Towards a Synthesis of Vigabatrin Using Glycal Chemistry

Department of Chemistry, Indian Institute of Technology, Madras, India
*
Author to whom correspondence should be addressed.
Molecules 2005, 10(8), 871-883; https://doi.org/10.3390/10080871
Submission received: 24 October 2004
/
Revised: 8 December 2004
/
Accepted: 7 January 2005
/
Published: 31 August 2005
{kind=link}
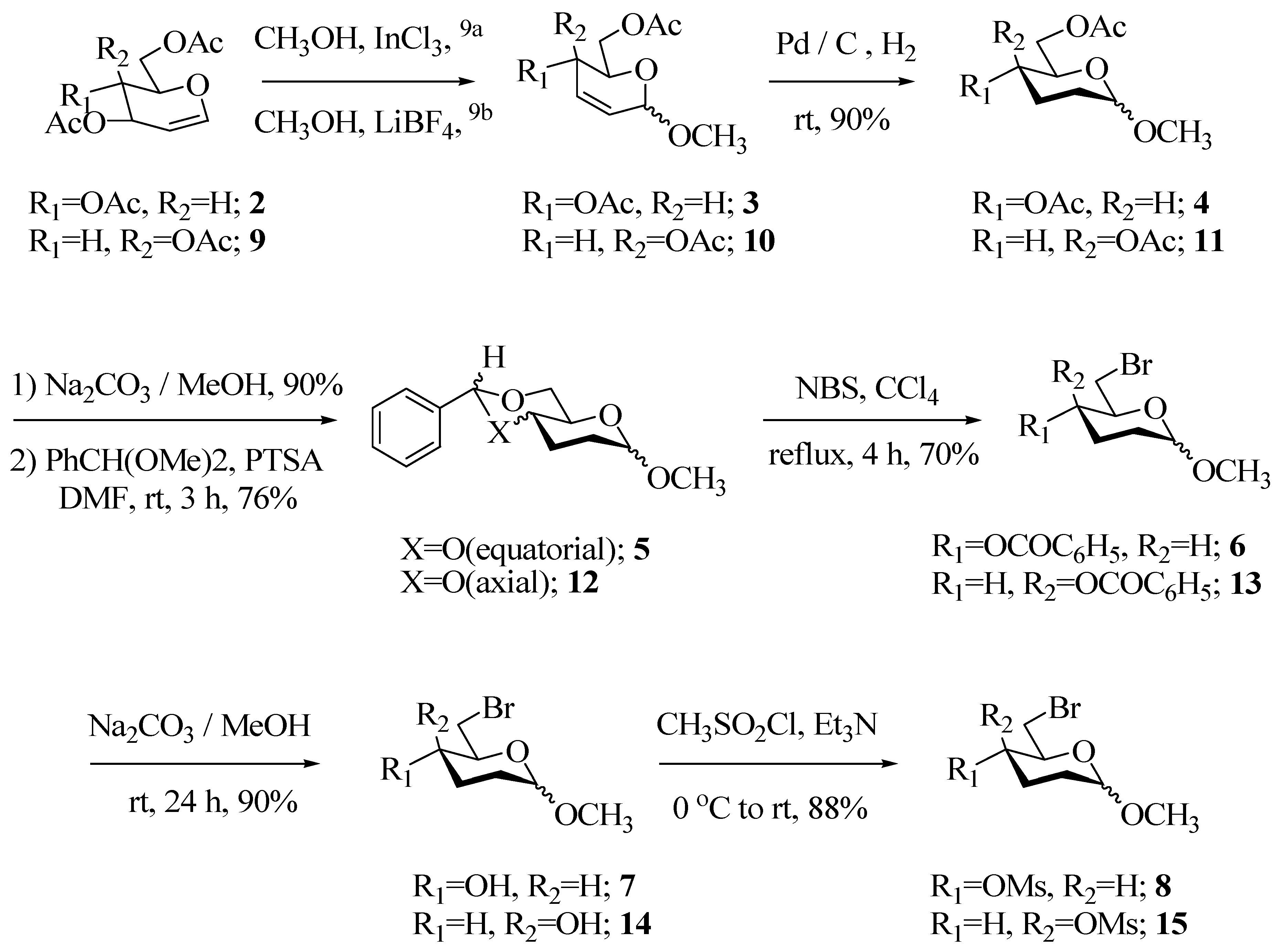{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Application of the Ferrier rearrangement led to a novel carbohydrate based synthetic route to 4-aminohexenoic acid viz. (R) and (S)-vigabatrin. The potential of D- glucose or D-galactose as the chiral starting materials for the synthesis of (R) and (S)- vigabatrin has been explored.
Introduction
γ-Aminobutyric acid (GABA) is the vital inhibitory neurotransmitter in the mammalian central nervous system [1]. It was found that 4-amino-5-hexenoic acid (γ-vinyl GABA, vigabatrin, 1) is one of the most effective and selective catalytic inhibitors of GABA-T.
Inhibition of this enzyme results in an increase in the levels of GABA and could have therapeutic applications in neurological disorders including epilepsy [2], Parkinson’s disease [3], Huntington’s chorea [4] and Alzheimer’s disease [5]. Recently it has been found that an increase in GABA also blocks the effect of drug addiction to nicotine and cocaine [6]. Vigabatrin (1), not a complex chiral molecule, is already marketed in its racemic form as Sabril® in Europe [7], and has evoked considerable interest among organic chemists and medicinal chemists as an attractive synthetic target.
Since vigabatrin has a chiral center at C-4, two enantiomers are possible viz. (R)-vigabatrin and (S)-vigabatrin. It has been found that the (S)-enantiomer is pharmacologically active. Several methods have been reported for the synthesis of enantiomerically pure 1 and most of these employ natural α- amino acids viz., L-glutamic acid, regioselective ring opening of an enantiomerically enriched epoxy alcohol (obtained through Sharpless epoxidation), the Pd(0)-catalyzed deracemization reaction and Sharpless asymmetric aminohydroxylation [8]. The vinyl moiety was then introduced by pyrolysis of an N-oxide or an ester or through a Wittig reaction. While many syntheses of racemic and enantiomerically pure vigabatrin are known, the potential of D-glucose or D-galactose as the chiral starting materials for the synthesis of (R) and (S)-vigabatrin has not been explored. Herein we disclose our results in this area.
