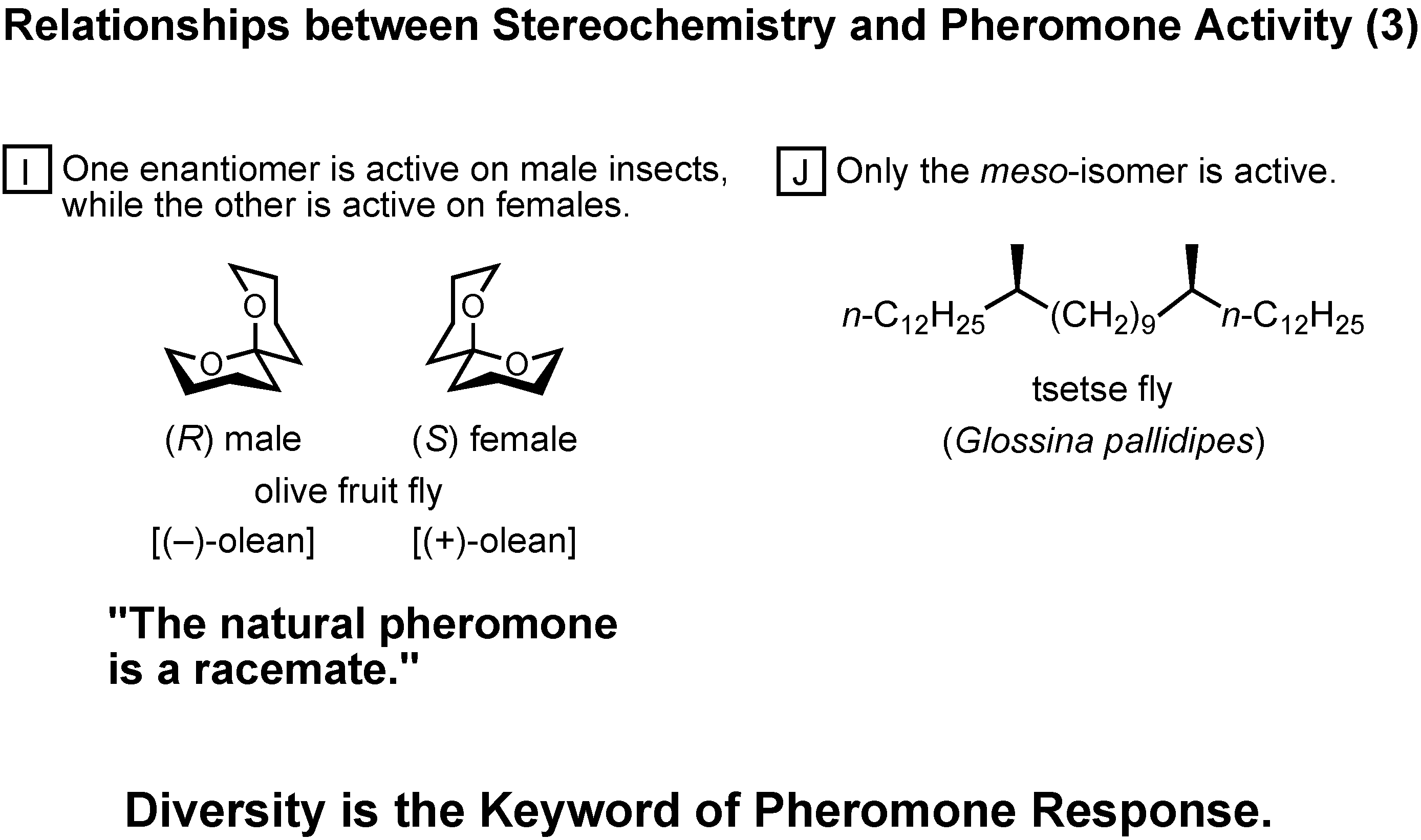Thank you for your kind introduction. I also thank Profs. Tkachev and De Kimpe for their kind invitation to attend this conference. This is my first visit to Siberia and I really expect to see something new in Altai mountains after the Conference. May I have the first PowerPoint picture, please…
Today I would like to tell you about these three aspects in organic synthesis related to pheromone science: No. 1; Synthesis can prove or disprove the proposed structure of a pheromone. No. 2: Synthesis can provide a substantial amount of a useful pheromone. No. 3: Synthesis can clarify the structure-activity relationships among pheromones (
Figure 1).
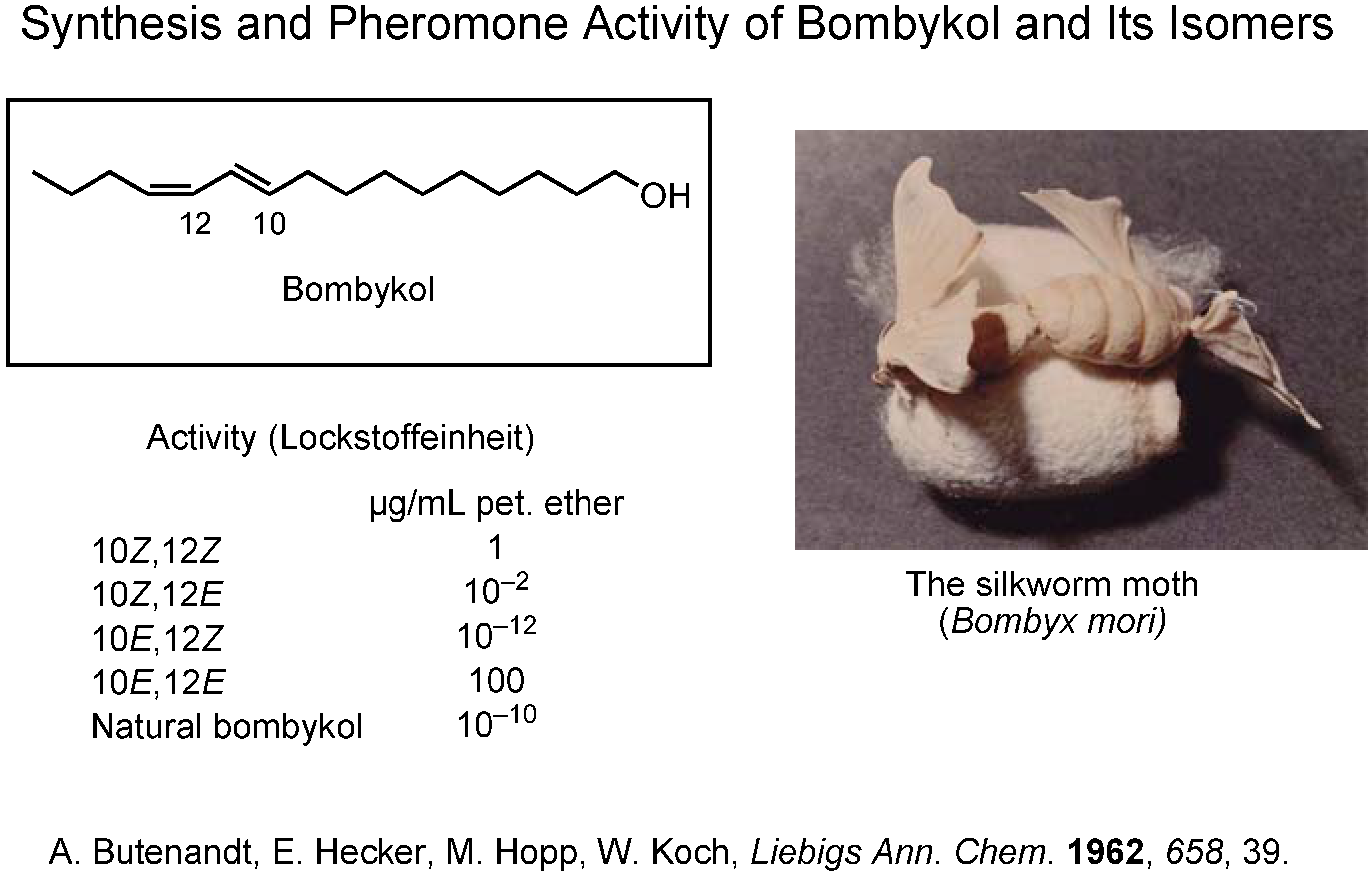
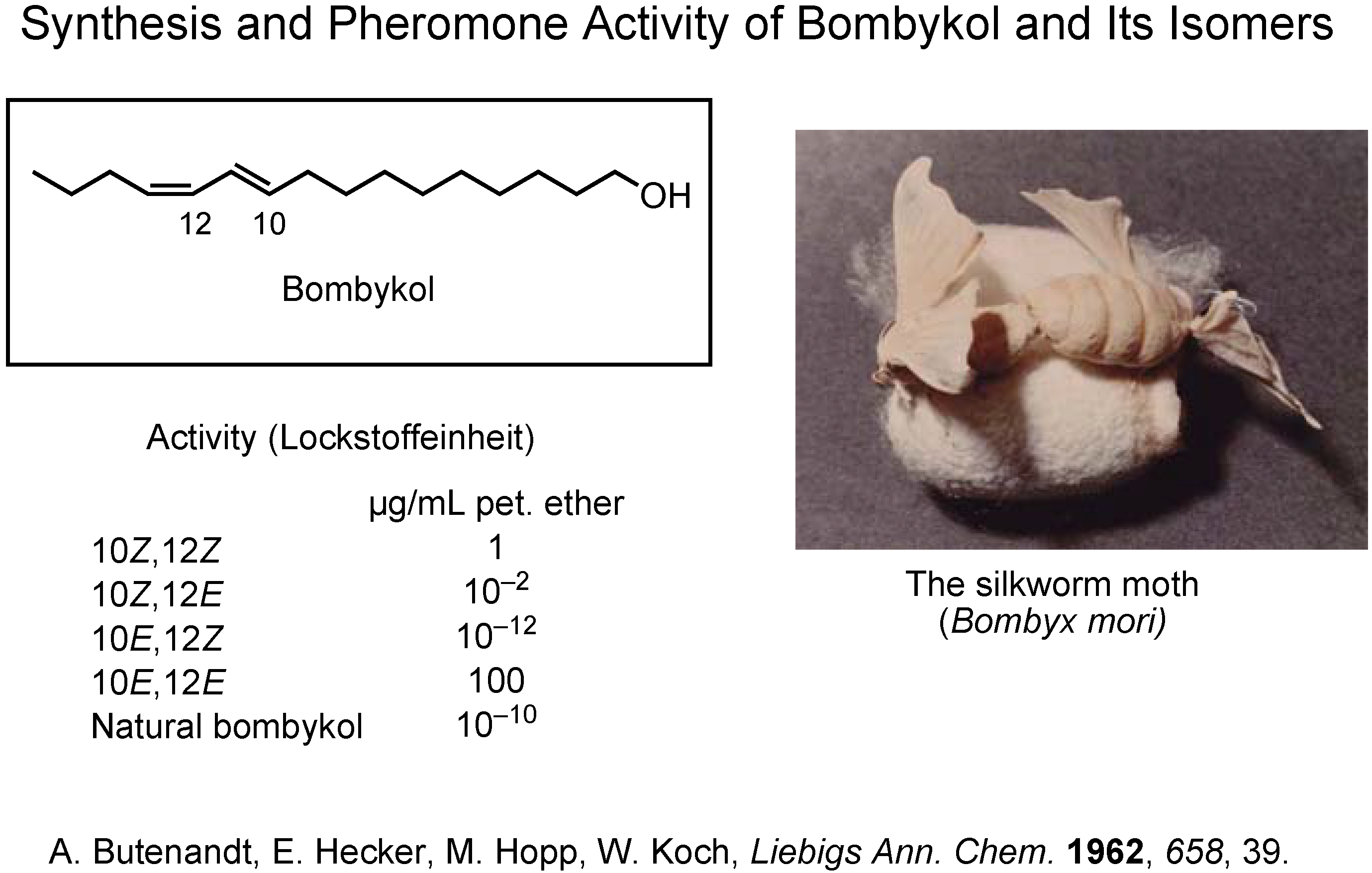
Since the beginning of pheromone chemistry, synthesis has been one of the very important methodologies. Professor Butenandt isolated and identified bombykol as the female-produced sex pheromone of the silkworm moth,
Bombyx mori (
Figure 2)
. The final stage of his investigation was to synthesize the four
cis/
trans isomers of bombykol. Because only the (10
E,12
Z)-isomer was as active as the pheromone itself, the structure of bombykol was determined as (10
E,12
Z)-10,12-hexadecadien-1-ol.
