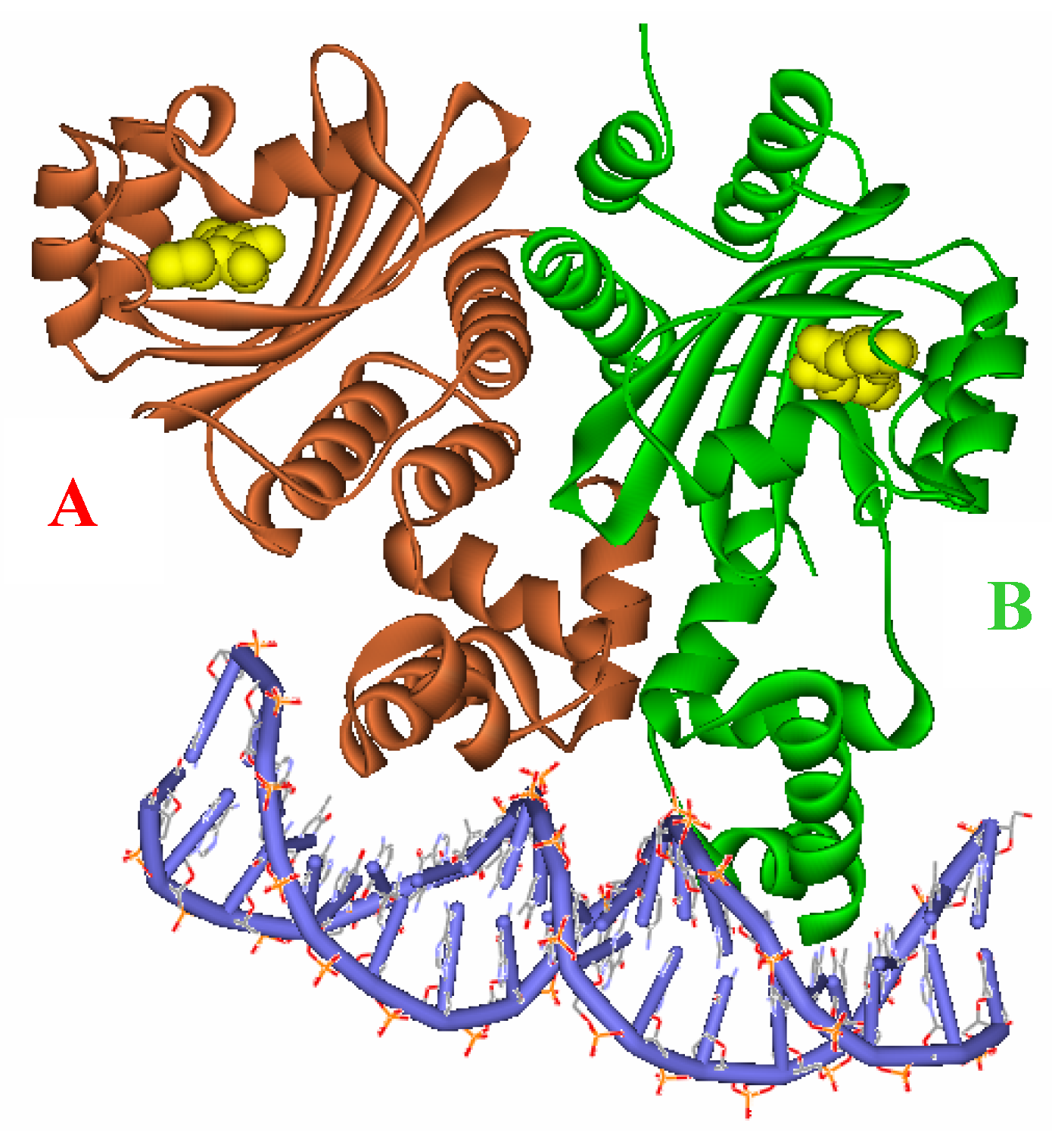
Figure 3.