![Molecules 10 00871 i001]()

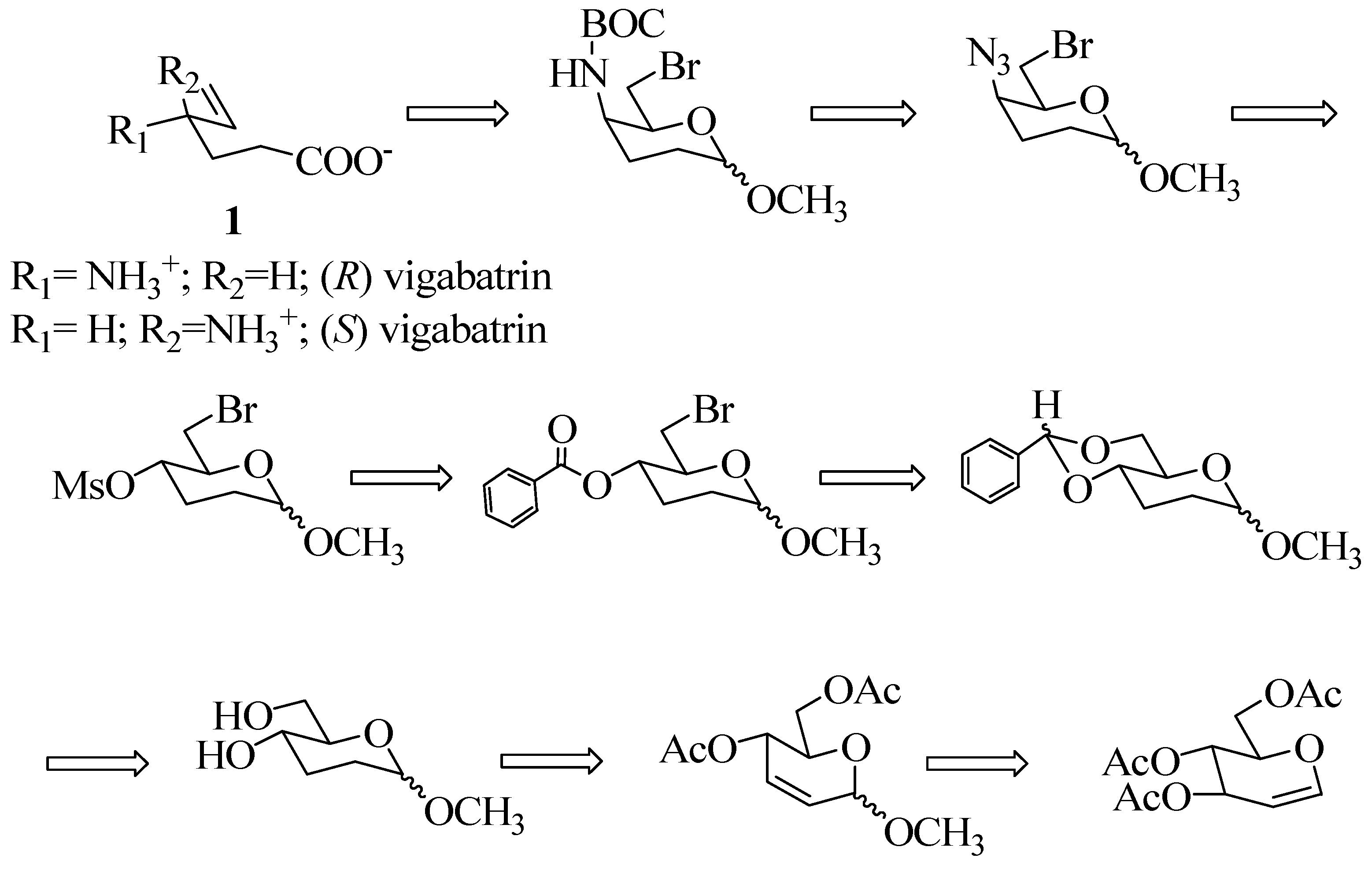
The synthesis of the target molecule vigabatrin (1) was visualized as starting from the 2,3- unsaturated methylglycoside (3), obtained via Ferrier rearrangement of tri-O-acetyl-D-glucal (2) [9]. The 5-amino functionality could be introduced by a SN2 displacement of the mesylate by azide ion and subsequently the vinyl group could be constructed by fragmentation of 6-bromo-6-deoxyglycosides with Zn or n-BuLi - the Vasella reaction [10]. The complete retrosynthetic analysis of (R)-vigabatrin is outlined in Scheme 1.
Scheme 1.

Results and Discussion
Methyl-4,6-di-O-acetyl-2,3-dideoxy-α-D-erythro-hex-2-enopyranoside (3), obtained from 3,4,6-tri- O-acetylglucal 2 [9a] was hydrogenated using Pd/C in MeOH for 1h to afford the saturated derivative 4 in 90% yield. After purification and subsequent hydrolysis of 4, the diol obtained was directly converted to the benzylidene derivative 5 in 76% yield using benzaldehyde dimethylacetal/cat. PTSA in DMF. The benzylidene derivative was fully characterized by spectroscopic techniques. The signal due to the benzylidene proton appeared as a singlet at δ 5.55 in the 1H-NMR spectrum and that of the acetal carbon at δ 101.66 in the 13C-NMR spectrum. Compound 5 was subjected to the Hannesian- Huller reaction [11] using NBS in refluxing CCl4, to furnish the bromobenzoate 6 in 70% yield (Scheme 2).
Scheme 2.

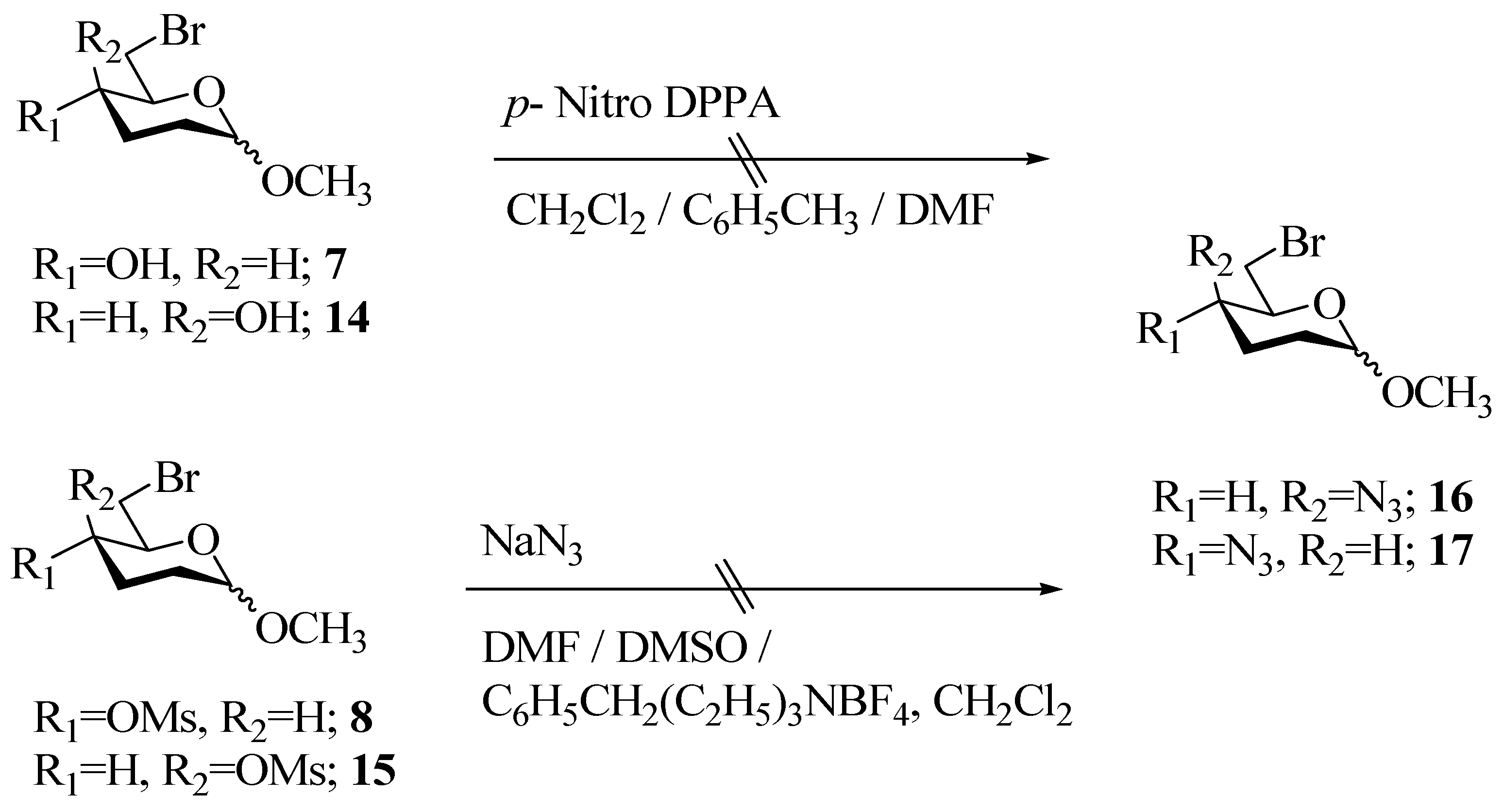
The benzoate 6 was hydrolyzed using Na2CO3/MeOH to give methyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (7) in 80% yield as a homogeneous gum (by TLC). The 1H-NMR and 13C-NMR spectral data were in accordance with the proposed structure. An attempt was made to introduce the azide group at C-4 with an inversion by a modified Mitsonobu reaction (Scheme 3) using bis-(p-nitrophenyl)phosphorazidate as reagent [12], however, there was no reaction at either ambient or elevated temperature (110 °C) and the starting material 7 was recovered.