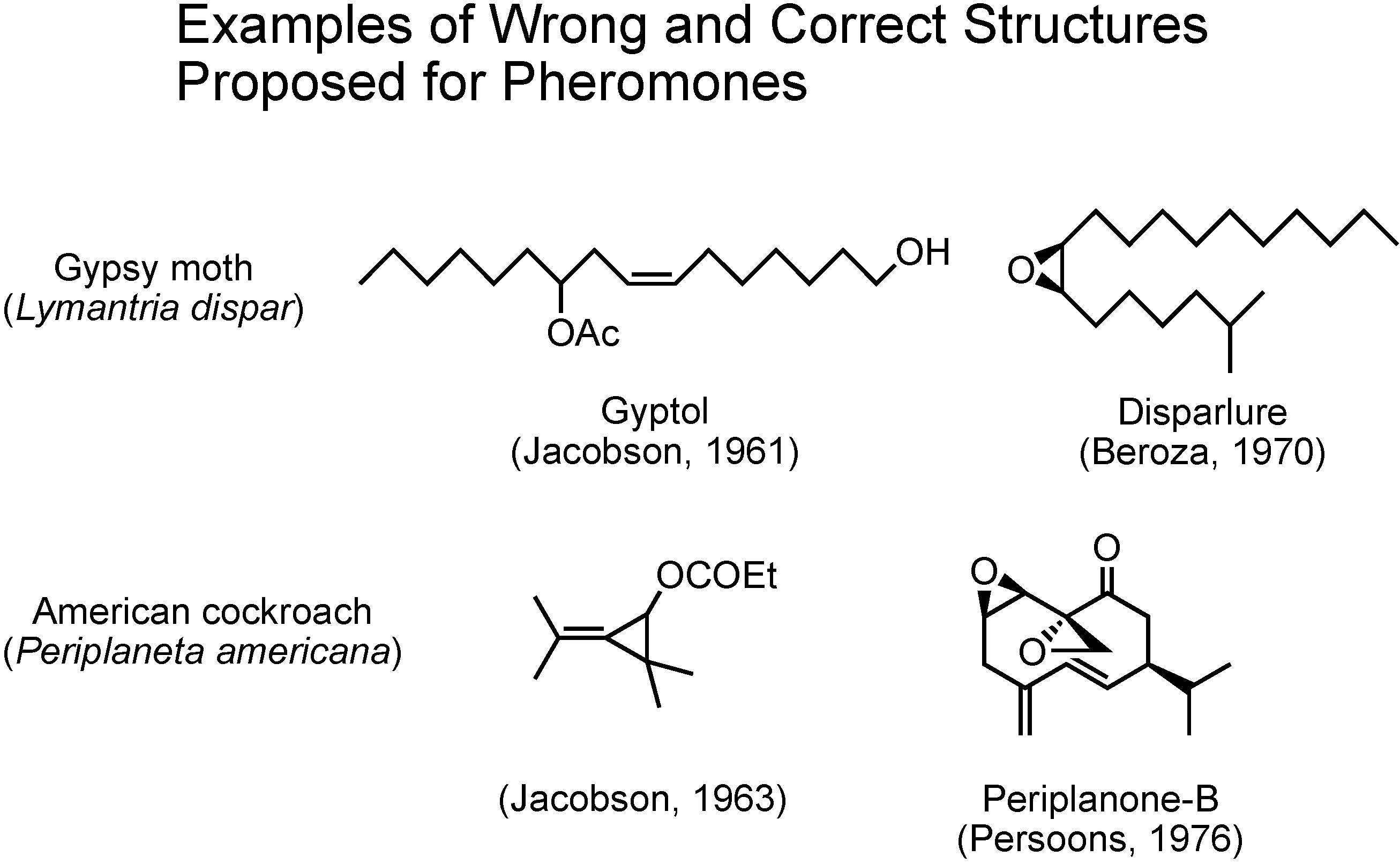
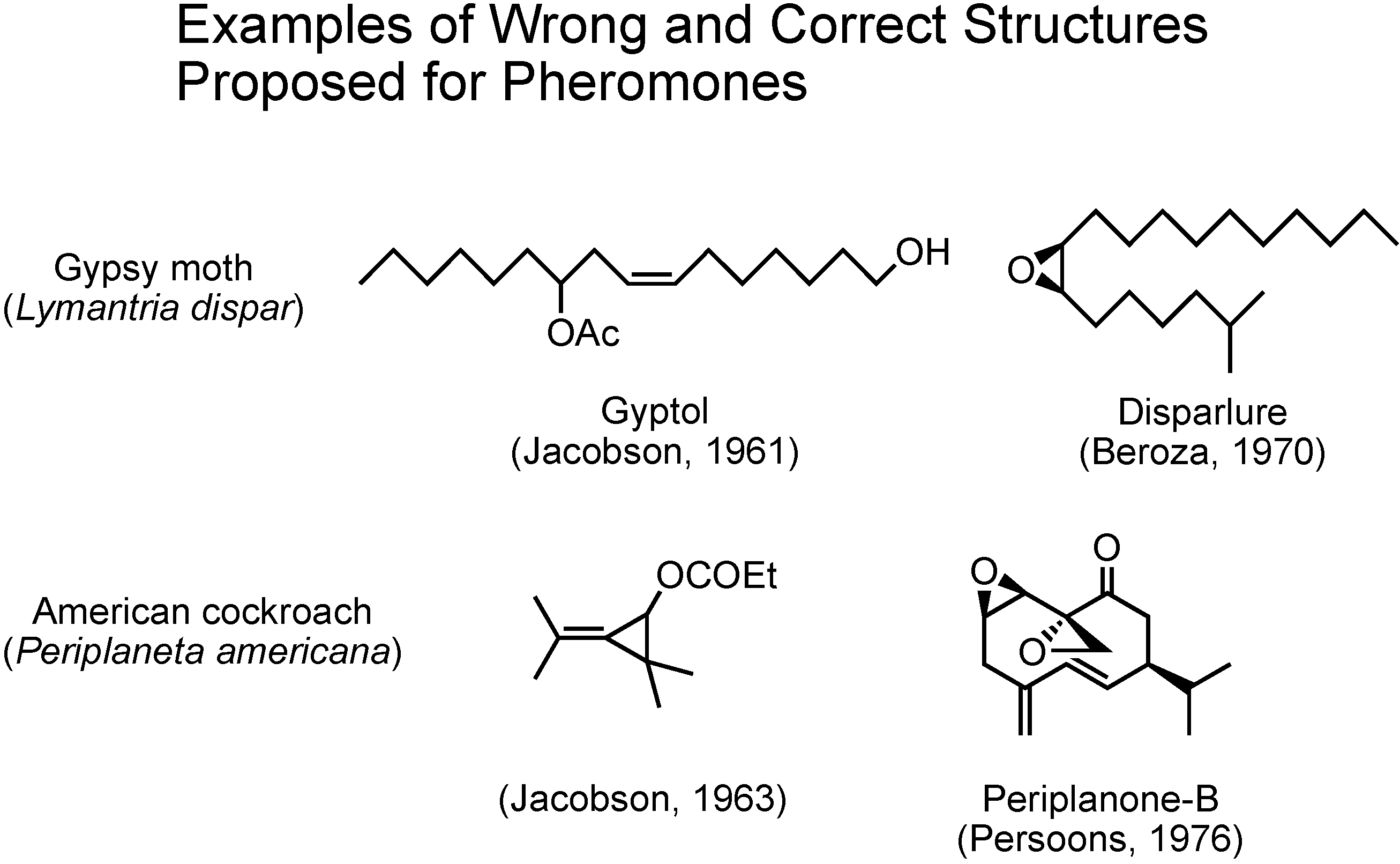
In the past, at the beginning of pheromone research, there were some cases in which incorrect structures were proposed for pheromones (
Figure 3). In 1961 Jacobson claimed gyptol to be the pheromone of the gypsy moth, and in 1963 he claimed this unusual cyclopropane compound to be the pheromone of the American cockroach. These two compounds were soon synthesized by others and the synthetic compounds did not work as pheromones at all. Further studies on these two insects later enabled Beroza and Persoons to identify disparlure and periplanone B as the true pheromones of gypsy moth and American cockroach, respectively. Again, synthesis played the major role.
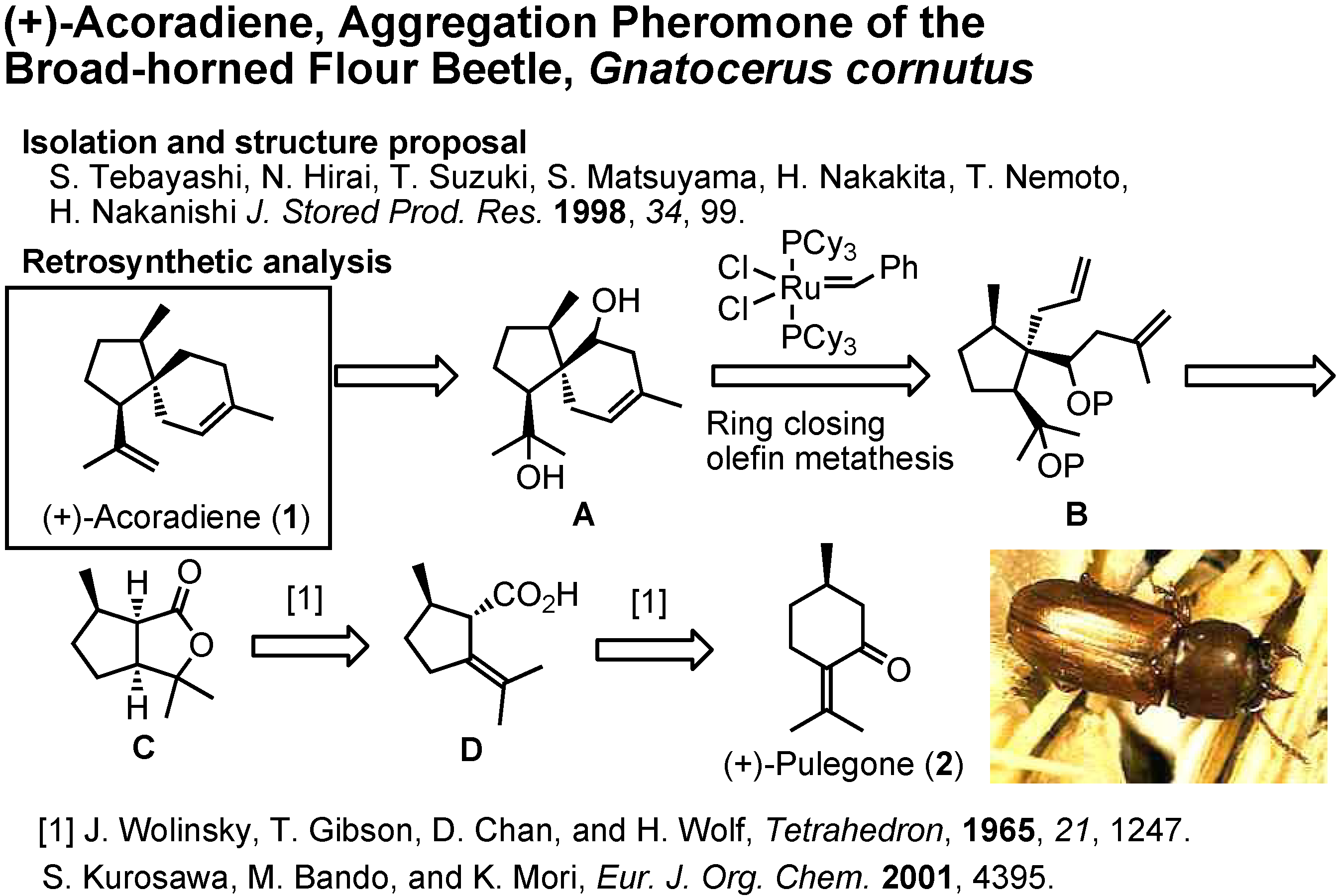
Now I shall tell you how we have determined the correct stereostructure of the aggregation pheromone of the broad-horned flour beetle (
Figure 4). Isolation and structure proposal of this pheromone was reported in 1998 by Tebayashi
et al. at the University of Tsukuba in Japan.
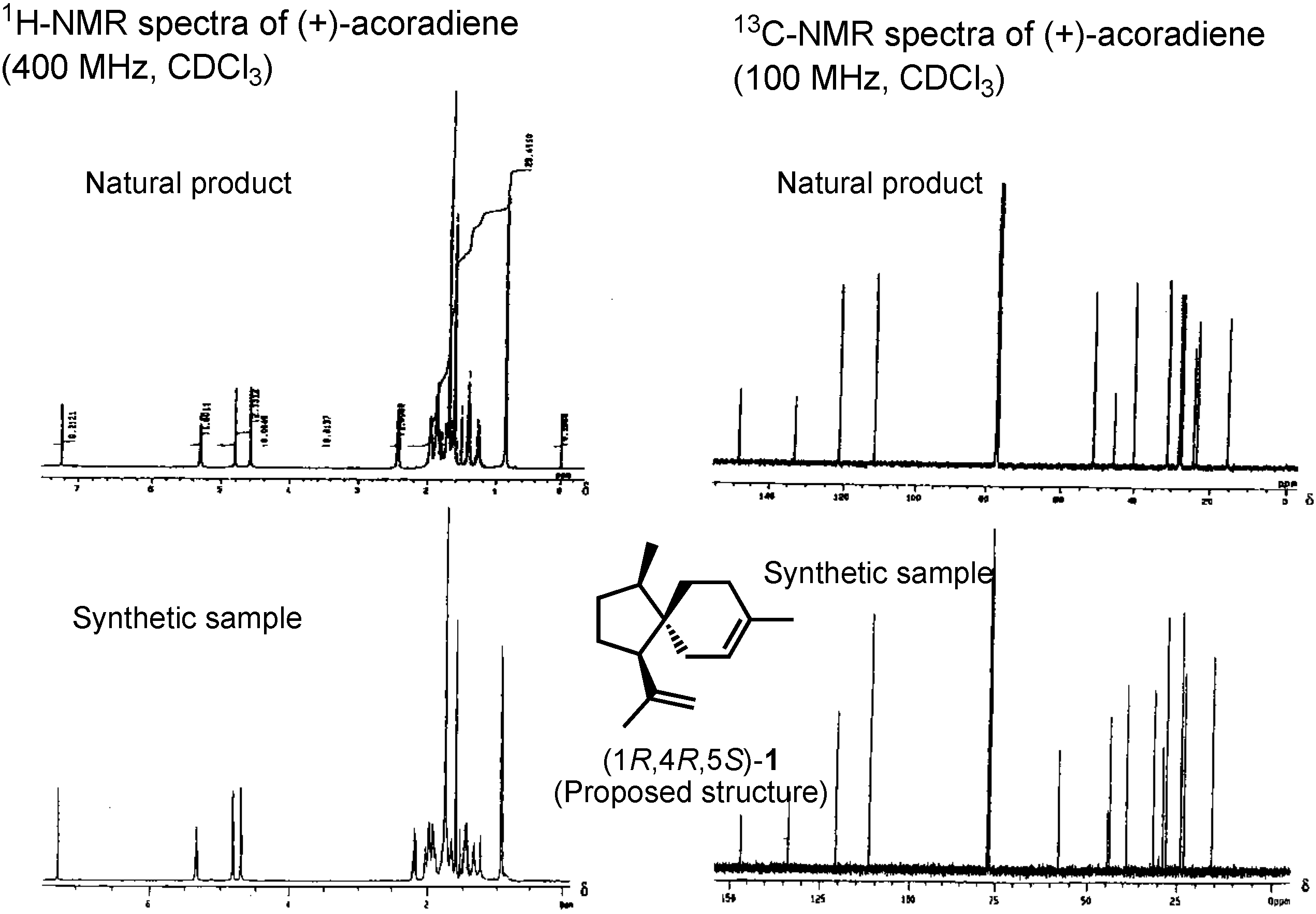
The proposed structure, (+)-acoradiene, was unique as the only pheromone with a spirosesquiterpene structure. We therefore synthesized it in 2001. Ring-closing olefin metathesis (Β→Α) was the key-step. Because diol A was nicely crystalline, its structure could be determined beyond doubt by X-ray analysis, conducted by Dr. Bando. When we synthesized (+)-acoradiene, Dr. Kurosawa carefully compared the NMR spectrum of the synthetic product with that of the pheromone (
Figure 5). Here you can see that the NMR spectra of these two samples are not identical. The proposed structure, therefore, must be incorrect. We hypothesized that the natural pheromone was one of the possible isomers of
1, but this hypothesis must be proven. We decided to synthesize a stereoisomeric mixture of all the possible isomers of
1.