Ribbon diagram of the two subunits, A (red) and B (green), complexed with the autoinducer (yellow) of the asymmetric unit.
Figure 3.
Ribbon diagram of the two subunits, A (red) and B (green), complexed with the autoinducer (yellow) of the asymmetric unit.
Docking: The active site of Monomer A was first identified and defined using an eraser size of 5.0 Å which resulted in a cavity size of 1709 point units. The ligands were docked into the active site (TraR:A) using the ‘Flexible Fit’ option. The ligand-receptor site complex was subjected to ‘in situ’ ligand minimization which was performed using the in-built CHARMm forcefield calculation. The nonbond cutoff and the distance dependence was set to 11 Å and (ε = 1R) respectively. The determination of the ligand-binding affinity was calculated using the shape-based interaction energies of the ligand with the protein. Consensus scoring with the top tier of s=10% using two scoring methodologies, LigScore and PLP1 were used to estimate the ligand-binding energies.
Results and Discussion
Crystal Structure. The crystal structure of the TraR protein complexed with its autoinducer
N-(3-oxooctanoyl)-
l-HSL, (3-oxooctanoyl-HSL) (
Figure 2), and target DNA was retrieved from the Protein Data Bank (entries: 1h0m). The protein was found to exist as two crystallographically independent complexes in an asymmetric unit attached to a single duplex DNA. One of the asymmetric unit dimers (A:B) bound to the DNA duplex with its individual 3-oxooctanoyl-HSL, coloured yellow, is shown in
Figure 3. The cognate autoinducer was identified as those of
N-(3-oxooctanoyl)-HSL which promotes agonist behaviour.
Figure 4.
Chemical structures of AHL compounds used in the docking study modified from [
15].
Figure 4.
Chemical structures of AHL compounds used in the docking study modified from [
15].
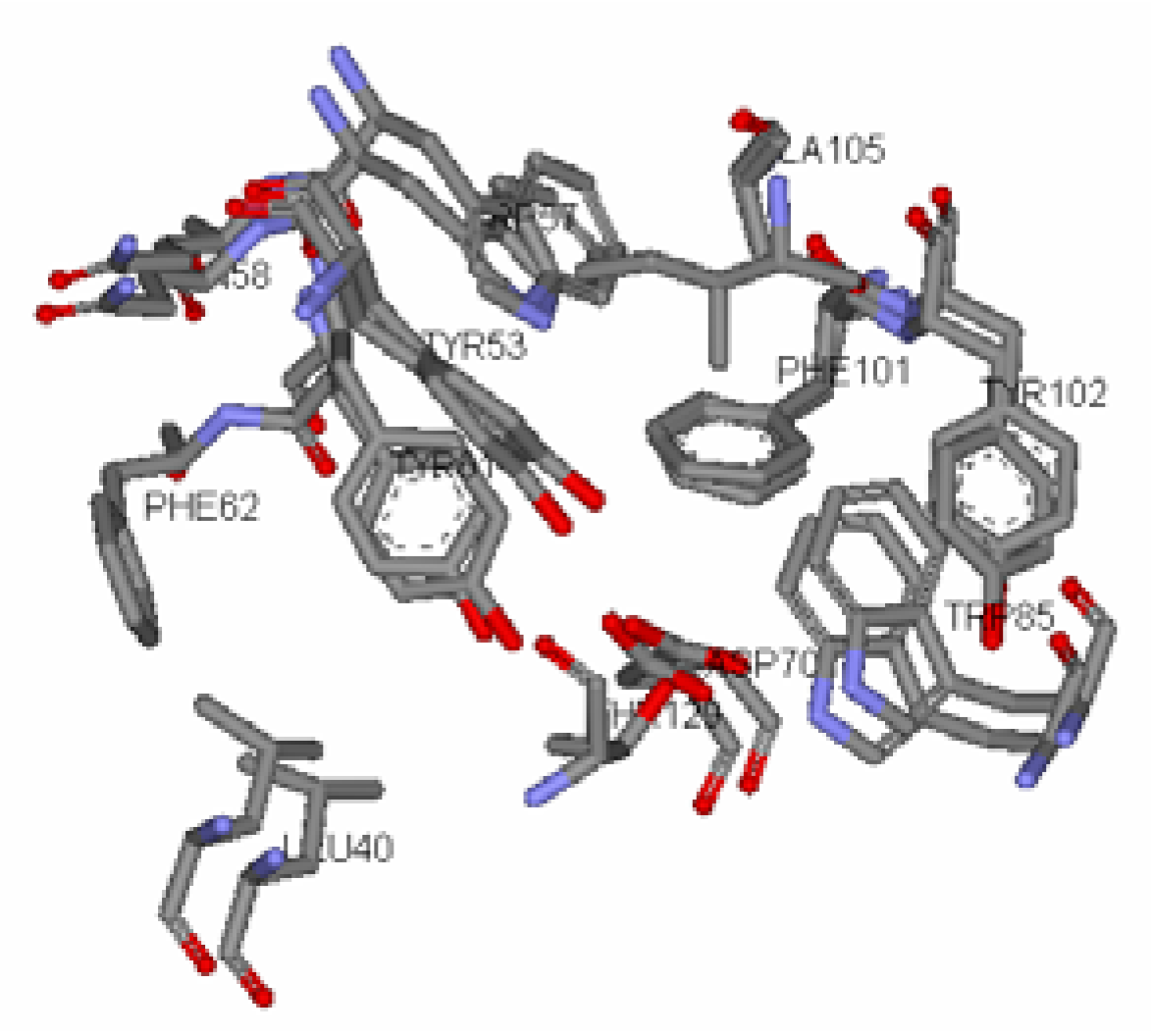
The TraR (A:B) dimer and (C:D) dimer are structural analogues where the conformation of the active sites are conserved [
18]. Furthermore, the monomers A and B were found to be structural analogues of each other, differing only in their conformation around the active site (
Figure 5) when superimposed together. For our study, only monomer A was used as the active receptor site as it gives a fairly good representation of the entire receptor site present in TraR.