Having failed to introduce the C-4 azido group by the above methodology, the bromo derivative 7 was converted to the mesylate using mesyl chloride in Et3N. The mesylate 8 was obtained as a viscous liquid in 82% yield. Again considerable efforts were directed to effect exclusive displacement of the mesylate group with azide ion to obtain 16, but all were in vain. These reactions were attempted at different temperatures ranging from 0 °C to 110 °C, and in different solvents viz. DMF and DMSO, as well as under phase transfer catalyst conditions, but without any success, as invariably, a mixture of monoazide, diazide and other products was obtained (Scheme 3).
Scheme 3.

From the above observations, it was apparent that there was not much difference in the reactivity of bromo as a leaving group at C-6 and mesylate at C-4 towards nucleophilic substitution with azide ion. We then considered the synthesis of the C-4 epimer of the mesylate 8, in the hope that there would be some steric hindrance for substitution of the Br at C-6 due to the axial mesylate group and this would result in the selective displacement of the mesylate by azide furnishing the desired 17, a potential precursor for pharmacologically active (S)-vigabatrin. We thus synthesized the epimeric mesylate 15 via a similar synthetic sequence as described for the mesylate 8 (Scheme 2). The synthesis proceeded well in an overall yield of 42% over six steps. All the intermediates leading to 15 were characterized by IR, 1H-NMR and 13C-NMR spectroscopy. Unfortunately, our attempts to bring about a selective displacement of the mesylate group in 15 by azide ion to form 17 were also unsuccessful. Though TLC analysis revealed that the product was homogeneous, giving a close moving non-polar spot but the 1H-NMR and 13C-NMR spectra indicated it to be a mixture of 4,6-diazide and 6-azide derivatives.
In retrospect, we realized that selective displacement of the secondary mesylate with azide ion at C-4, be it axial or equatorial, in the presence of the primary bromide was neither practical nor feasible. Initial incorporation of the azido group at C-4 and later appropriate functionalisation for the Vasella reaction [10] should serve as a viable alternative. Accordingly, the synthetic strategy was slightly modified (Scheme 4).
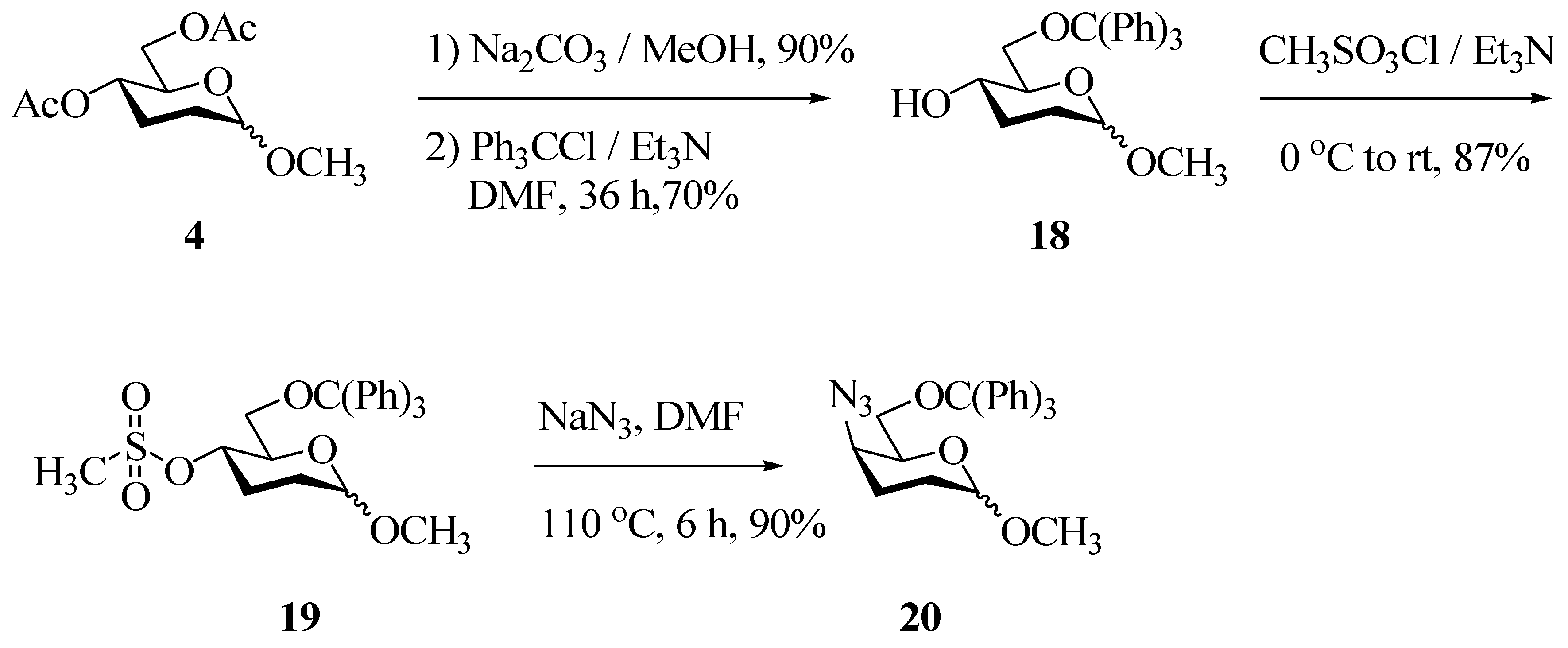
In the proposed alternative route diacetate 4 was hydrolyzed and the C-6 hydroxyl was selectively protected as the corresponding trityl ether using Ph3CCl/Et3N in DMF, thus furnishing compound 18 in 70% yield. The trityl ether 18 was again characterized using IR, 1H- and 13C-NMR spectroscopy. Since we had learned from previous experience that bis (p-nitrophenyl)phophorazidate was not successful in replacing the C4-OH by an azido group, we did not attempt this reaction on 18. Instead 18 was directly converted to the mesylate 19 in 80% yield using methanesulfonyl chloride/Et3N in CH2Cl2. The mesylate 19 underwent a smooth nucleophilic substitution reaction upon treatment with NaN3 in DMF at 110 °C and afforded the crystalline (m.p. 69 °C), methyl 4-azido-6-triphenylmethyl-2,3-dideoxy-D-threo-hexopyranoside (20) in 90% yield.
Scheme 4.

While the anomeric proton resonated at δ 4.67(d), the signal due to H-4 of 20 was shifted upfield δ 3.87(td). The stereochemistry at C-4 was established from the coupling constants of H-4. The signal due to H-4 in 19 appeared as a triplet of doublets with J = 9.99Hz and 5.51Hz, whereas the signal due to H-4 in the azide 20 appeared as a triplet of doublets with smaller coupling constants of 6.1 Hz and 1.47 Hz. The large coupling constant between J4,5 = J3a,4 = 9.99 Hz for H-4 in 19, due to the diaxial nature of H3a and H5 at C-3 and C-5 disappeared, giving rise to a coupling constant J4,5 and J3a,4 = 6.1 Hz due to the vicinal axial-equatorial relationship existing in 20. Based on this, we infer that nucleophilic substitution by the azide has taken place with complete inversion at C-4, thus leading to the threo series.