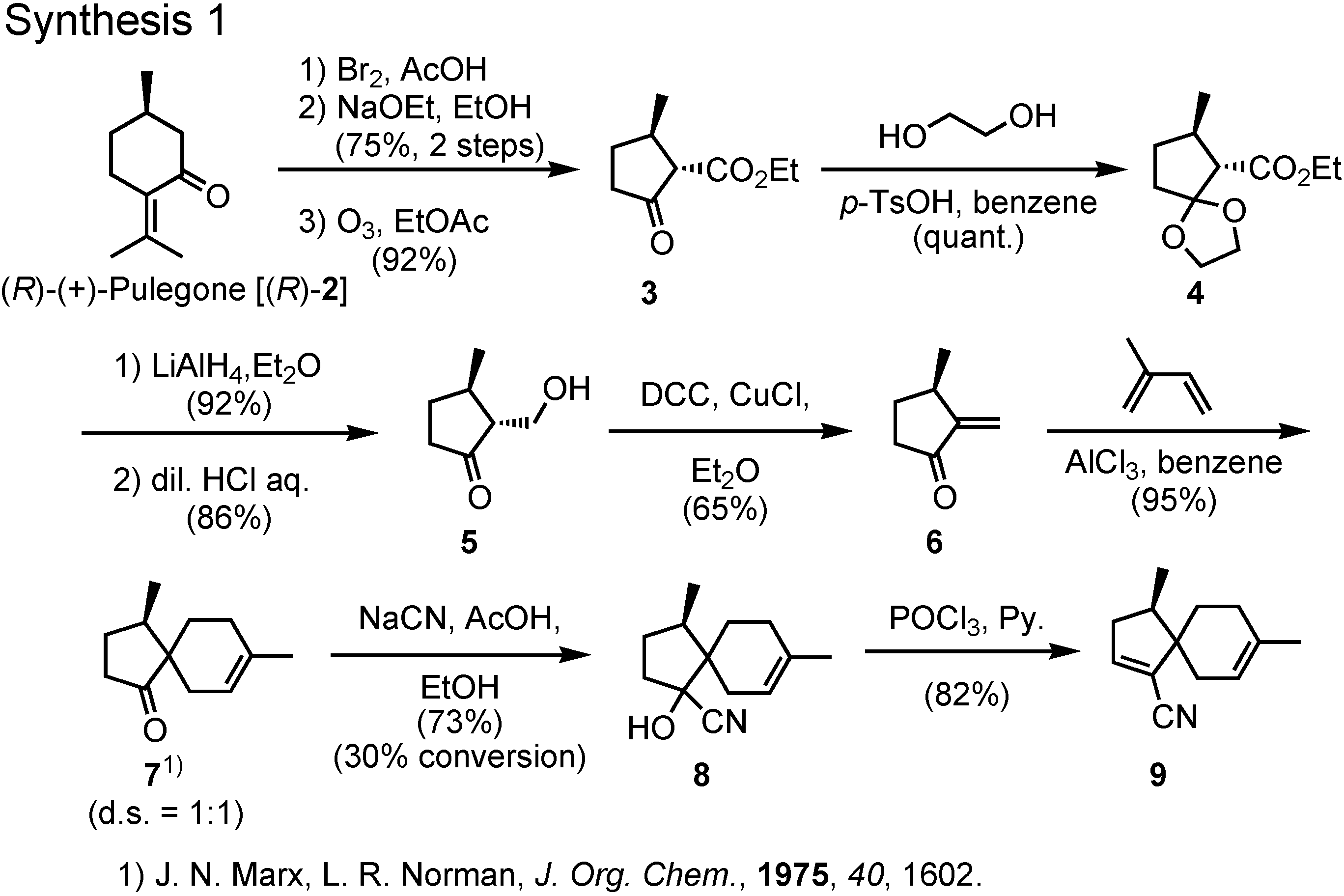
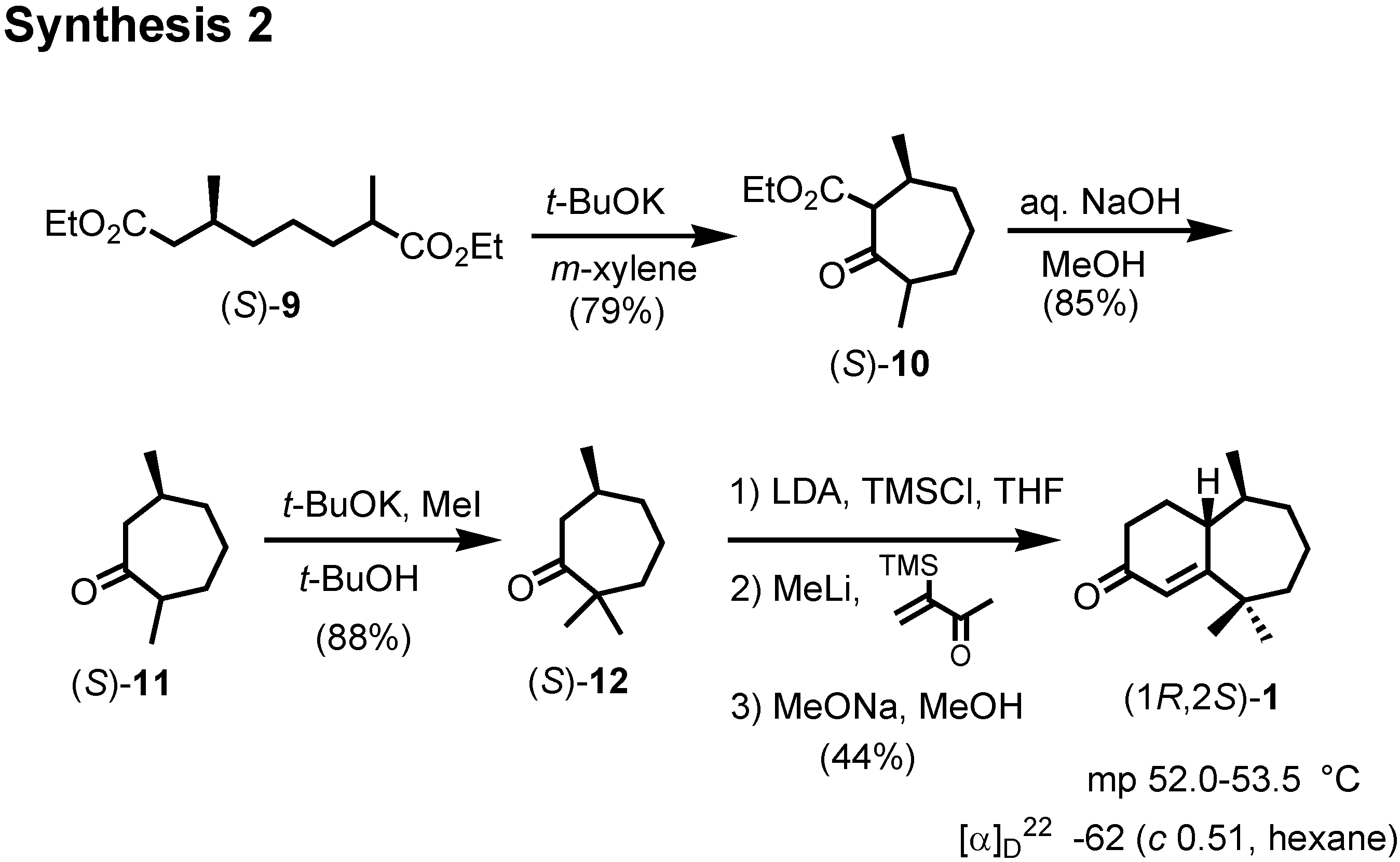
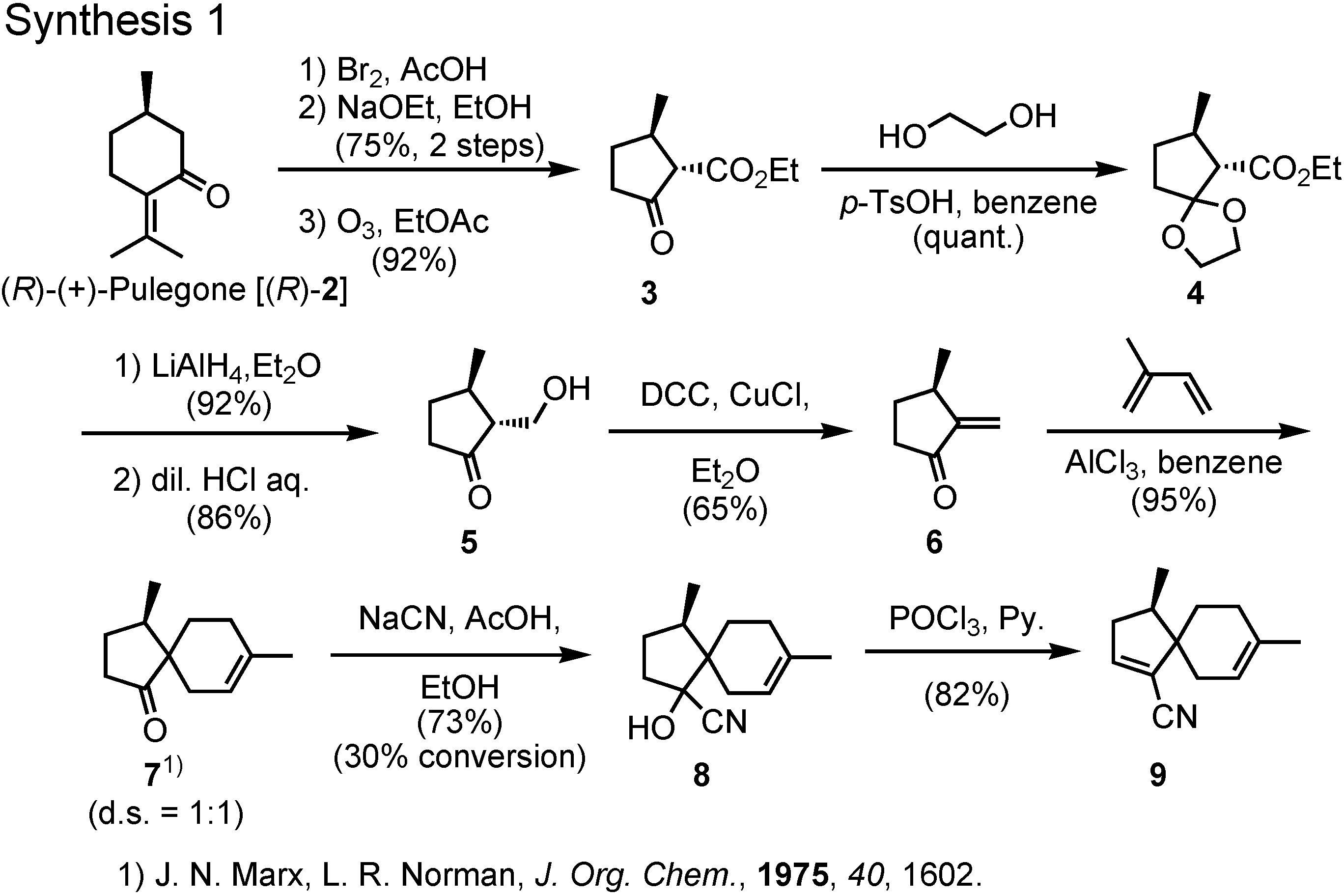
The synthesis of a stereoisomeric mixture of acoradiene started from (
R)-(+)-pulegone to give spiro ketone
11 (
Figure 6). Hydrocyanation of
7 furnished cyanohydrin
8, which was dehydrated to give
9.
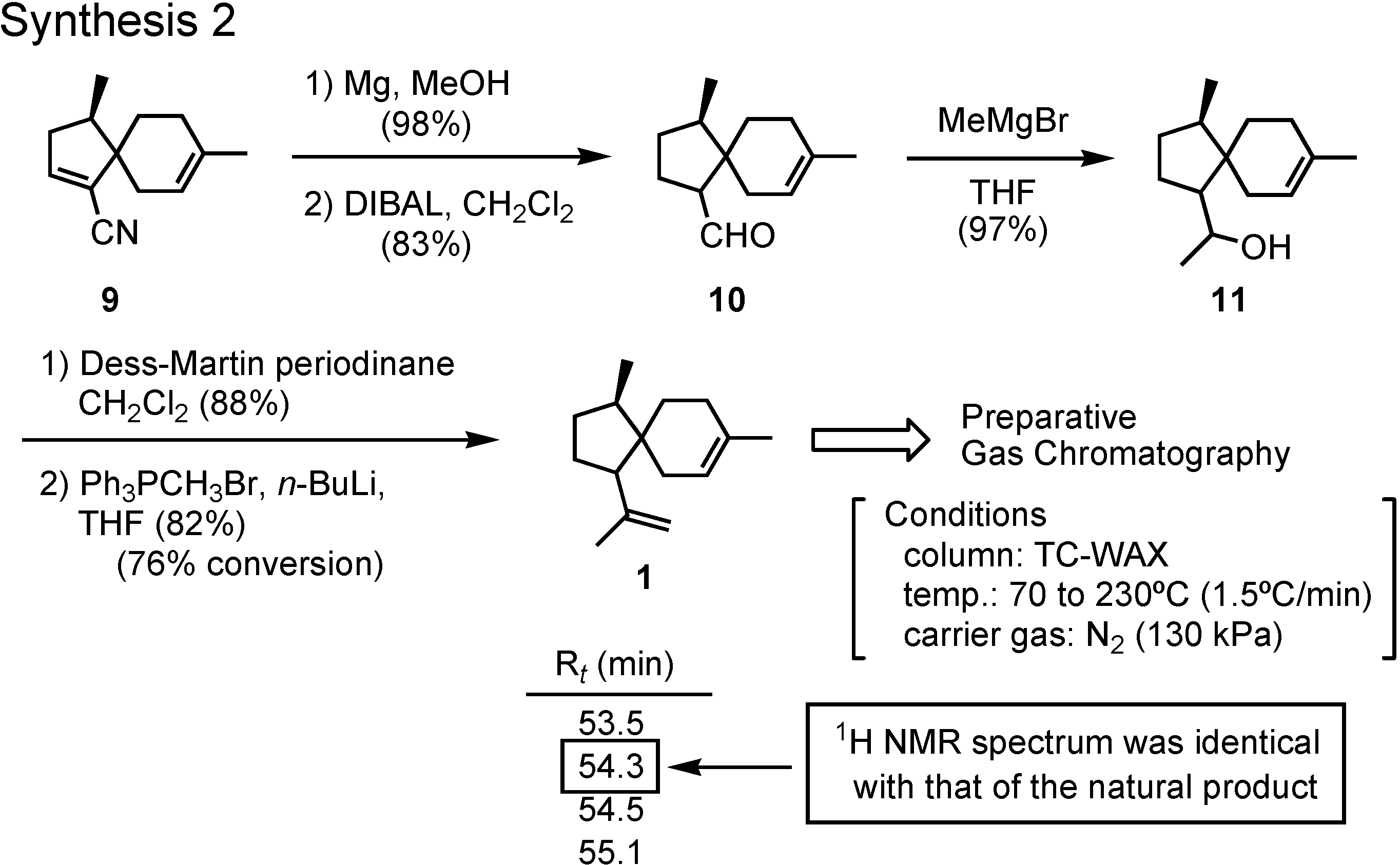
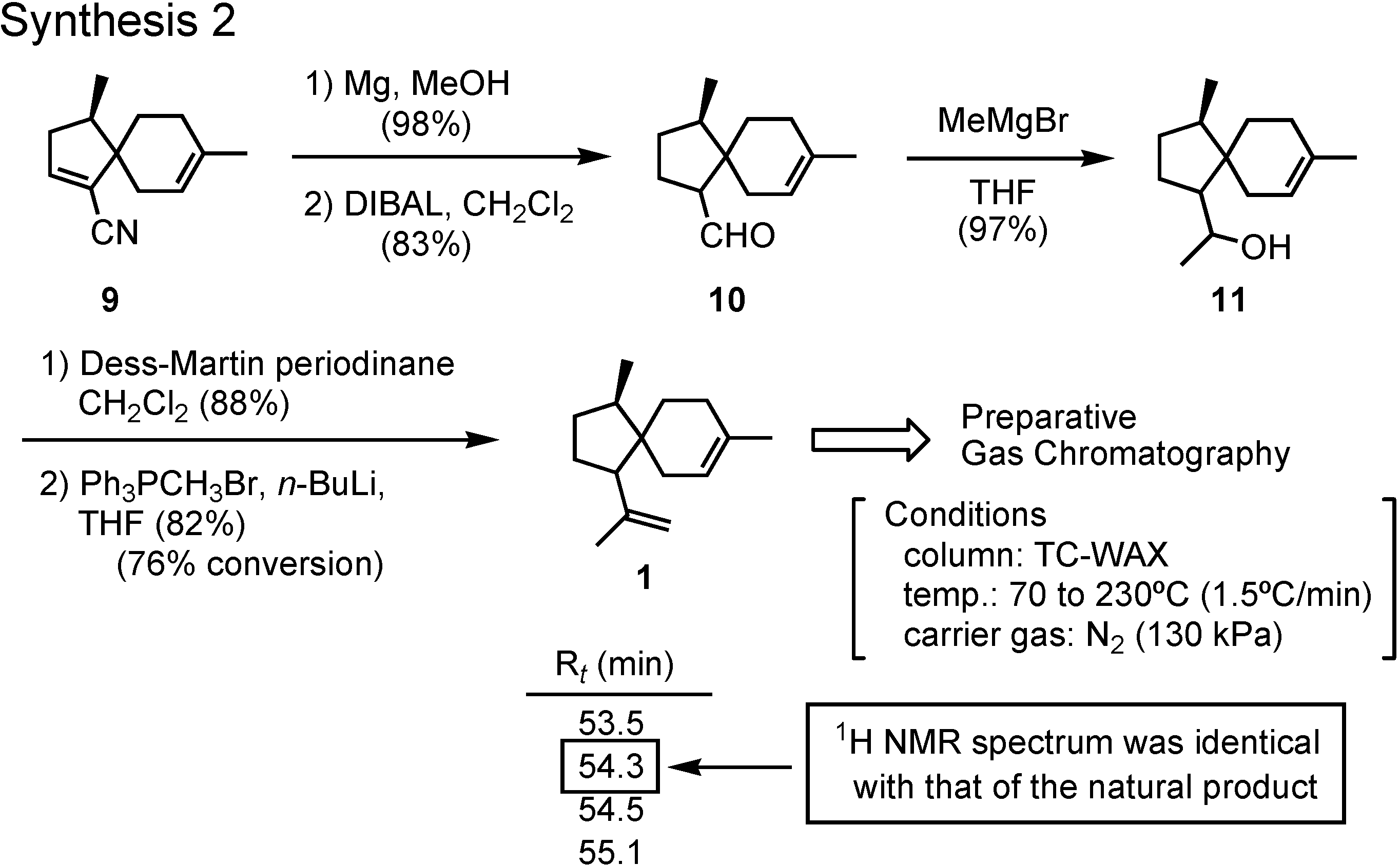
Reduction of
9 with magnesium and methanol gave the saturated nitrile, which was further reduced with DIBAL to give aldehyde
10 (
Figure 7). The remaining two carbons were attached to
10 by Grignard and Wittig reactions, respectively, to give
1 as a stereoisomeric mixture. The mixture could be separated by preparative GC, and a fraction with a retention time of 54.3 min showed an NMR spectrum identical with that of the natural product. The pheromone must be a stereoisomer of