A range of synthetic AHLs, preserving the HSL moiety as its ‘head’ but varying in the chain lengths for the fatty acyl group with different saturation levels and oxidation states (
Figure 4) have been synthesized by Zhu
et al [
15]. These compounds were assayed for the ability to induce β-galactosidase activity in strain WCF47(pCF372) and to also competitively displace the cognate AHL, 3-oxooctanoyl-HSL (
5).
Figure 5.
Superimposed Active Site of Monomer A & B
Figure 5.
Superimposed Active Site of Monomer A & B
Table 1.
Biological activity and scoring function values of the ligands docked in the Monomer A receptor site
Table 1.
Biological activity and scoring function values of the ligands docked in the Monomer A receptor site
| Inhibitor Liganda | A/Azb,c | LigScored | PLP1e |
| 3-Oxobutanoyl-HSL (1) | 106 | 5.02 | 58.49 |
| 3-Oxopentanoyl-HSL (2) | 103 | 5.43 | 67.90 |
| 3-Oxohexanoyl-HSL (3) | 100 | 5.11 | 71.37 |
| 3-Oxoheptanoyl-HSL (4) | 62 | 5.91 | 90.48 |
| 3-Oxoundecanoyl-HSL (6) | 82 | 5.01 | 95.58 |
| 3-Oxododecanoyl-HSL (7) | 74 | 4.72 | 93.05 |
| Butanoyl-HSL (8) | 118 | 5.13 | 66.01 |
| Pentanoyl-HSL (9) | 103 | 5.06 | 64.45 |
| Hexanoyl-HSL (10) | 112 | 5.40 | 74.02 |
| Heptanoyl-HSL (11) | 53 | 6.09 | 81.68 |
| Octanoyl-HSL (12) | 28 | 6.11 | 91.51 |
| Decanoyl-HSL (13) | 109 | 5.39 | 90.17 |
| Dodecanoyl-HSL (14) | 121 | 4.40 | 93.37 |
| 2-Butenoyl-HSL (15) | 94 | 4.66 | 53.76 |
| 2-Pentenoyl-HSL (16 | 112 | 4.83 | 67.13 |
| 2-Hexenoyl-HSL (17) | 97 | 4.95 | 74.06 |
| 2-Octenoyl-HSL (18) | 100 | 5.07 | 72.08 |
| 2-Nonenoyl-HSL (19) | 109 | 4.99 | 90.08 |
| 2-Decenoyl-HSL (20) | 106 | 5.26 | 86.45 |
| 2-Butynoyl-HSL (21) | 100 | 4.39 | 56.30 |
| 2-Hexenoyl-HSL (22) | 106 | 4.88 | 78.36 |
| 2-Octynoyl-HSL (23) | 94 | 5.24 | 82.09 |
| 3-Hydroxynonanoyl-HSL (24) | 65 | 6.11 | 100.75 |
| 3-Hydroxydodecanoyl-HSL (25) | 103 | 3.88 | 84.37 |
| diHSL decandioate (26) | 100 | 0.00 | 0.00 |
| diHSL-3,12-dioxotetradecandioate (27) | 100 | 0.00 | 0.00 |
| 3-Oxooctanoyl-HSL (5) | 100 | 5.77 | 90.72 |
The structures listed in