Scheme 5.

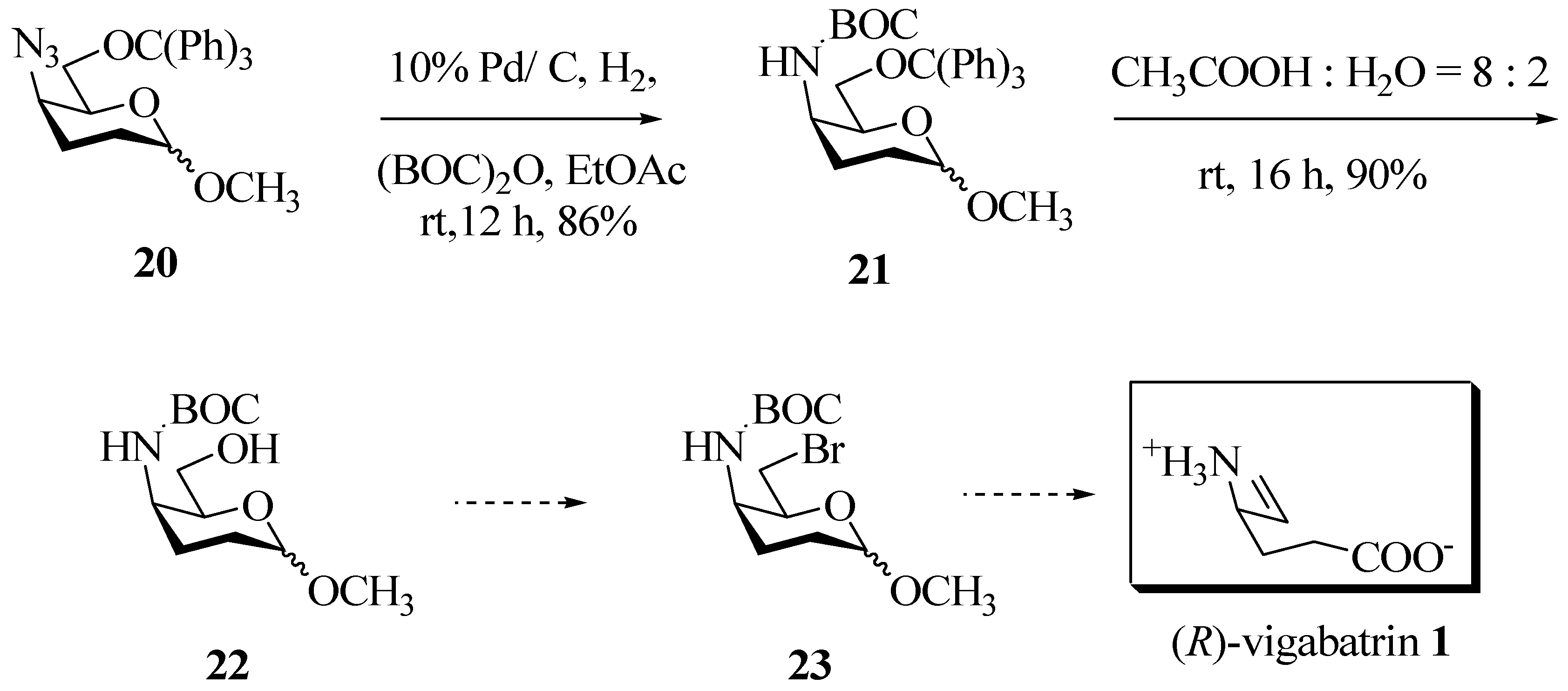
Catalytic reduction of azide 20 over 10% Pd/C and in the presence of (BOC)2O/EtOAc furnished the protected amine 21 in 86% yield as a semi solid (Scheme 5). Detritylation of 21 proceeded without any complications in aq. 80% acetic acid at ambient temperature to furnish methyl 4-(N-t-butoxycarbonyl)-2,3-dideoxy-D-threo-hexopyranoside (22) in 90% yield.
After successful incorporation of the protected amino functionality at C-4, the conversion of 22 to the bromo derivative 23 was necessary for effecting the Vasella reaction and arrive at (R)-vigabatrin (1). This appears to be a straightforward reaction, but various attempts to convert the 6-OH to the corresponding bromide by CBr4/PPh3 or PBr3/pyridine were all in vain. The lack of reactivity of the 6-OH in the above reaction may be due to the presence of a bulky axial 4-(N-t-butoxycarbonyl) group at C-4. The same steric hindrance should be absent in the erythro series, which therefore offers a rationale and motivation to pursue the synthesis of pharmacologically active (S)-vigabatrin. These synthetic efforts are currently underway.
Conclusions
Both methyl-4-methanesulfonyl-6-bromo-2,3-dideoxy-D-erythro-hex-2-eno-pyranoside (8) and methyl-4-methanesulfonyl-6-bromo-2,3-dideoxy-D-threo-hex-2-eno-pyranoside (15) failed to undergo exclusive mono nucleophilic substitution reaction at C-4 with azide under various conditions. The successful incorporation of the amino group and arriving at 20 was finally achieved by adopting a different synthetic sequence, wherein C6-OH is temporarily protected and its derivatization to bromo derivative is postponed to a later stage. Completion of the synthesis of (R)-vigabatrin and (S)-vigabatrin is currently underway.
Experimental
General
1H- and 13C-NMR spectra were obtained using a Varian Gemini 200 NMR and were recorded at 200 and 50 MHz respectively. All reagents and chemicals were obtained from Aldrich Chemical Company (USA) and were used as received unless otherwise noted.
General procedure for the hydrogenation of methyl-4,6-di-O-acetyl-2,3-dideoxy-D-erythro-hex-2- enopyranosid (3) and methyl-4,6-di-O-acetyl-2,3-dideoxy-D-threo-hex-2-enopyranoside(10).
Compounds 3 or 10 (1 mmol) was placed in a 25 mL round bottomed flask. The substrate was dissolved in methanol (5 mL) and a catalytic amount of 10% Pd/C catalyst was added. Hydrogen gas was passed through the solution using a balloon. When there was no more uptake of hydrogen (5 h to 6 h), the reaction was stopped and the catalyst filtered. The solvent was evaporated in a rotary evaporator under reduced pressure to afford methyl-4,6-di-O-acetyl-2,3-dideoxy-D-erythro-hexopyranoside (4) and methyl-4,6-di-O-acetyl-2,3-dideoxy-D-threo-hexopyranoside (11).
Methyl-4,6-di-O-acetyl-2,3-dideoxy-D-erythro-hexopyranoside (4). Semi-solid; TLC: Rf: 0.7 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2960, 2848, 1734, 1452, 1360, 1270, 1126, 1084, 1062, 995, 969, 956, 899, 857; 1H-NMR δ (ppm): 1.44-1.99 (m, 4H, H-2a, H-2e, H-3a, H-3e), 1.96 (s, 3H, OCOCH3), 1.98 (s, 3H, -OCOCH3), 3.26 (s, 3H, OCH3), 3.92-4.06 (m, 3H, H-5, H-6a, H-6b), β), 4.65-4.81 (m, 1H, H-4), 5.20 (bs, 1H, H-1); 13C-NMR δ (ppm): 20.60 (q, -OCOCH3), 20.77 (q, -OCOCH3), 22.51 (t, C-3), 24.16 (t, C-2), 54.70 (q, -OCH3), 63.61 (t, C-6), 66.67 (d, C-5), 66.96 (d, C-4), 97.88 (d, C-1), 170.48 (s, -OCOCH3), 170.65 (s, -OCOCH3).