1. But how about the stereochemistry of that isomer?
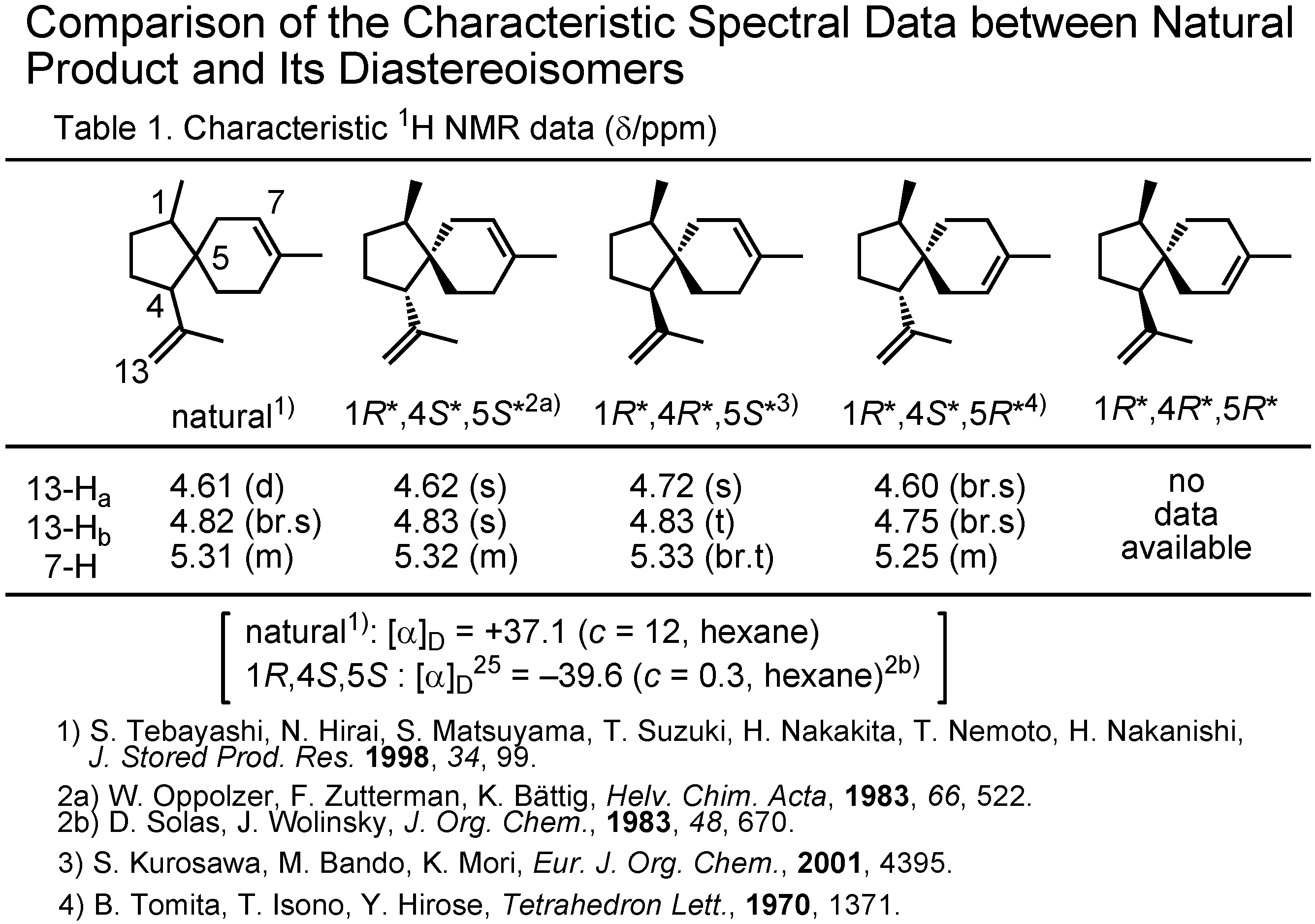
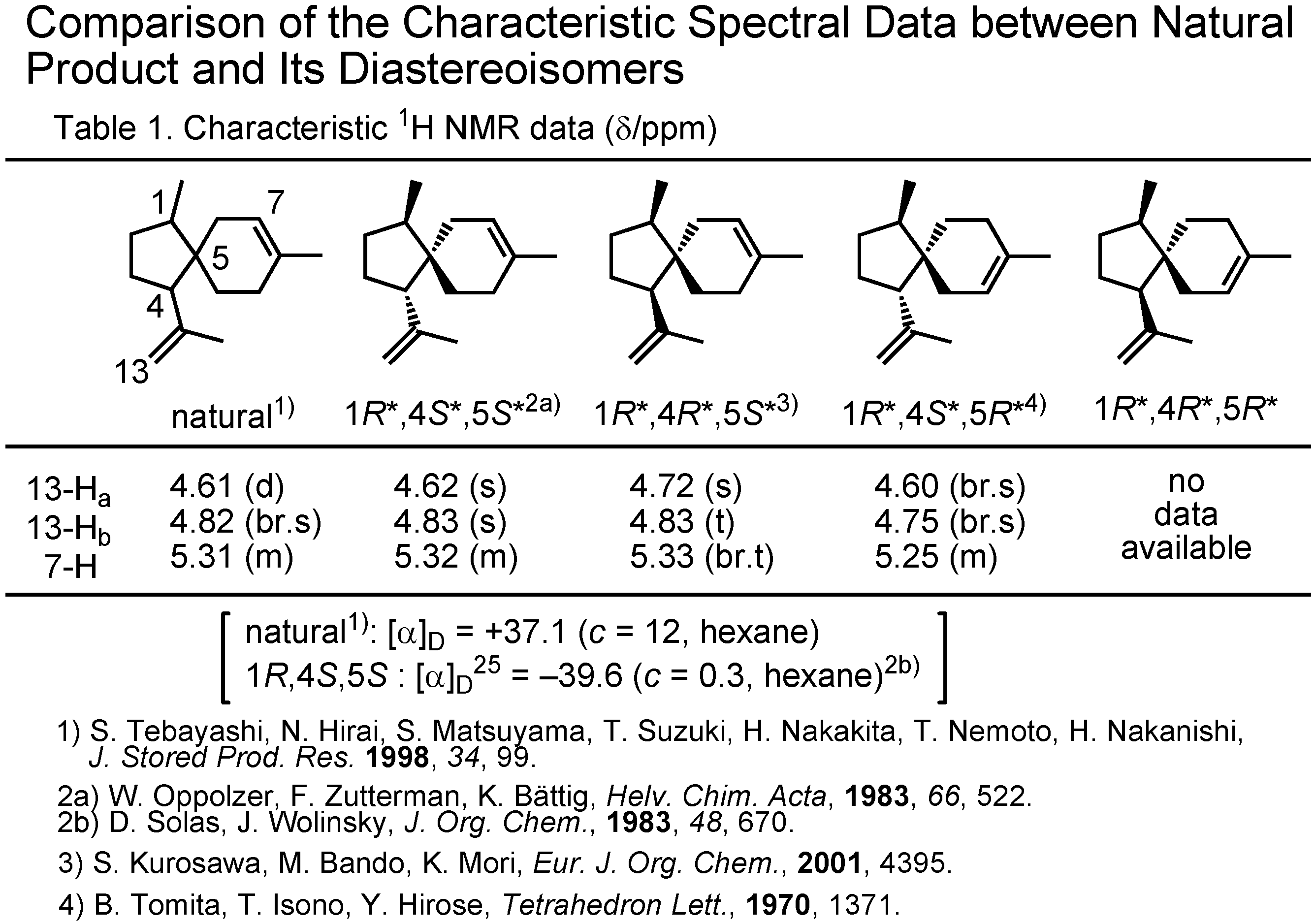
At this stage Dr. Tashiro surveyed the literature through SciFinder, and collected the NMR data of the stereoisomers of acoradiene (
Figure 8). This table shows the chemical shift values of the olefinic protons of the acoradiene stereoisomers. Although the (1
R,4
R,5
R)-isomer was unknown, the data of other three stereoisomers could be compared with those of the pheromone. The δ-values of the natural pheromone were in good accord with those of (1
R,4
S,5
S)-isomer. In 1983 Wolinsky had reported the specific rotation of that isomer to be –39.6, while that of the natural pheromone was +37.1. The natural pheromone therefore must be (1
S,4
R,5
R)-isomer. This conclusion has to be proven by a synthesis.