Figure 4 were docked according to a molecular mechanics/dynamic protocol allowing side chain flexibility to the receptor site region. To evaluate the docking protocol without water molecules, all water molecules present in the crystal structure and the native AHL (3-oxooctanoyl-HSL) were removed from the active site and the native AHL was redocked into the active site to investigate the water solvent effect. A similar AHL conformation, polar interactions and position relative to the active site was observed for the native AHL indicating the absence of any solvent effects during ligand-binding. This assumption is supported by the observation that the discrete hydrophobic cavity opening of the receptor site probably precludes any solvent [
18] and the conformation of the AHL in the receptor cavity is likely to depend on the interactions of the ligand and the receptor.
Docking Results. To correlate the biological activity of the different autoinducers and the shape-directed docking of ligands, we scored our models using two scoring functions, LigScore (Protein-Ligand Affinity Energy) and PLP1 (Steric and H-bonding intermolecular function) as implemented in the LigandFit module [
20,
21,
22]. These scoring functions have been used to estimate the ligand-binding affinity to screen out active and inactive compounds during the process of virtual screening [
23]. Previous work has also shown that a correlation does exist between binding affinities and the dock scores [
24]. Scores of the docked ligands are tabulated in
Table 1, (larger score value indicates better ligand-binding affinity).
When the biological activities were compared to the two scoring functions, a similar rank order was observed for six ligands that exhibited competitive antagonist behaviour: 3-oxoheptanoyl-HSL (4); 3-oxoundecanoyl-HSL (6); 3-oxododecanoyl-HSL (7); heptanoyl-HSL (11); octanoyl-HSL (12); and 3-hydroxynonanoyl-HSL (24). Ligands 4, 6, and 7 have similar binding affinities and also demonstrated similar reductions in β-galactosidase activity. Heptanoyl-HSL (11) and octanoyl-HSL (12) were more potent inhibitors of β-galactosidase activity and have higher receptor affinities based on their LigScore. Ligand-binding affinities were unavailable for diHSL-decandioate (26) and diHSL-3,12-dioxotetradecan-dioate (27) as the docking protocol failed to run due to the dimeric nature of these ligands, the ligands being larger than the binding pocket. However, diHSL-3,12-dioxotetradecan-dioate (27) was shown to have antagonist activity at higher concentration (>100 nM) suggesting it may access the binding pocket and could bind in a dimeric fashion with the adjoining monomer e.g. TraR dimer (A:B).