Methyl-4,6-di-O-acetyl-2,3-dideoxy-D-threo-hexopyranoside (11). Semi solid; TLC: Rf: 0.7 (hexane- EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2962, 2848, 1735, 1458, 1368, 1272, 1124, 1088, 1060, h992, 966; 1H-NMR δ (ppm): 1.50-2.00 (m, 4H, H-2a, H-2e, H-3a, H-3e), 2.06 (s, 3H, OCOCH3), 2.11 (s, 3H, -OCOCH3), 3.38 (s, 3H, OCH3), 4.00-4.25 (m, 3H, H-5, H-6a, H-6b), 4.78 (apparent d, J = 1.18Hz, H-4), 4.93 (bs, 1H, H-1); 13C-NMR δ (ppm): 20.53 (q, -OCOCH3), 20.82 (q, -OCOCH3), 22.24 (t, C-3), 23.89 (t, C-2), 54.46 (q, -OCH3), 63.37 (t, C-6), 66.42 (d, C-5), 66.69 (d, C-4), 97.67 (d, C-1), 170.23 (s, -OCOCH3), 170.41 (s, -OCOCH3).
General procedure for the hydrolysis and subsequent benzylidene protection of methyl-4,6-di-O- acetyl-2,3-dideoxy-D-erythro-hexopyranoside (4) and methyl-4,6-di-O-acetyl-2,3-dideoxy-D-threo- hexopyranoside (11) to obtain the corresponding benzylidene acetals
Compounds 4 or 11 (1 mmol) was dissolved in absolute methanol (50 mL) and stirred for 3h at room temperature with anhydrous sodium carbonate (3 mol). The solids were removed by filtration and the filtrate was evaporated under reduced pressure to give the crude product, which can be used as such for benzylidene protection with out any purification. Diols were dissolved in dry DMF (5 mL) in a two necked round bottomed flask. To this was added benzaldehyde dimethylacetal (1.2 mmol), a catalytic amount of p-toluene-sulfonic acid (15-25 mg) and the contents stirred at room temperature (for 3 h in the case of gluco-4, 14 h in the case of galacto-11). The reaction mixture was poured into cold water (5 mL), stirred for an additional 10 min and the product extracted with hexane (3 x 25 mL), dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain methyl 4,6-O-benzylidene-2,3-dideoxy-D-erythro-hexopyranoside (5) and methyl-4,6-O-benzylidene-2,3-dideoxy-D-threo-hexopyranoside (12).
Methyl-4,6-O-benzylidene-2,3-dideoxy-D-erythro-hexopyranoside (5). Solid (m.p. = 63 °C); TLC: Rf: 0.7 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2944, 2864, 1452, 1369, 1315, 1289, 1126, 1097, 1052, 995, 976, 915; 1H-NMR δ (ppm): 1.64-2.12 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.36 (s, 3H, OCH3), 3.44-3.70 (m, 3H, H-5, H-6a, H-6b), 4.19-4.22 (m, 1H, H-4), 4.67 (d, J1,2a = 2.55Hz, 1H, H-1), 5.55 (s, 1H, ArCHO2), 7.23-7.49 (m, 3H, Ar-H), 7.61-7.88 (m, 2H, Ar-H); 13C-NMR δ (ppm): 23.97 (t, C-3), 29.45 (t, C-2), 55.30 (q, -OCH3), 64.00 (t, C-6), 67.00 (d, C-5), 67.63 (d, C-4), 96.92 (d, C-1), 101.66 (d, -CH-Ar), 124.96 (d, Ar-CH), 127.01 (d, Ar-CH), 127.36 (d, Ar-CH), 137.71 (s, Ar-C).
Methyl-4,6-O-benzylidene-2,3-dideoxy-D-threo-hexopyranoside (12). Semi-solid; TLC: Rf: 0.7 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2948, 2866, 1458, 1366, 1310, 1288, 1128, 1086, 1057, 998; 1H-NMR δ (ppm): 1.62-2.15 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.39 (s, 3H, OCH3), 3.31-3.59 (m, 2H, H-6a, H-6b), 3.83-4.24 (m, 2H, H-4, H-5), 4.83 (bs, 1H, H-1), 5.54 (s, 1H, PCHO2), 7.24-7.62 (m, 4H, Ar-H), 7.85 (d, J = 7.27Hz, 1H, Ar-H); 13C-NMR δ (ppm): 22.18 (t, C-3), 24.69 (t, C-2), 55.30 (q, -OCH3), 64.00 (t, C-6), 67.63 (d, C-5), 67.92 (d, C-4), 98.25 (d, C-1), 101.52 (d, -O2CHAr), 126.25 (d, Ar-CH), 127.66 (d, Ar-CH), 128.92 (d, Ar-CH), 138.30 (s, Ar-C).
General procedure for the opening of benzylidene acetals of methyl-4,6-O-benzylidene-2,3-dideoxy-D- erythro-hexopyranoside (5) and methyl-4,6-O-benzylidene-2,3-dideoxy-D-threo-hexopyranoside (12) using N-bromosuccinimide
Compound 5 or 12 (1 mmol) was dissolved in dry CCl4 (5 mL) in a two necked round bottomed flask. To this solution, N-bromosuccinimide (1.2 mmol) and barium carbonate (0.55 mol) were added. The reaction mixture is heated to reflux for the required period (4 h in the case of 5, 2.5 h in the case of 12). During the initial period of heating, a reddish orange color developed but faded before completion of the reaction. The yellowish gummy residue was washed with CCl4 (2 x 25 mL), and the filtrate and washings were evaporated under reduced pressure. The residue was dissolved in ether (50 mL) and the solution washed with water (3 x 25 mL), dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain methyl-4-O-benzoyl-6- bromo-2,3-dideoxy-D-erythro-hexopyranoside (6) and methyl-4-O-benzoyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (13).
Methyl-4-O-benzoyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (6). Viscous liquid; TLC: Rf: 0.8 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2944, 1718, 1596, 1446, 1360, 1312, 1267, 1126, 1113, 1062, 1027, 998, 944 ; 1H-NMR δ (ppm): 1.69-2.13 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.39 (s, 3H, OCH3), 3.42-3.62 (m, 2H, H-6a, H-6b), 4.03-4.09 (m, 1H, H-5), 4.76 (bs, 1H, H-1), 4.86-4.94 (m, 1H, H-4), 7.39-7.57 (m, 3H, Ar-H), 7.99 (d, J = 7.22Hz, 2H, Ar-H); 13C-NMR δ (ppm): 24.01 (t, C-3), 28.76 (t, C-2), 32.95 (t, C-6), 54.80 (q, -OCH3), 70.07 (d, C-5), 70.98 (d, C-4), 97.63 (d, C-1), 128.43 (d, Ar-CH), 129.60 (d, Ar-CH), 129.72 (d, Ar-CH), 133.25 (s, Ar-C), 165.37 (s, -OCO-Ar).