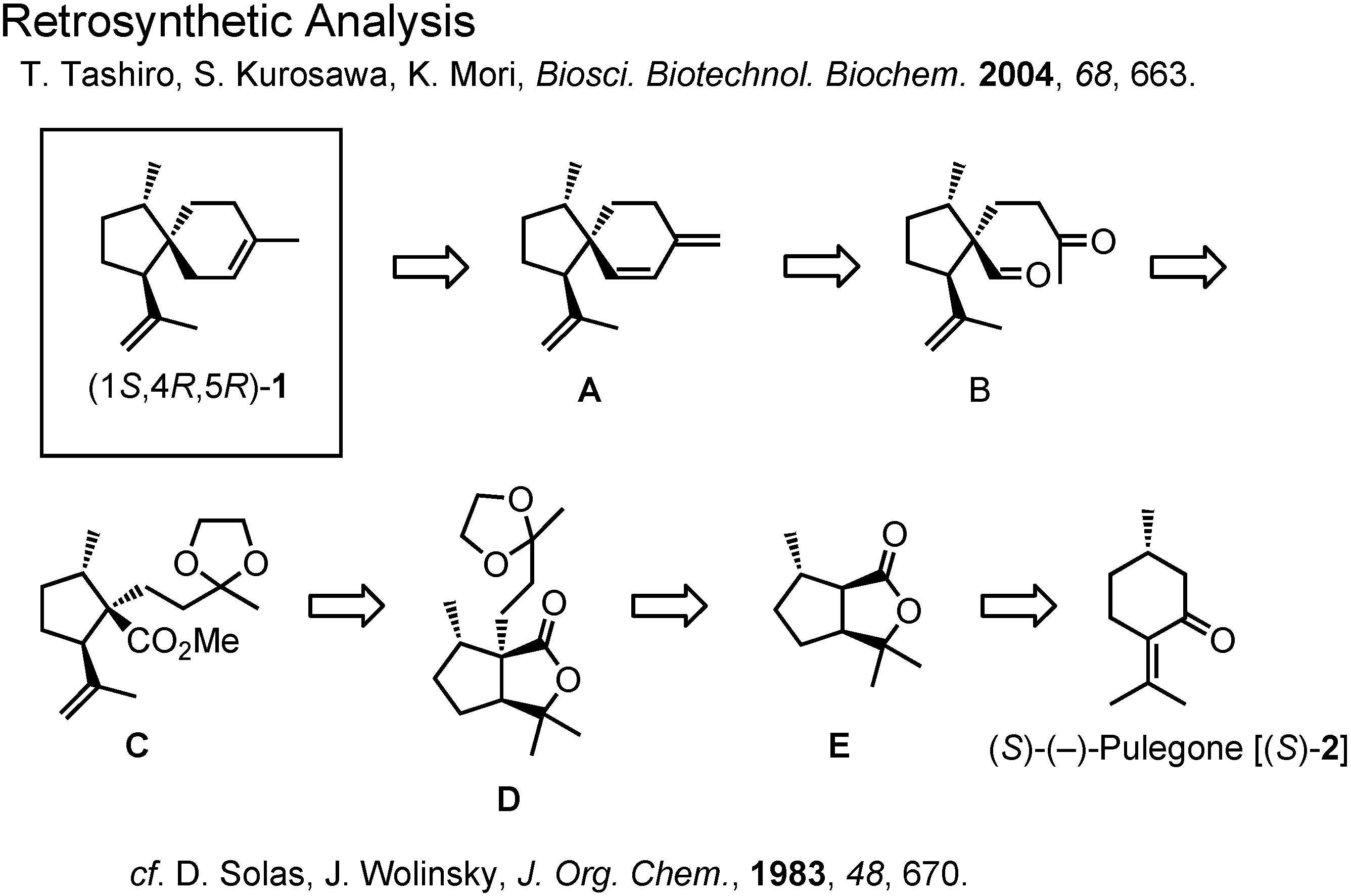
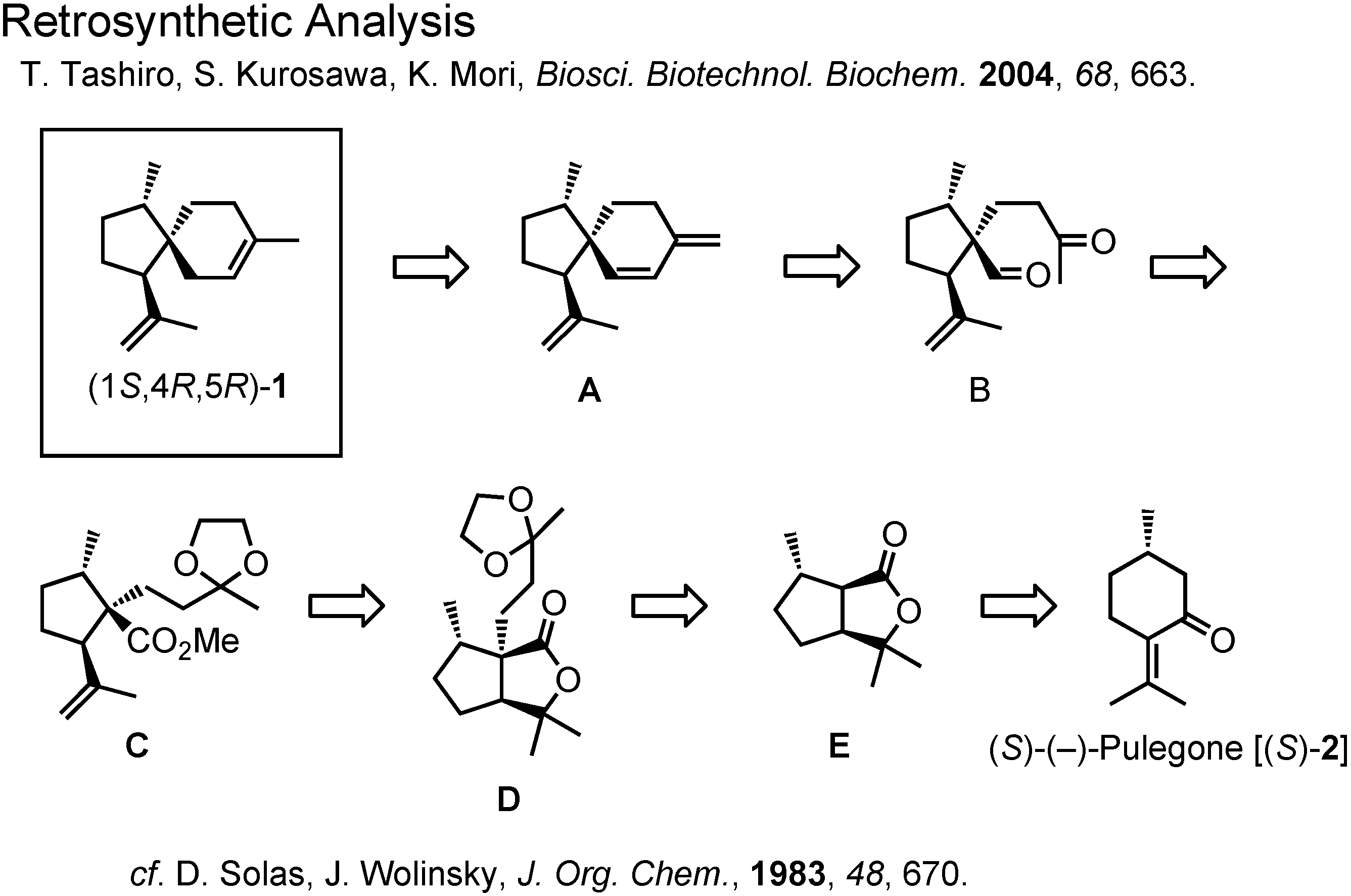
Figure 9 shows the retrosynthetic analysis of (1
S,4
R,5
R)-acoradiene Our analysis is a slight modification of Wolinsky’s synthesis of the (1
R,4
S,5
S)-isomer.
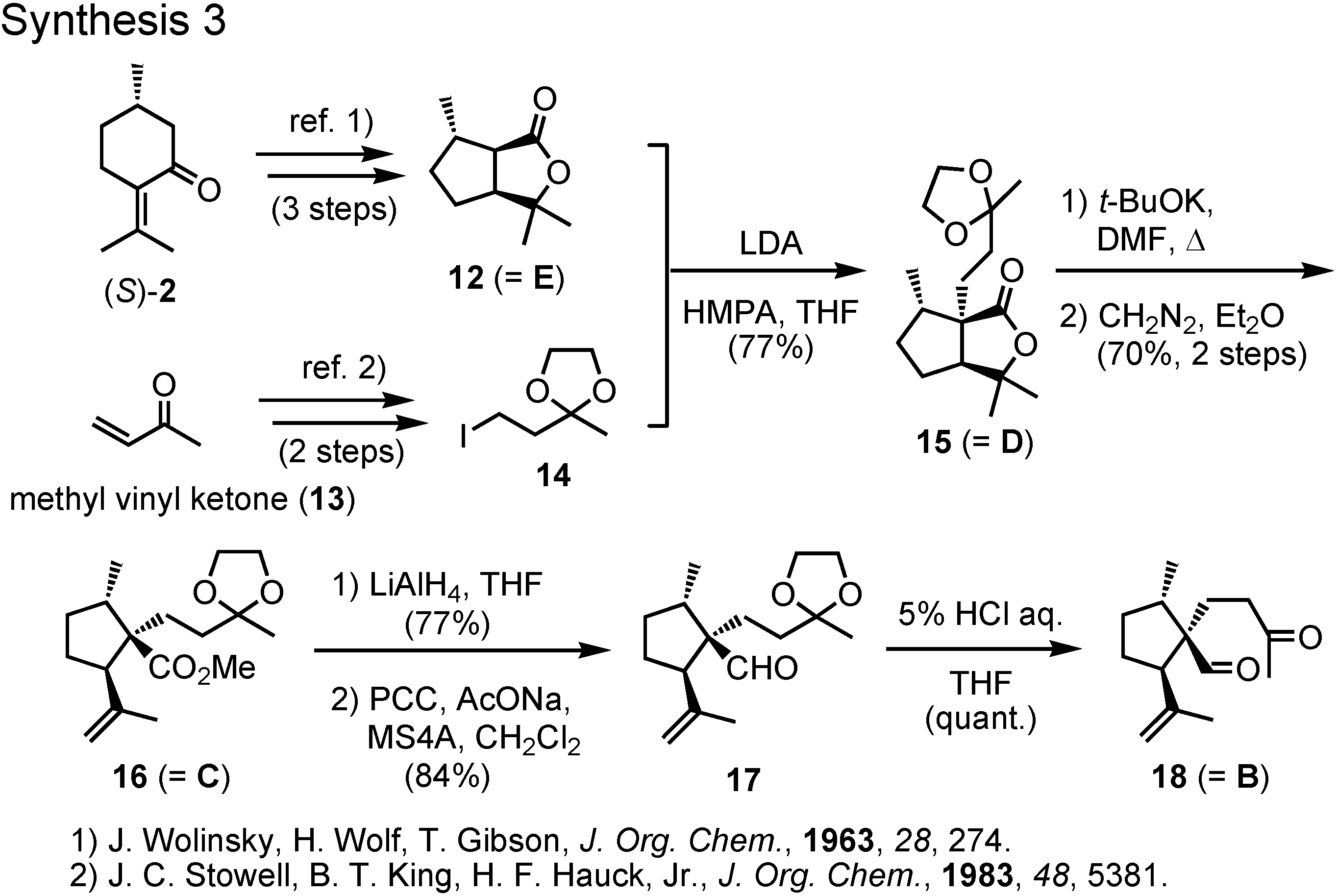
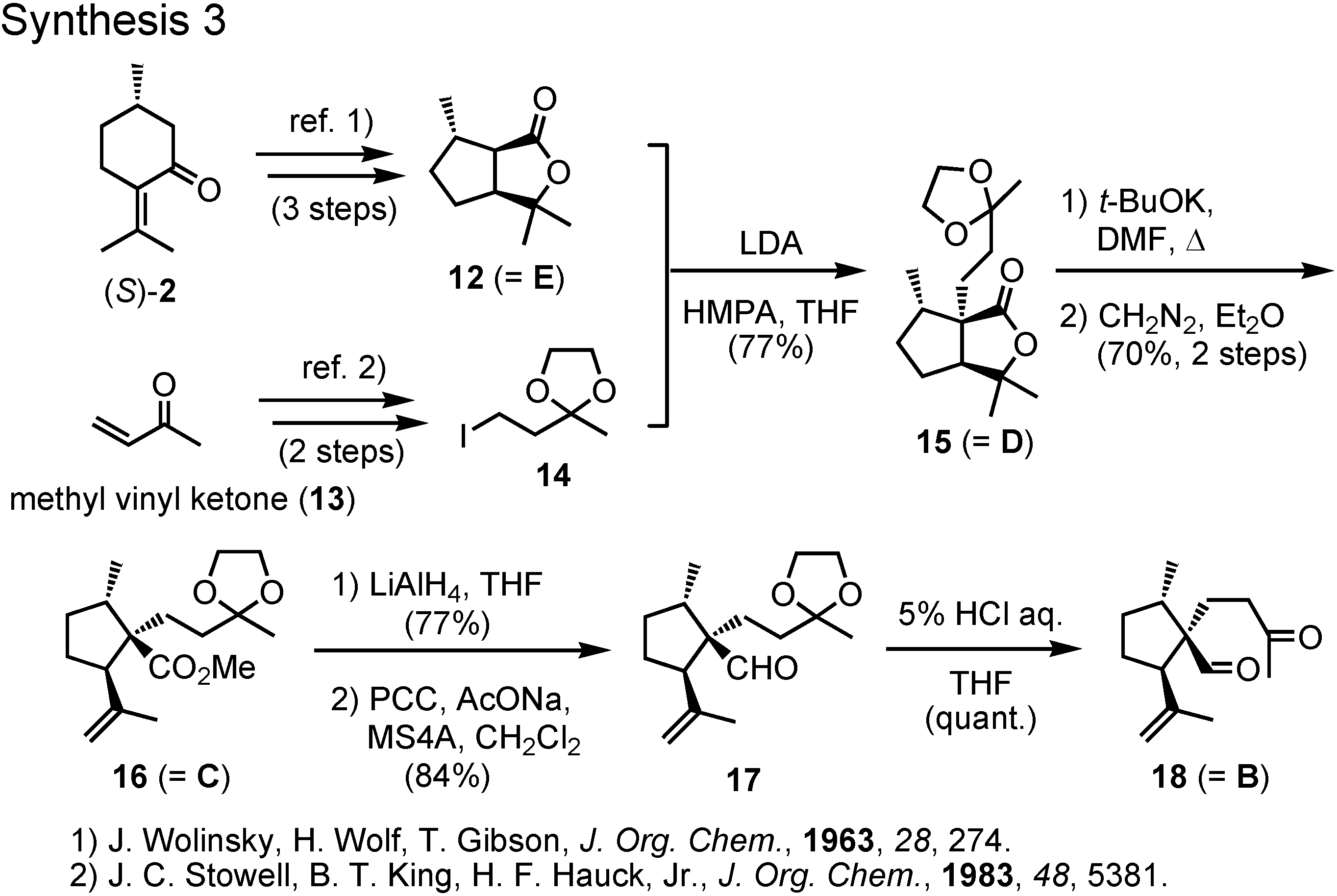
(
S)-Pulegone, the starting material, was converted to lactone
12 according to Wolinsky (
Figure 10). The alkylating agent
14 was prepared from methyl vinyl ketone. Alkylation of the lactone
12 with iodide
14 afforded
15, which was treated with potassium
t-butoxide to cleave the lactone ring. The corresponding methyl ester
16 was reduced with lithium aluminum hydride and the generated alcohol was oxidized with PCC to furnish aldehyde
17. Treatment of
17 with dilute hydrochloric acid gave
18, the precursor for aldol cyclization.