It was observed that the scoring range required for antagonist behaviour was a LigScore >5 and a PLP1 score >90. However, for the ligand to be a good antagonist they must also have higher scoring values compared to the native AHL, 3-oxooctanoyl-HSL (5), as demonstrated by the six ligands which have either a higher LigScore or PLP1 or both, compared to the native AHL, (5). Ligands such as decanoyl-HSL (13) and 2-nonenoyl-HSL (19) have lower affinity/docking scores than (5), and did not display inhibition activity. The remaining 18 ligands have poor scoring values and similarly, did not exhibit antagonist behaviour.
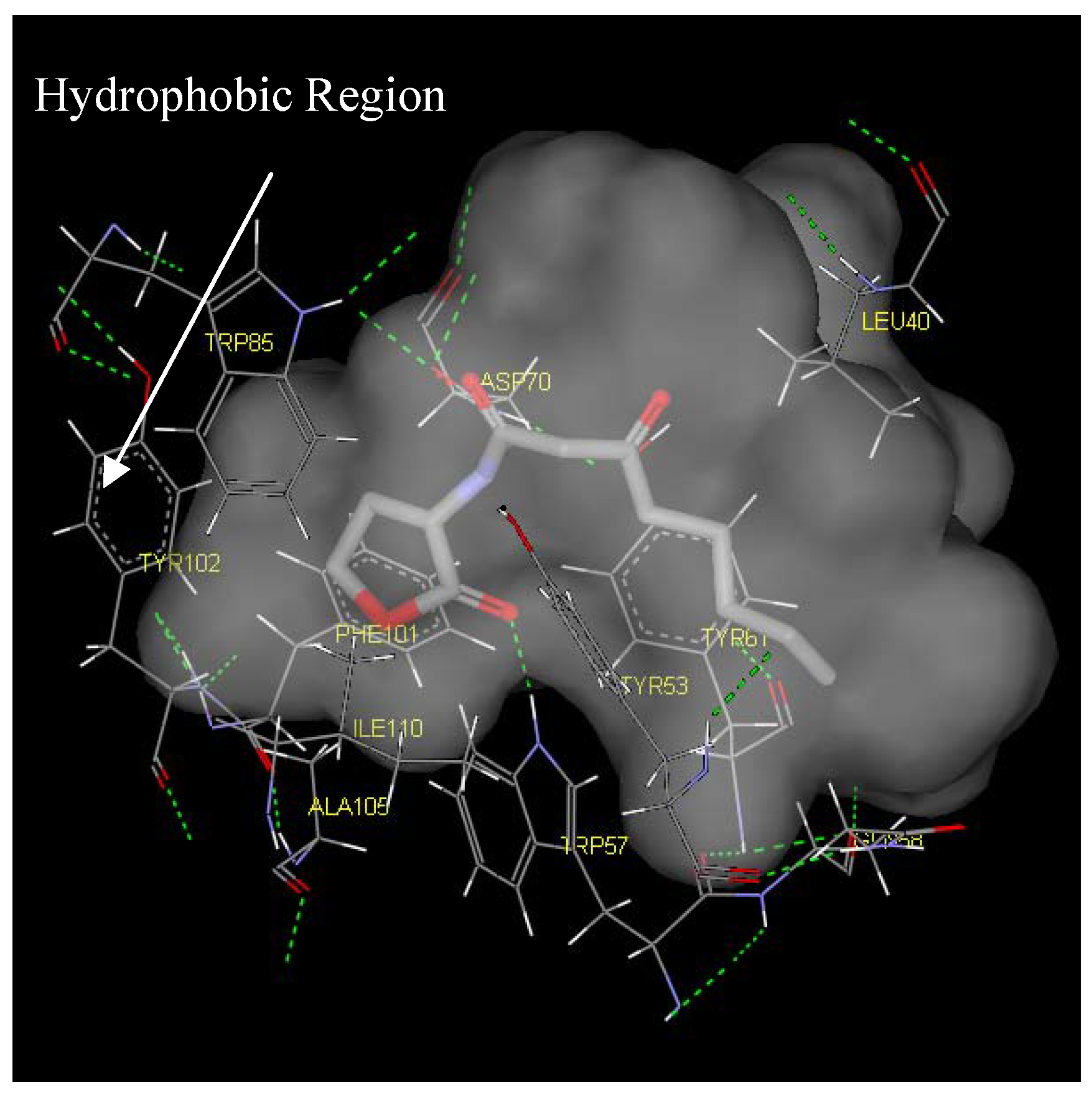
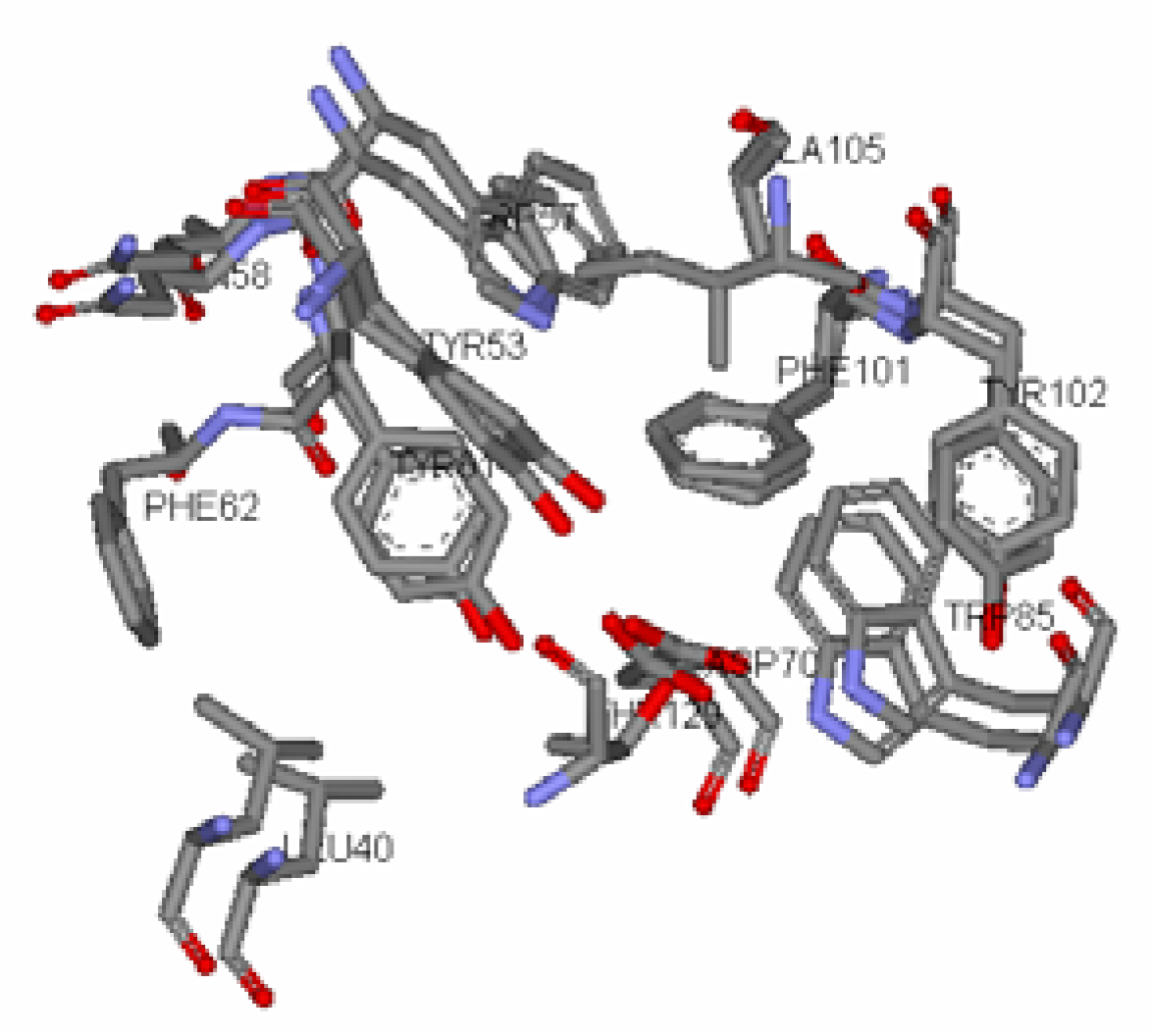
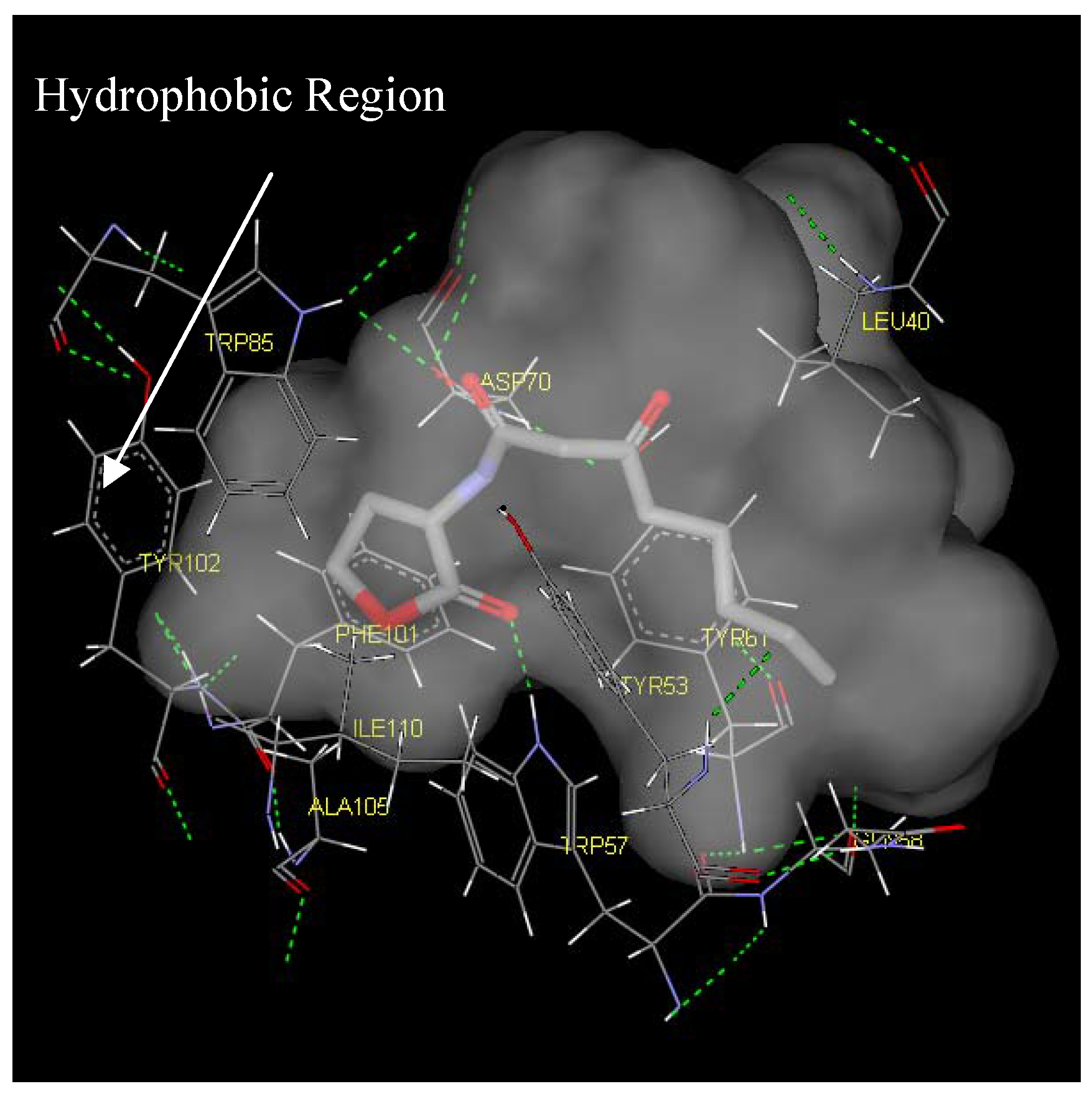
Antagonist selectivity. Although flexibility of the ligand side chains was allowed in all docking protocols, the initial binding pattern of the ‘head’ and ‘tail’ of the ligand to the receptor site remains conserved for all docked ligands. When the docked ligands were superimposed onto the traces of the co-crystallized native autoinducer of TraR, we noticed that they shared the same conformation with the ‘head’ docked into the hydrophilic region bound by Trp52, Trp57, Asp70, Phe101, Tyr102, Ala105 and Ile110 and the tail extended into the hydrophobic region bound by Leu40, Tyr53, Gln58, Tyr61 and Phe62 (