Methyl-4-O-benzoyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (13). Viscous liquid; TLC: Rf: 0.8 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2948, 1712, 1603, 1444, 1366, 1312, 1276, 1171, 1126, 1120, 1072, 1024, 979, 951, 905; 1H-NMR δ (ppm): 1.57-2.16 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.35 (s, 3H, OCH3), 3.32-3.60 (m, 2H, H-6a, H-6b), 4.14 (t, J5,6a = J5,6b = 6.81Hz, 1H, H-5), 4.84 (bs, 1H, H-1), 5.25 (bs, 1H, H-4), 7.24-7.58 (m, 3H, Ar-H), 8.04 (d, J = 7.38Hz, 2H, Ar-H); 13C-NMR δ (ppm): 21.81 (t, C-3), 24.15 (t, C-2), 31.28 (t, C-6), 55.44 (q, -OCH3), 69.26 (d, C-5), 70.18 (d, C-4), 97.99 (d, C-1), 128.42 (d, Ar-CH), 129.30 (d, Ar-CH), 129.94 (d, Ar-CH), 133.19 (s, Ar-C), 165.68 (s, - OCOAr).
General procedure for the hydrolysis of methyl-4-O-benzoyl-6-bromo-2,3-dideoxy-D-erythro- hexopyranoside (6) and Methyl-4-O-benzoyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (13).
Compounds 6 or 13 (1 mmol) were dissolved in absolute methanol (50 mL) and stirred for 3h at room temperature with anhydrous sodium carbonate (3 mol). The solids were removed by filtration and the filtrate was evaporated under reduced pressure. The residue was further purified by flash column chromatography on silica gel to obtain methyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (7) and methyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (14).
Methyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (7). Viscous liquid; TLC: Rf: 0.3 (hexane- EtOAc = 7:3); IR (CHCl3) υ (cm-1): 3408,2928, 2864, 1372, 1340, 1315, 1123, 1094, 1014, 982, 956, 902, 864 ; 1H-NMR δ (ppm): 1.69-2.01 (m, 4H, H-2a, H-2e, H-3a, H-3e), 2.20 (bs, 1H, 4-OH), 3.40 (s, 3H, OCH3), 3.50-3.70 (m, 3H, H-5, H-6a, H-6b), 3.77 (d, J4,5 = 9.09 Hz, H-4), 4.75 (bs, 1H, H-1); 13C-NMR δ (ppm): 27.45 (t, C-3), 29.13 (t, C-2), 34.21 (t, C-6), 54.64 (q, -OCH3), 68.38 (d, C-4), 72.41 (d, C-5), 97.50 (d, C-1).
Methyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (14). Viscous liquid; TLC: Rf: 0.3 (hexane- EtOAc = 7:3); IR (CHCl3) υ (cm-1): 3410, 2928, 2866, 1376, 1343, 1316, 1128, 1098, 1017, 986, 962; 1H-NMR δ (ppm): 1.67-2.10 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.42 (s, 3H, OCH3), 3.37-3.55 (m, 3H, 4-OH, H-6a, H-6b), 3.90 (bs, 1H, H-4), 3.99 (distorted t, J = 6.20Hz, 1H, H-5), 4.76 (bs, 1H, H-1); 13C-NMR δ (ppm): 23.37 (t, C-3), 25.68 (t, C-2), 31.87 (t, C-6), 54.94 (q, -OCH3), 65.04 (d, C-4), 70.58 (d, C-5), 98.45 (d, C-1).
General procedure for the mesitylation of methyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (7) and methyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (14).
Compound 7 (or 14) (1 mmol) was dissolved in dry dichloromethane (5 mL) in a two-necked round-bottomed flask. To this solution, methansesulfonyl chloride (1.2 mmol) and triethyl amine (2 mmol) were added at 0 °C. The reaction mixture was stirred for 3 h at ambient temperature. The reaction mixture was diluted with CH2Cl2 (2 x 25 mL), washed with water (3 x 25 mL), dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain methyl-4-O-methanesulfonyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (8) and methyl-4-O-methanesulfonyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (15).
Methyl-4-O-methanesulfonyl-6-bromo-2,3-dideoxy-D-erythro-hexopyranoside (8). Viscous liquid; TLC: Rf: 0. 7 (hexane-EtOAc = 1:1); IR (CHCl3) υ (cm-1): 2944, 2864, 1356, 1174, 1129, 1094, 905, 960, 908, 889, 867, 832 ; 1H-NMR δ (ppm): 1.72-2.15 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.06 (s, 3H, -SO2CH3), 3.36 (s, 3H, OCH3), 3.45-3.70 (m, 2H, H-6a, H-6b), 3.87 (d, J = 2.5Hz, 1H, H-5), 4.51 (d, J = 5.5Hz, 1H, H-4), 4.70 (bs, 1H, H-1); 13C-NMR δ (ppm): 25.32 (t, C-3), 28.89 (t, C-2), 32.68 (t, C-6), 38.98 (q, -OSO2CH3), 54.94 (q, -OCH3), 68.97 (d, C-5), 76.61 (d, C-4), 97.43 (d, C-1).
Methyl-4-O-methanesulfonyl-6-bromo-2,3-dideoxy-D-threo-hexopyranoside (15). Vscous liquid; TLC: Rf: 0. 5 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 2948, 2864, 1358, 1176, 1123, 1098, 908, 958, 908; 1H-NMR δ (ppm): 1.72-2.24 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.10 (s, 3H, -SO2CH3 of α), 3.15 (s, 3H, -SO2CH3 of β), 3.41 (s, 3H, OCH3 of α), 3.44 (s, 3H, OCH3 of β), 3.28-3.70 (m, 2H, H-6a, H-6b), 4.06 (t, J5,6a = J5,6b = 6.07Hz, 1H, H-5), 4.78 (bs, 1H, H-1), 4.92 (bs, 1H, H-4); 13C-NMR δ (ppm): 23.35 (t, C-3), 23.85 (t, C-2), 30.34 (t, C-6), 38.65 (q, -OSO2CH3), 54.94 (q, -OCH3), 69.12 (d, C-5), 74.53 (d, C-4), 98.05 (d, C-1).
Methyl-6-O-triphenylmethyl-2,3-dideoxy-D-erythro-hexopyranoside (18). Methyl-2,3-dideoxy-D-erythro-hexopyranoside (4) (1 mmol) was dissolved in dry DMF (5 mL) in a two necked round bottomed flask. To this solution, triphenylmethyl chloride (1.1 mmol), triethyl amine (2 mmol) and catalytic amount DMAP (25 mg) were added. The reaction mixture was stirred for 36 h at room temperature. The solution was poured in to ice water (25 mL) and extracted with dichloromethane (3x 25 mL). The organic layer was washed successively with saturated ammonium chloride solution (3 x 25 mL) and water (3 x 25 mL), dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain (18). Semi solid; TLC: Rf: 0.5 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 3504(b), 2928, 2862, 1600, 1484, 1443, 1369, 1321, 1126, 1081, 988, 944, 899; 1H-NMR δ (ppm): 1.69-1.83 (m, 4H, H-2a, H-2e, H-3a, H-3e), 2.74 (bs, 1H, 4-OH), 3.41 (s, 3H, OCH3), 3.43-3.60 (m, 4H, H-4, H-5, H-6a, H-6b), 4.61 (bs, 1H, H-1), 7.20-7.32 (m, 9H, Ar-H), 7.43-7.46 (m, 6H, Ar-H); 13C-NMR δ (ppm): 28.79 (t, C-3), 29.96 (t, C-2), 54.39 (q, -OCH3), 66.05 (t, C-6), 69.05 (d, C-5), 70.88 (d, C-4), 87.48 (s, -OC(Ph)3), 97.18 (d, C-1), 127.15 (d, Ar-CH), 127.92 (d. Ar-CH), 128.56 (d, Ar-CH), 143.38 (s, Ar-C), 143.57 (s, Ar-C).