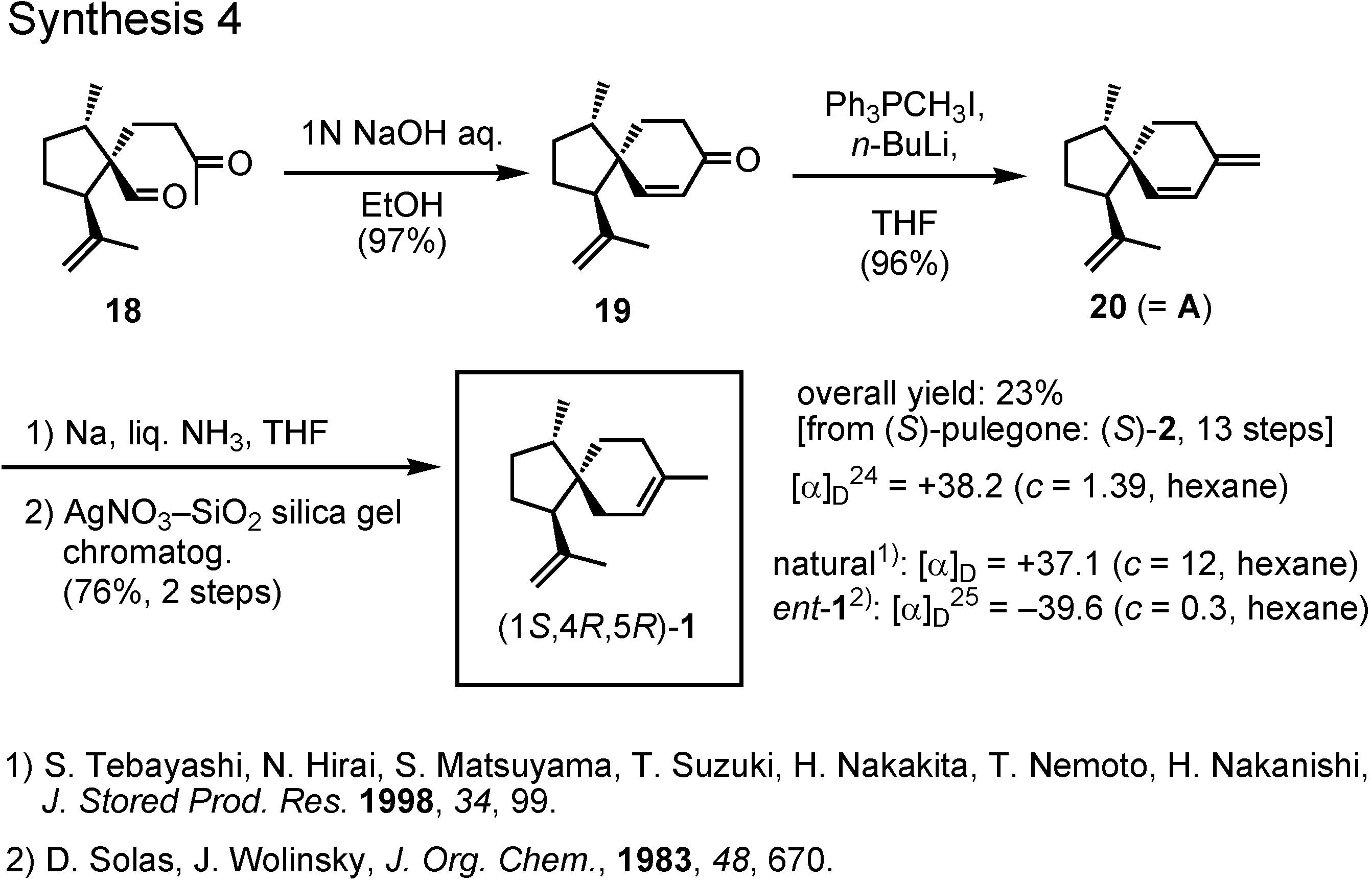
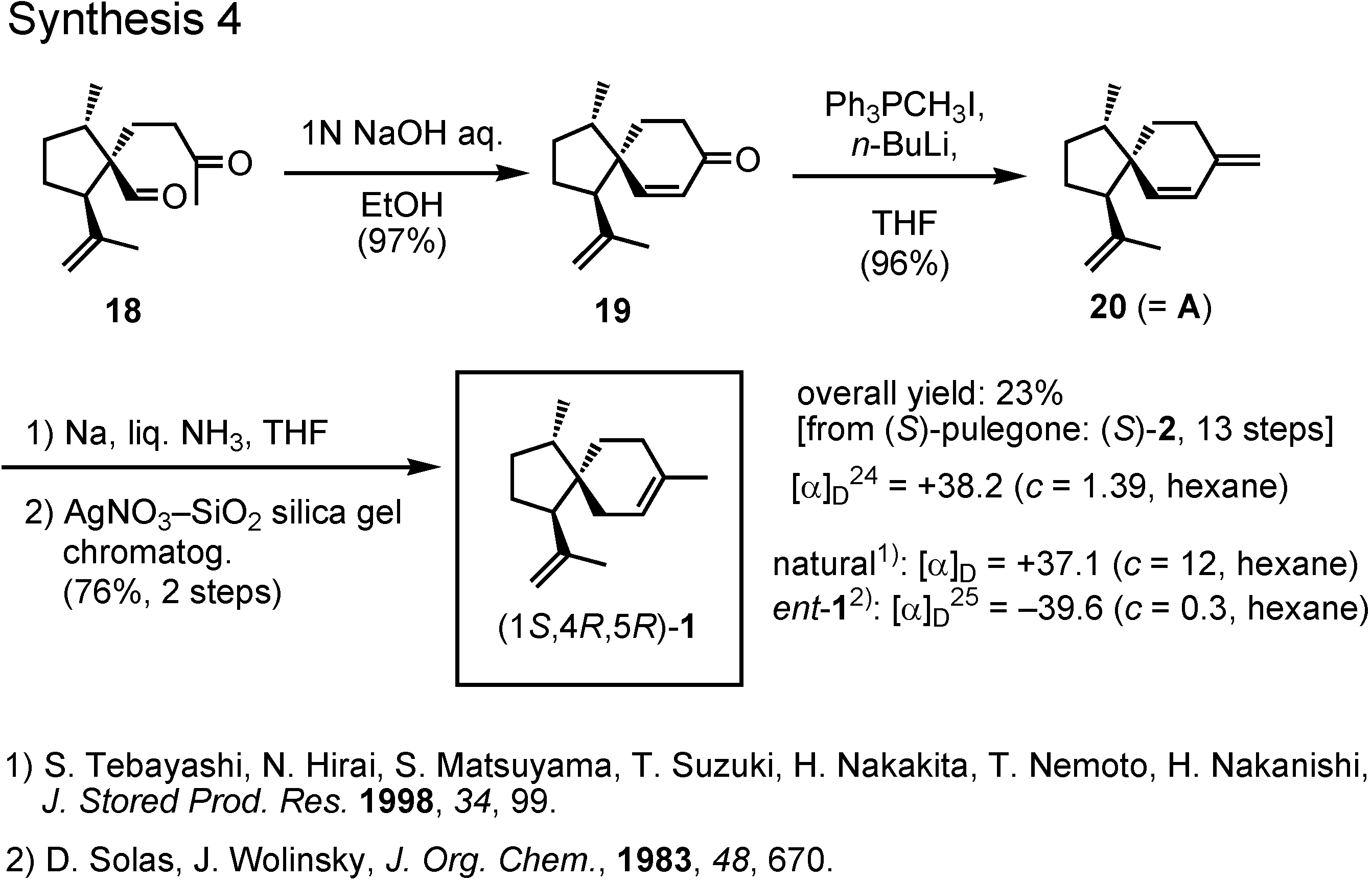
Treatment of the keto aldehyde
18 with dilute sodium hydroxide provided spiroketone
19, to which a methylene group was added by Wittig reaction to give
20 (
Figure 11). Finally, 1,4-reduction of diene
20 with sodium in liquid ammonia yielded (1
S,4
R,5
R)-acoradiene
1 in 23% overall yield. Its specific rotation value was in good agreement with that of the pheromone. Then, how about the NMR spectrum ?
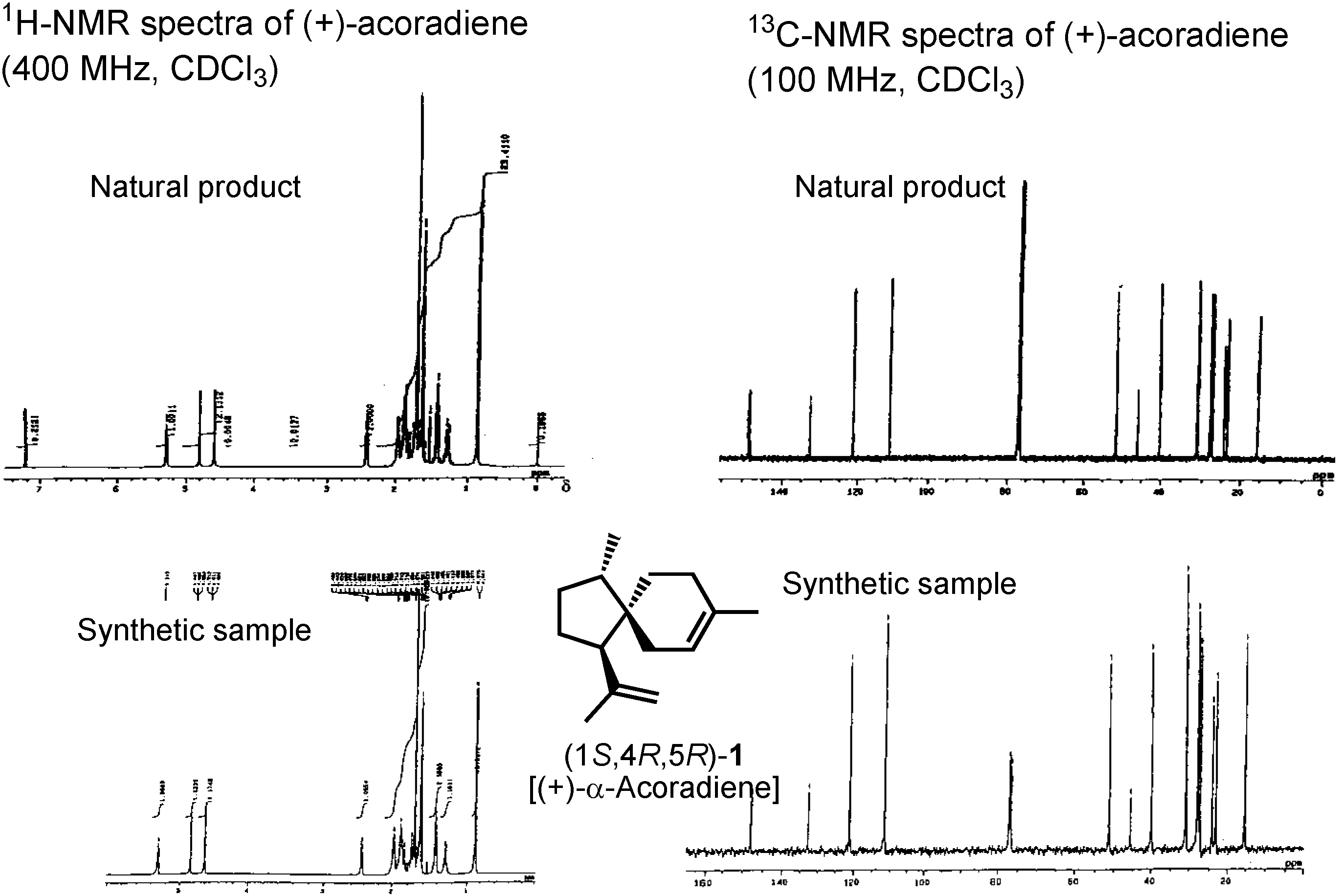
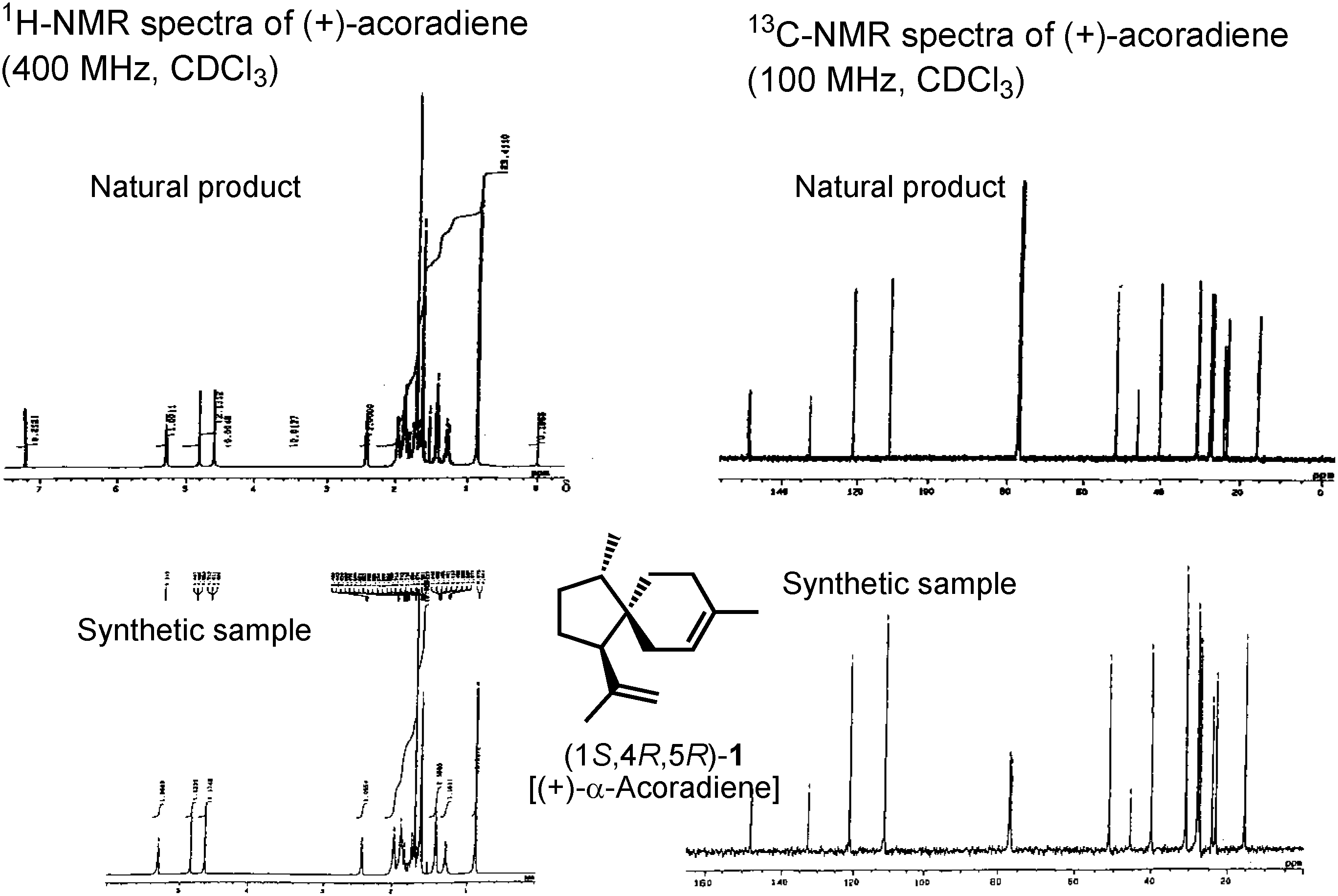
As you can see in
Figure 12, both proton and carbon NMR spectra of the synthetic product were identical with those of the natural pheromone In the case of this sesquiterpene pheromone, our synthesis could finally establish its true structure.