Figure 6). With competitive antagonists, we would assume that ligands with the higher binding affinities for the receptor site would be able to displace the cognate autoinducer and that this activity would be observed at concentration ratios that are closer to the native AHL, (
5). The 6 ligands (ligand
4,
6,
7,
11,
12, and
24,
Figure 4) that exhibited antagonist behaviour have higher binding affinities compared to the native AHL (
5), as determined from both scoring functions.
Figure 6.
Binding pocket surface representation of Monomer A (TraR) with the native autoinducer (E) docked into the receptor site. The ‘transparent cloud’ indicates the binding pocket while atoms of the autoinducer are displayed as thick lines. The thin lines are representative of the different residues surrounding the active site. Green dotted line indicates H-bonding
Figure 6.
Binding pocket surface representation of Monomer A (TraR) with the native autoinducer (E) docked into the receptor site. The ‘transparent cloud’ indicates the binding pocket while atoms of the autoinducer are displayed as thick lines. The thin lines are representative of the different residues surrounding the active site. Green dotted line indicates H-bonding
From the ligand-binding affinities, it can be concluded that there exists a notable ‘tail’ length threshold of about eight carbons extending from the nitrogen atom that is most favorable for ligand-binding affinity. Any change in the length resulted in low scores and unfavourable binding as observed in ligands
7 and
14 with ‘tail’ lengths of eleven carbons. This observation was supported by the available biological data showing a decrease in competitive induction (
Table 1).
Two main classes of acyl chains were studied closely, one having two carbonyl moieties present in the ‘tail’ at the C1 and C3 positions (ligand 1-7), while the other having a single carbonyl moiety at C1 (ligand 8-14). A comparison of both classes of compounds revealed that the ‘tail’ system with only one carbonyl moiety offered a competitive edge over the compounds with two carbonyl moieties. These compounds have stronger ligand-binding affinities and were confirmed to be better antagonists in biological assays. This could largely be due to the conformation of the ligand and possibly the existence of a hydrophilic and hydrophobic region in the receptor cavity. The AHLs, when docked to the cavity, assume the bent-conformation, where the ‘head’ is stabilized in the hydrophilic region through H-bonding between the NH of the homoserine lactone and the ε oxygen of the buried Asp70, and between the carbonyl oxygen of the lactone moiety and the ε nitrogen of Trp57. This stability is probably further enhanced by the H-bonding of the first carbonyl moiety adjacent the nitrogen in the acyl chain with ε nitrogen of Trp85. The acyl carbon chain extends into the cavity and is further stabilized by the hydrophobic interactions with Leu40, Tyr53, Gln58 and Tyr61.
The presence of the second carbonyl moiety (ligand 1-7) extending into hydrophobic region probably disrupts the hydrophobic interactions and thus, an overall lowering of the binding affinity for the native signal. The series of the 3-oxo-C1-9-HSL (ligand 1-7) has a generally lower score compared to the C2-11-HSL (ligand 8-14) but activation is better in the former series. We hypothesize that the 3- oxo moiety might be essential for activation of the genes but may not be suitable for antagonist activity.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}