Methyl-4-O-methanesulfonyl-6-triphenylmethyl-2,3-dideoxy-D-erythro-hexopyranoside (19). Methyl-6-O-triphenylmethyl-2,3-dideoxy-D-erythro-hexopyranoside (18) (1 mmol) was dissolved in dry dichloromethane (5 mL) in a two-necked round-bottomed flask. To this solution, methansesulfonyl chloride (1.2 mmol) and triethyl amine (2 mmol) were added at 0 °C. The reaction mixture was brought to room temperature over a period of 1.5 h. The reaction mixture was diluted with CH2Cl2 (2 x 25 mL), washed with water (3 x 25 mL), dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain (19). Semi solid; TLC: Rf: 0.6 (hexane-EtOAc = 8:2); IR (CHCl3) υ (cm-1): 2928, 1600, 1488, 1446, 1356, 1174, 1129, 972, 956; 1H-NMR δ (ppm): 1.69-2.27 (m, 4H, H-2a, H-2e, H-3a, H-3e), 2.55 (s, 3H, 4-OSO2CH3), 3.40 (s, 3H, OCH3), 3.26-3.57 (m, 2H, H-6a, H-6b), 3.86 (dd, J4,5 = 9.7Hz, J5,6a = 2.96Hz, 1H, H-5), 4.64 (td, J4,5 = J3a,4 = 9.99Hz, J3e,4 = 5.51Hz, 1H, H-4), 4.77 (s, 1H, H-1), 7.05-7.28 (m, 9H, Ar-H), 7.45-7.48 (m, 6H, Ar-H); 13C-NMR δ (ppm): 25.89 (t, C-3), 28.81 (t, C-2), 37.93 (q, -OSO2CH3), 54.50 (q, -OCH3), 62.57 (t, C-6), 69.28 (d, C-5), 75.61 (d, C-4), 86.58 (s, -OC(Ph)3), 97.14 (d, C-1), 125.73 (d, Ar-CH), 127.04 (d, Ar-CH), 127.74 (d, Ar-CH), 128.70 (d, Ar-CH), 143.57 (s, Ar-C).
Methyl-4-azido-6-O-triphenylmethyl-2,3-dideoxy-D-threo-hexopyranoside (20). Methyl-4-O- methanesulfonyl-6-triphenylmethyl-2,3-dideoxy-D-erythro-hexopyranoside (19) (1 mmol) was dissolved in dry DMF (3 mL) in a two necked round bottomed flask. To this solution were added sodium azide (1.2 mmol) and one or two drops of water, just to solubilise the azide. The reaction mixture was heated at 110 °C for 6 h, then poured in to ice water (25 mL) and extracted with dichloromethane (5 x 25 mL). The organic layer was dried over anhydrous sodium sulphate and concentrated. The residue was purified by flash column chromatography on silica gel to obtain (20). Solid (m.p. = 69 °C); TLC: Rf: 0.6 (hexane-EtOAc = 8:2); IR (CHCl3) υ (cm-1): 3056, 2928, 2112, 1484, 1443, 1366, 1331, 1264, 1206, 1180, 1126, 1078, 1033, 995, 953, 921, 899, 707, 630; 1H-NMR δ (ppm): 1.69-2.13 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.15 (dd, J6a,6b = 13.18Hz, J5,6a = 5.86Hz, 1H, H-6a), 3.20-3.39 (m, 1H, H-6b), 3.33 (s, 3H, OCH3), 3.73 (s, 1H, H-5), 3.87 (td, J4,5 = J3a,4 = 6.1Hz, J3e,4 = 1.47Hz, 1H, H-4), 4.67 (d, J1,2e = 2.93Hz, 1H, H-1), 7.20-7.31 (m, 9H, Ar-H), 7.42-7.82 (m, 6H, Ar-H); 13C-NMR δ (ppm): 22.60 (t, C-3), 24.40 (t, C-2), 54.67 (q, -OCH3), 56.77 (d, C-4), 63.67 (t, C-6), 68.13 (d, C-5), 86.91 (s, -OC(Ph)3), 97.07 (d, C-1), 127.06 (d, Ar-CH), 127.82 (d, Ar-CH), 128.64 (d, Ar-CH), 128.74 (d, Ar-CH), 143.91 (s, Ar-C).
Methyl-4-(N-t-butoxycarbonyl)-6-O-triphenylmethyl-2,3-dideoxy-D-threo-exopyranoside (21). Methyl-4-azido-6-O-triphenylmethyl-2,3-dideoxy-D-threo-hexopyranoside (20) (1 mmol) was dissolved in EtOAc (5 mL) in a two necked round bottomed flask. To this solution, a catalytic amount of 10% Pd / C catalyst (25mg) and di-t-butylcarbonate (1.2 mmol) were added. Hydrogen gas was passed through the solution using a balloon. When there was no more uptake of hydrogen (~ 12h), the reaction was stopped and the catalyst filtered. The solvent was evaporated in a rotary evaporator under reduced pressure and the residue was purified by flash column chromatography on silica gel to obtain pure product (21). Semi solid; TLC: Rf: 0.5 (hexane-EtOAc = 8:2); IR (CHCl3) υ (cm-1): 3440(s), 2992, 2960, 1708, 1484, 1449, 1385, 1356, 1328, 1161, 1116, 1068, 1030, 1001, 905, 630; 1H-NMR δ (ppm): 1.36 (s, 9H, C(CH3)3), 1.60-2.03 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.09-3.17 (m, 2H, H-6a, H-6b), 3.42 (s, 3H, OCH3), 3.71 (bs, 1H, H-5), 4.05 (bs, H-4), 4.72 (bs, 1H, H-1), 5.13 (d, J 8.3Hz, 1H, -NH), 7.20-7.30 (m, 9H, Ar-H), 7.44-7.84 (m, 6H, Ar-H); 13C-NMR δ (ppm): 23.87 (t, C-3), 24.49 (t, C-2), 28.35 (q, -OC(CH3)3), 45.88 (d, C-4), 54.62 (q, -OCH3), 64.54 (t, C-6), 69.00 (d, C-5), 79.11 (s, OC(CH3)3 of α), 86.65 (s, OC(CH3)3 of β), 98.07 (d, C-1), 126.95 (d, Ar-CH), 127.77 (d, Ar-CH), 128.68 (d, Ar-CH), 143.98 (s, Ar-C), 155.35 (s, -OCO(CH3)3).
Methyl-4-(N-t-butoxycarbonyl)-2,3-dideoxy-D-threo-hexopyranoside (22). To methyl-4-(N-t- butoxycarbonyl)-6-O-triphenylmethyl-2,3-dideoxy-D-threo-exopyranoside (21) (1 mmol) taken in a round bottomed flask, was added a solution of aq. 80% acetic acid (2 mL) and the contents stirred for 16 h at room temperature. The reaction mixture was poured into cold water (10 mL) and extracted with chloroform (5x 25 mL). The organic layer was washed repeatedly with water (3 x 25 mL), saturated hydrogen carbonate (3 x 25 mL) and again with water (3 x 25 mL). The organic layer was evaporated in a rotary evaporator under reduced pressure and the residue was purified by flash column chromatography on silica gel to obtain pure product (22). Viscous liquid; TLC: Rf: 0.2 (hexane-EtOAc = 7:3); IR (CHCl3) υ (cm-1): 3540(bs), 2992, 2960, 1706, 1488, 1449, 1386, 1361, 1328, 1164, 1110, 1066, 1028, 908; 1H-NMR δ (ppm): 1.36 (s, 9H, C(CH3)3), 1.67-2.17 (m, 4H, H-2a, H-2e, H-3a, H-3e), 3.19-3.70 (m, 2H, H-6a, H-6b), 3.35 (s, 3H, OCH3), 3.83-3.98 (m, 2H, H-4, H-5), 4.76 (bs, 1H, H-1), 5.15 (d, J = 8.68Hz, 1H, -NH); 13C-NMR δ (ppm): 23.06 (t, C-3), 24.70 (t, C-2), 28.23 (q, -OC(CH3)3), 44.33 (d, C-4), 54.67 (q, -OCH3), 61.75 (t, C-6), 69.33 (d, C-5), 80.17 (s, -OC(CH3)3), 97.74 (d, C-1), 157.03 (s, -OCO(CH3)3).