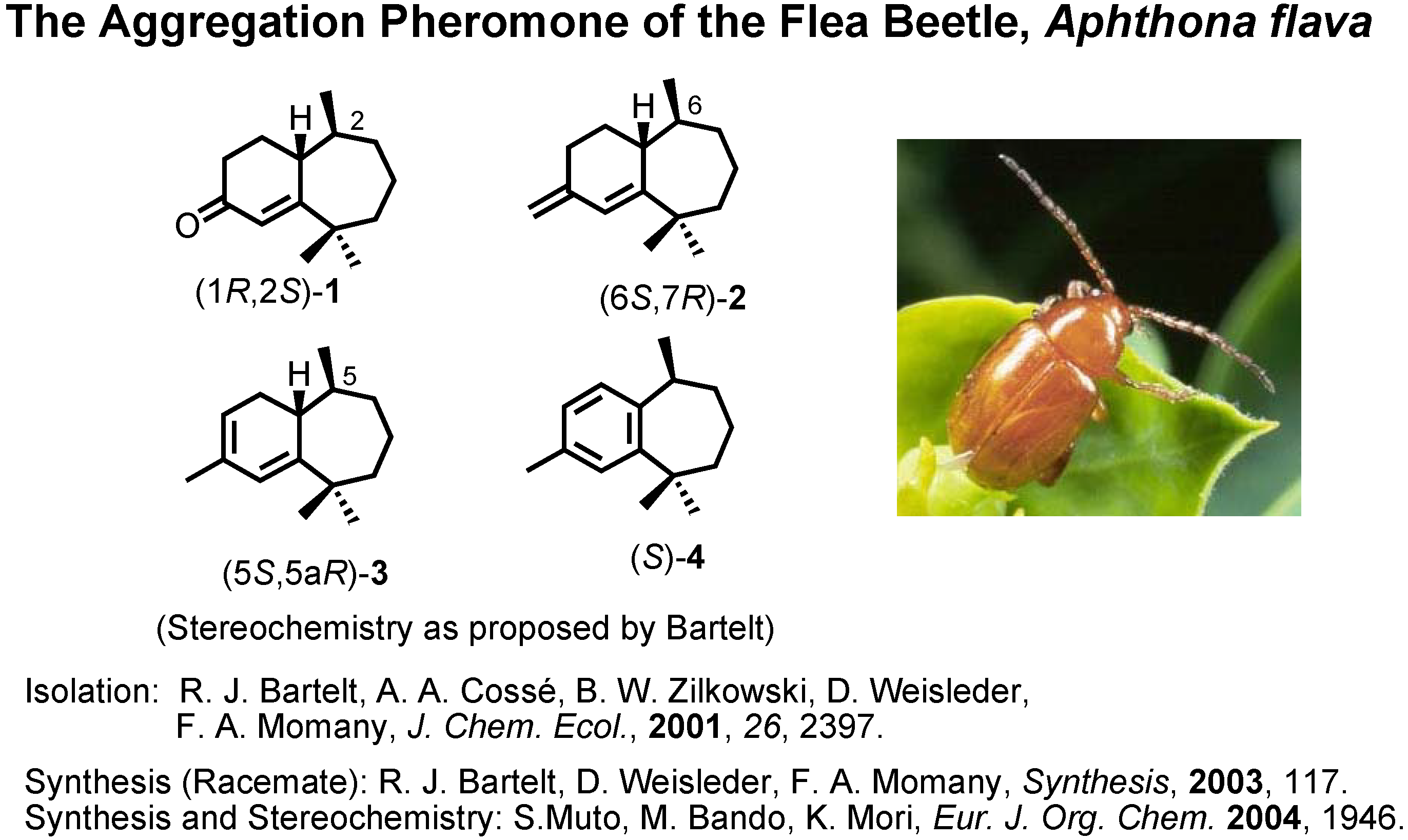
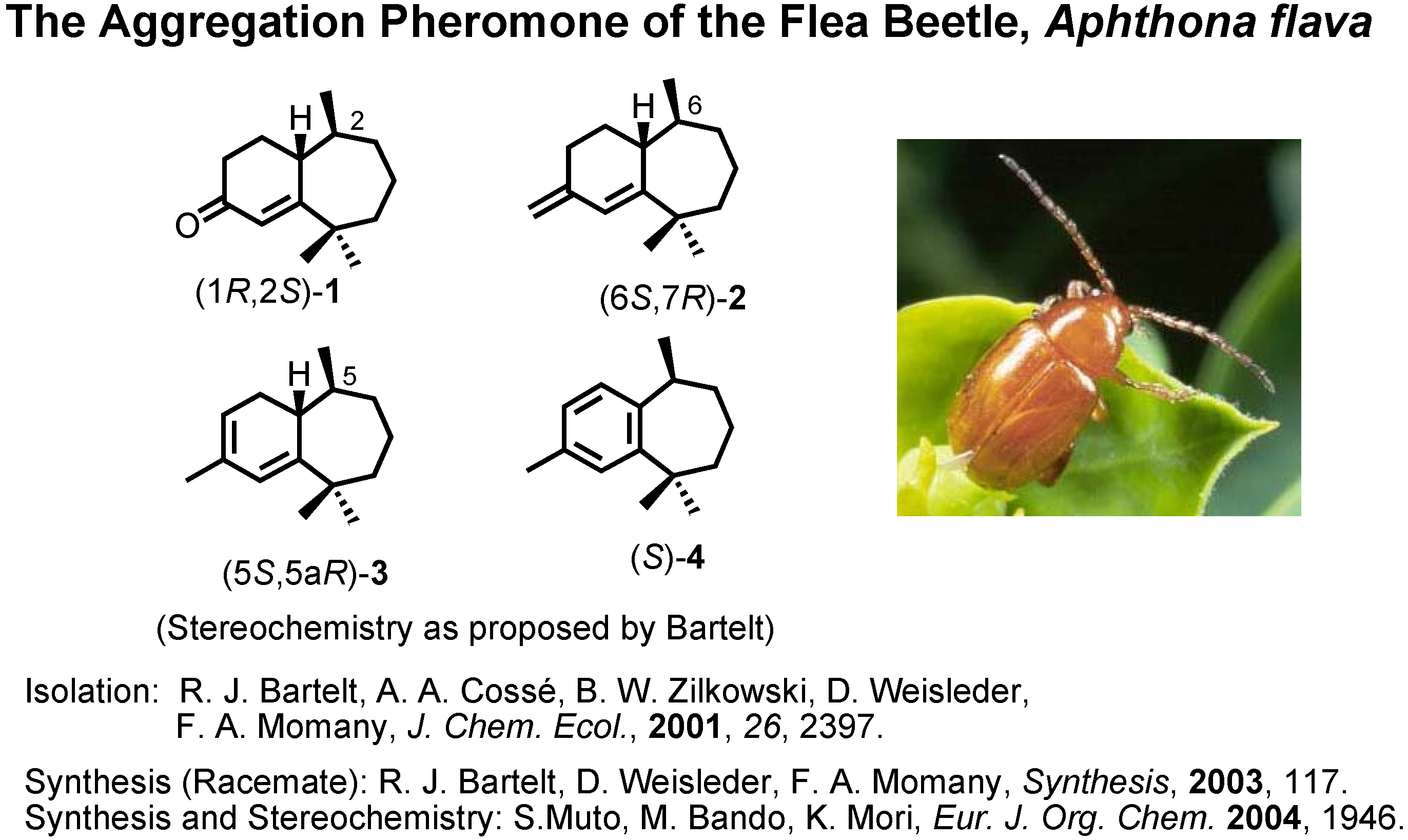
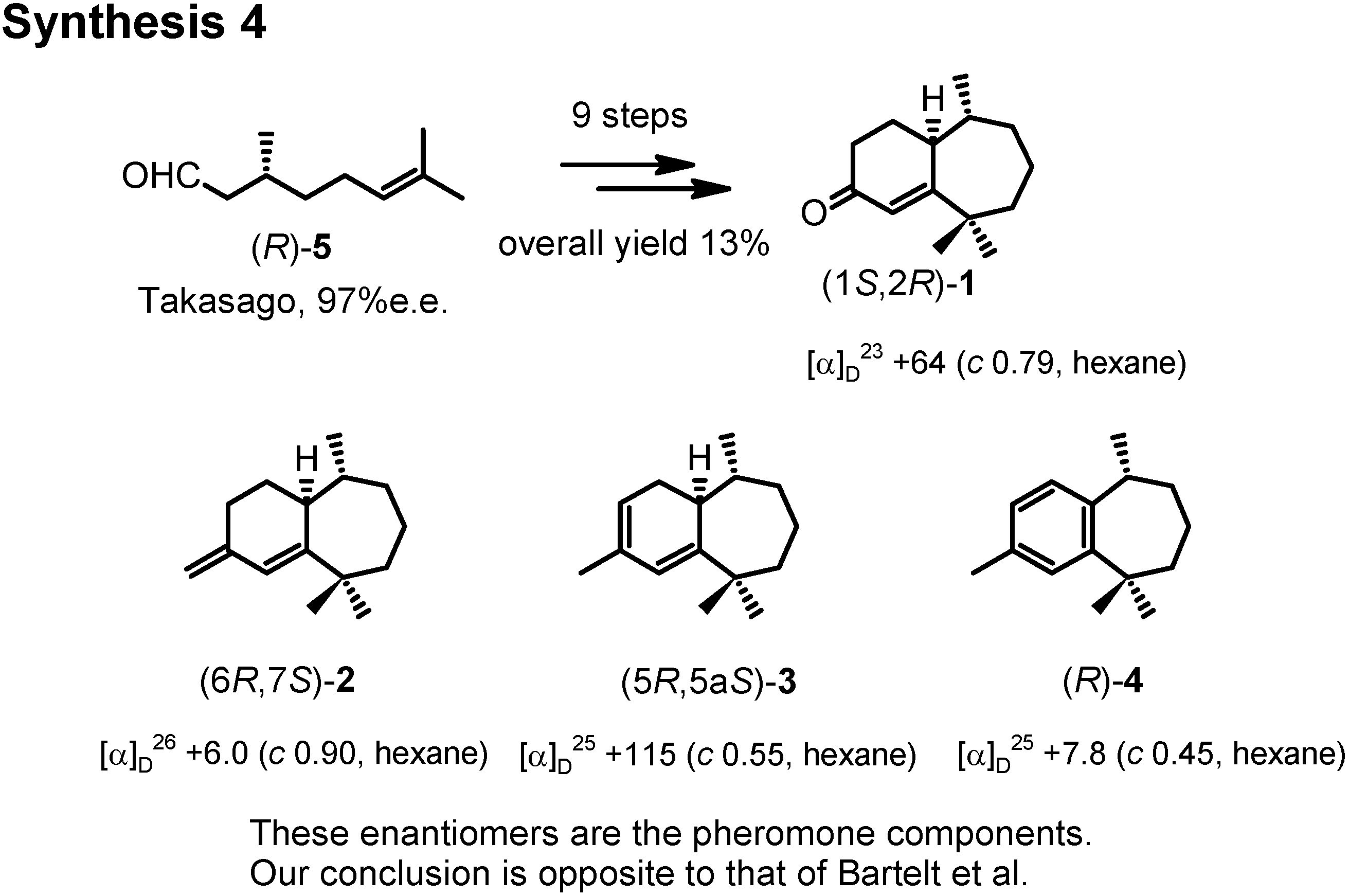
The next topic is the aggregation pheromone of the flea beetle,
Aphthona flava (
Figure 13). Bartelt
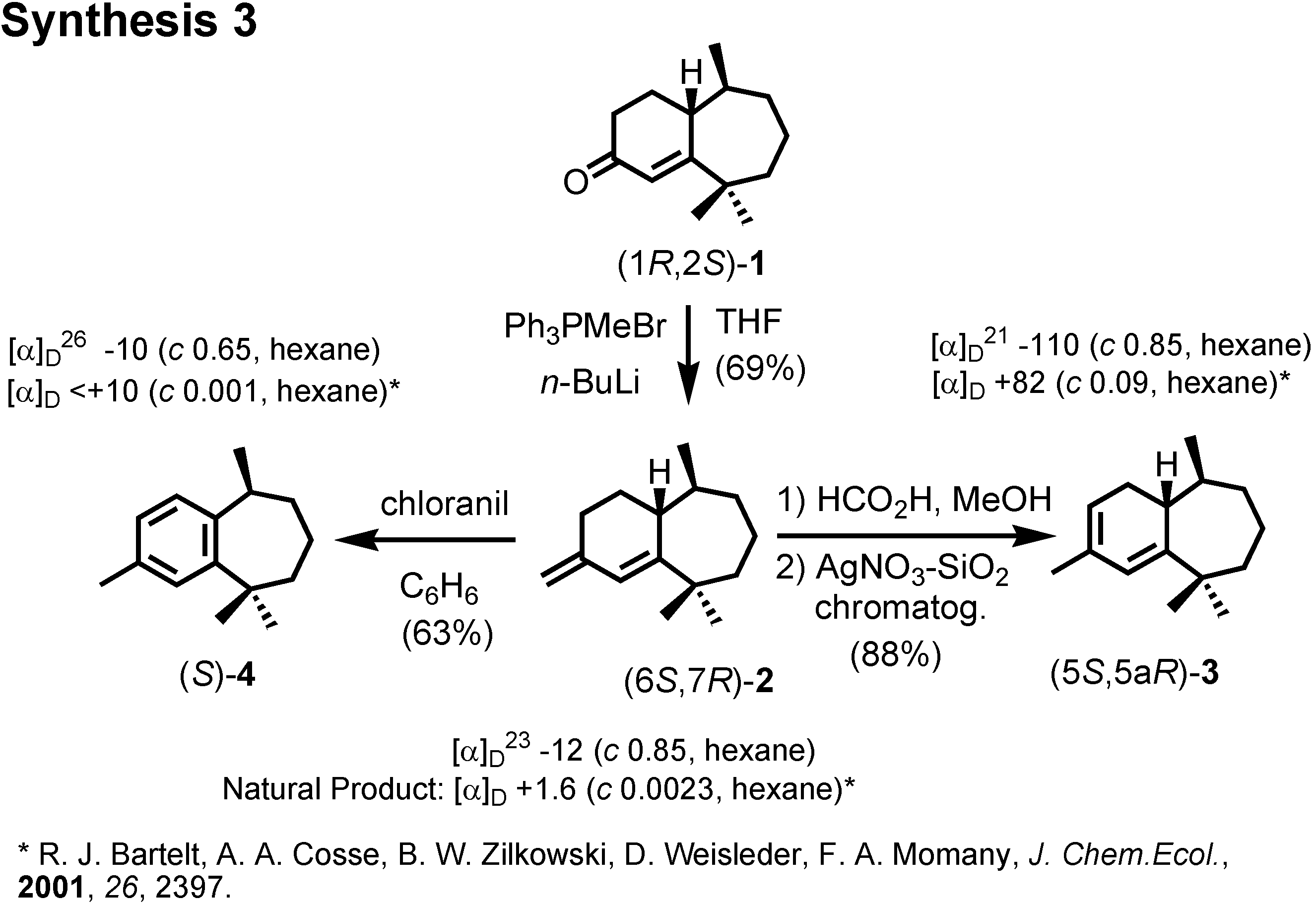
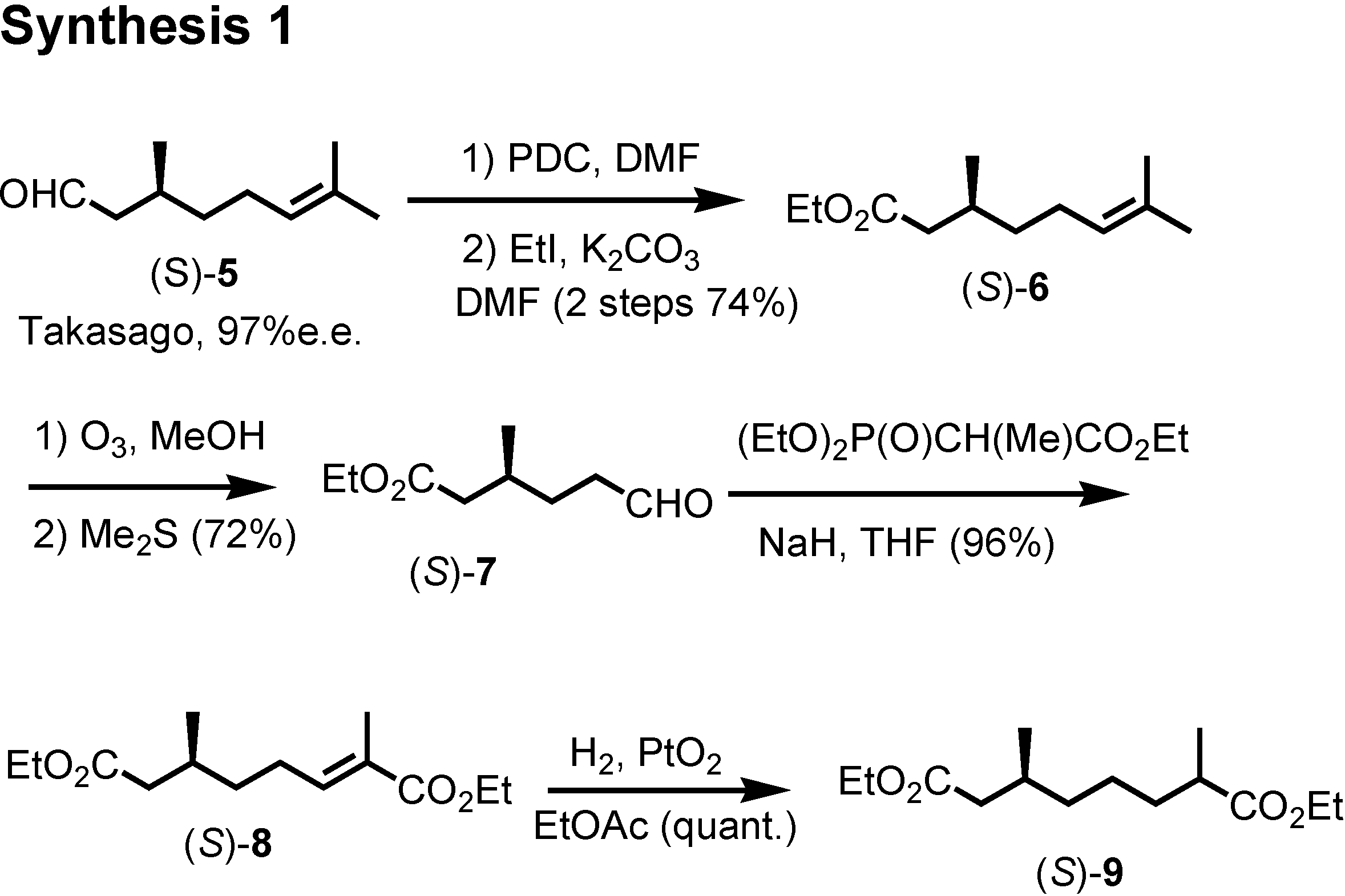
et al. at the U.S. Department of Agriculture isolated these himachalene-type sesquiterpenes as the male-produced sex pheromone of the flea beetle. They proposed the stereochemistry as depicted here. They also reported a synthesis of the racemic pheromones. We became interested in synthesizing these enantiomers to confirm the proposed stereochemistry. The results have been published recently. Our synthetic plan was to use citronellic acid so as to determine the stereochemistry at the methyl-branching.