References and Notes
- Krogsgaard-Larsen, P.; Scheel-Kruger, J.; Kotid, H. GABA-Neurotransmitters; Munksgard: Copenhagen, 1979. [Google Scholar] Lippert, B.; Metcalf, B. W.; Jung, M. J.; Casara, P. 4-amino-hex-5-enoic acid, a selective catalytic inhibitor of 4-aminobutyric-acid aminotransferase in mammalian brain. Eur. J. Biochem. 1977, 74, 441–445. [Google Scholar] Nanavati, S. M.; Silverman, R. B. Mechanisms of inactivation of γ-aminobutyric acid aminotransferase by the antiepilepsy drug γ-vinyl GABA (vigabatrin). J. Am. Chem. Soc. 1991, 11, 9341–9349. [Google Scholar]
- Gale, K. GABA in epilepsy: the pharmacologic basis. Epilepsia 1989, 30, S1–11. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O.; Lloyd, K. G.; Davidson, L. The GABA system, function of the basal ganglia, and Parkinson's disease. In (GABA in Nervous System Function); Roberts, E., Chase, T. N., Tower, D. B., Eds.; Raven Press: New York, 1976; pp. 479–485. [Google Scholar]
- Perry, T. L.; Hansen, S.; Kloster, M. Huntington's chorea. Deficiency of gamma-aminobutyric acid in brain. New Eng. J. Med. 1973, 28, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Aoyage, T.; Wada, T.; Nagai, M.; Kojima, F.; Harada, S.; Takeuchi, T.; Takahashi, H.; Kirokawa, K.; Tsumita, T. Increased gamma-aminobutyrate aminotransferase activity in brain of patients with Alzheimer's disease. Chem. Pharm. Bull. 1990, 38, 1748–1749. [Google Scholar] [CrossRef]
- Kushner, S. A.; Dewey, S. L.; Kornetsky, C. J. The irreversible γ-aminobutyric acid (GABA) transaminase inhibitor γ-vinyl-GABA blocks cocaine self-administration in rats. Pharmacol. Exp. Ther. 1999, 290, 797–802. [Google Scholar] Dewey, S. L.; Morgan, A. E.; Ashby, C. R., Jr.; Horan, B.; Kushner, S. A.; Logan, J.; Volkow, N. D.; Fowler, J. S.; Gardner, E. L.; Brodie, J. D. A novel strategy for the treatment of cocaine addiction. Synapse 1998, 30, 119–129. [Google Scholar] Brennan, M. Epilepsy drug could treat nicotine addiction. C&EN December 7. 1998, 13. [Google Scholar]
- Delgado-Escueta, A. V.; Ward, A. A., Jr.; Woodbury, D. M.; Porter, R. J. Basic Mechanisms of the Epilepsies; Raven Press: New York, 1986; p. 365. [Google Scholar]
- Kwon, T. W.; Keusenkothen, P. F.; Smith, M. B. Asymmetric synthesis of (S)-4-aminohex-5- enoic acid: a potent inhibitor of 4-aminobutyrate-2-oxoglutarate aminotransferase. J. Org.Chem. 1992, 57, 6169–6173. [Google Scholar] [CrossRef] Margolin, A. L. Synthesis of optically pure mechanism-based inhibitors of γ-aminobutyric acid aminotransferase (GABA-T) via enzyme-catalyzed resolution. Tetrahedron Lett. 1993, 34, 1239–1242. [Google Scholar] Wei, Z. Y.; Knaus, E. E. A short efficient synthesis of (S)-4-amino- 5-hexenoic acid [(S)-vigabatrin]. J. Org. Chem. 1993, 58, 1586–1588. [Google Scholar] Wei, Z. Y.; Knaus, E. E. Asymmetric synthesis of (S)-vigabatrin. An approach using methionine as the chiral pool. Synlett 1993, 295–296. [Google Scholar] Wei, Z.Y.; Knaus, E. E. One-step reduction-Wittig olefination of methyl pyroglutamate: chiral syntheses of (5S)-5-alkenyl-2-pyrrolidinones and (S)-vigabatrin. Synlett. 1994, 345–346. [Google Scholar] Wei, Z.-Y.; Knaus, E. E. Asymmetric synthesis of both enantiomers of vigabatrin: an approach using methionine as the chiral pool. Tetrahedron 1994, 50, 5569–5578. [Google Scholar] Trost, B. M.; Lemonine, R. C. An asymmetric synthesis of vigabatrin. Tetrahedron Lett. 1996, 37, 9161–9164. [Google Scholar] Alcon, M.; Poch, M.; Moyano, A.; Pericas, M. A.; Riera, A. Enantioselective synthesis of (S)-vigabatrin. Tetrahedron: Asymmetry 1997, 8, 2967. [Google Scholar] Chandrasekhar, S.; Mohapatra, S. Asymmetric synthesis of anti-convulsive drug (S)-vigabatrin. Tetrahedron Lett. 1998, 39, 6415–6418. [Google Scholar] Dagoneau, C.; Tomassini, A.; Denis, J.-N.; Vallée, Y. A short synthesis of γ-amino acids from nitrones; synthesis of vigabatrin. Synthesis 2001, 1, 150–154. [Google Scholar]
- Sobhana Babu, B.; Balasubramanian, K. K. Indium trichloride Catalyzed Glycosidation: An expeditious synthesis of 2,3-unsaturated glycopyranosides. Tetrahedron Lett. 2000, 41, 1271–1274. [Google Scholar] [CrossRef] Sobhana Babu, B.; Balasubramanian, K. K. Lithium tetrafluoborate catalyzed Ferrier rearrangement – Facile synthesis of alkyl 2,3-unsaturated glycopyranosides. Synth. Commun. 1999, 29, 4299–4305. [Google Scholar]
- Bernet, B.; Vasella, A. Fragmentation of 6-deoxy-6-halohexono-1,5-ortholactones: a concerted, nonstereospecific process. Helv. Chim. Acta. 1984, 67, 1328–47. [Google Scholar] [CrossRef]
- Hanessian, S.; Plessas, N. R. The reactions of O-benzylidene sugars with N-bromosuccinimide. II. Scope and synthetic utility in the methyl 4,6-O-benzylidenehexopyranoside series. J. Org. Chem. 1969, 34, 1035–1044. [Google Scholar] [CrossRef]
- Mizuno, M.; Shiori, T. Efficient method for the one-pot azidation of alcohols using bis(p- nitrophenyl)phosphorazidate. Chem. Commun. 1997, 2165–2166. [Google Scholar] [CrossRef]
- Sample availability: Contact the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Boga, S.; Aidhen, J.; Aidhen, I. Towards a Synthesis of Vigabatrin Using Glycal Chemistry. Molecules 2005, 10, 871-883. https://doi.org/10.3390/10080871
AMA Style
Boga S, Aidhen J, Aidhen I. Towards a Synthesis of Vigabatrin Using Glycal Chemistry. Molecules. 2005; 10(8):871-883. https://doi.org/10.3390/10080871
Chicago/Turabian StyleBoga, S., J. Aidhen, and I. Aidhen. 2005. "Towards a Synthesis of Vigabatrin Using Glycal Chemistry" Molecules 10, no. 8: 871-883. https://doi.org/10.3390/10080871