Citronellal manufactured by Takasago Corporation according to Noyori was our starting material (
Figure 14). Dr. Muto carried out the experimental work. (
S)-Citronellal was oxidized and esterified to give ethyl ester
6. Ozonolysis of
6 afforded aldehyde (
S)-
7, which was converted to unsaturated ester
8 by Horner-Wadsworth-Emmons reaction. Hydrogenaton of
8 over Adams’ platinum oxide gave saturated diester
9.
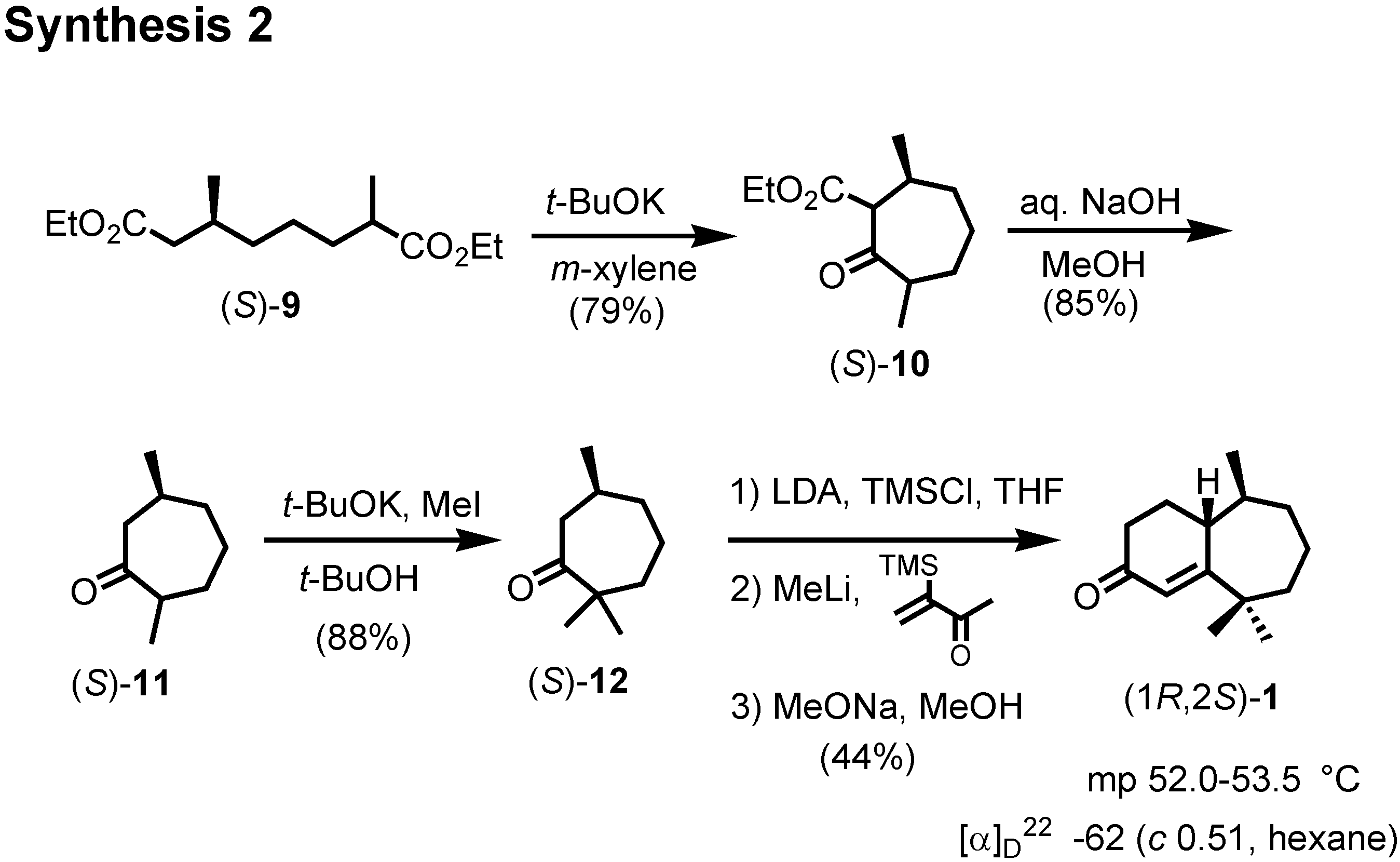
Dieckmann condensation of
9 by treatment with postassium
t-butoxide gave
10, which was hydrolyzed and decarboxylated to give (2
RS,6
S)-2,6-dimethylcycloheptanone
11 (
Figure 15). This dimethyl ketone
11 was further methylated to give
12. Finally (
S)-
12 was converted to crystalline (1
R,2
S)-ketone
1 by Stork modification of the Robinson annelation. This ketone
1 was also reported as a natural pheromone, but its chiroptical data were not reported.
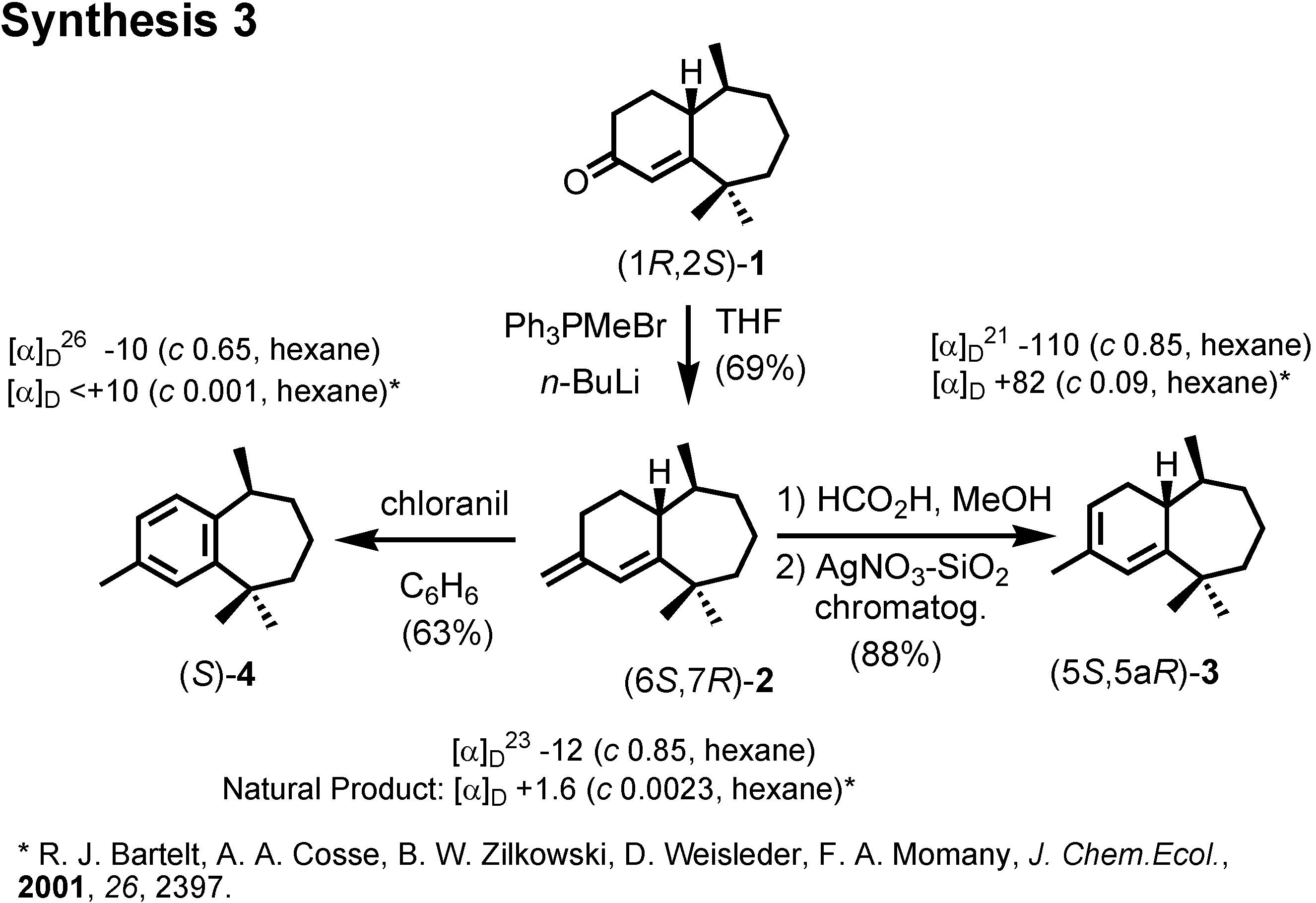
We therefore converted (1
R, 2
S)-
1 to three other pheromone components whose specific rotations had been reported by Bartelt (
Figure 16). When we compared the specific rotations of our synthetic products with the values reported by Bartelt, we were surprised, because our products were levorotatory, while the natural products had been reported as dextrorotatory.
We repeated the synthesis by starting from (
R)-citronellal (
Figure 17). Then, our products were all dextrorotatory like the natural products. Therefore these enantiomers are the pheromone components, and our conclusion is opposite to that of Bartelt. Indeed these compounds were active as aggregation pheromones against flea beetle in Hungary.
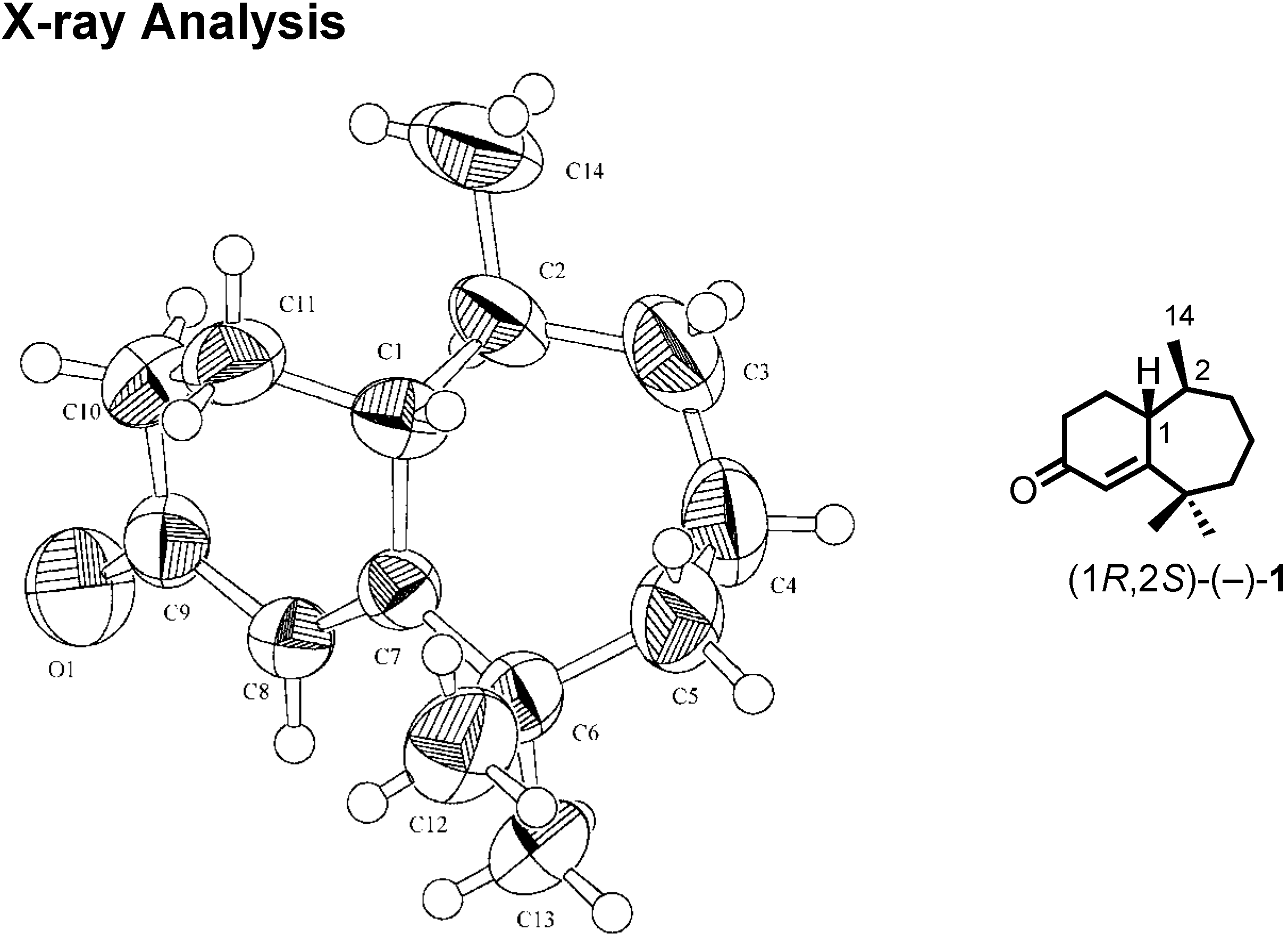
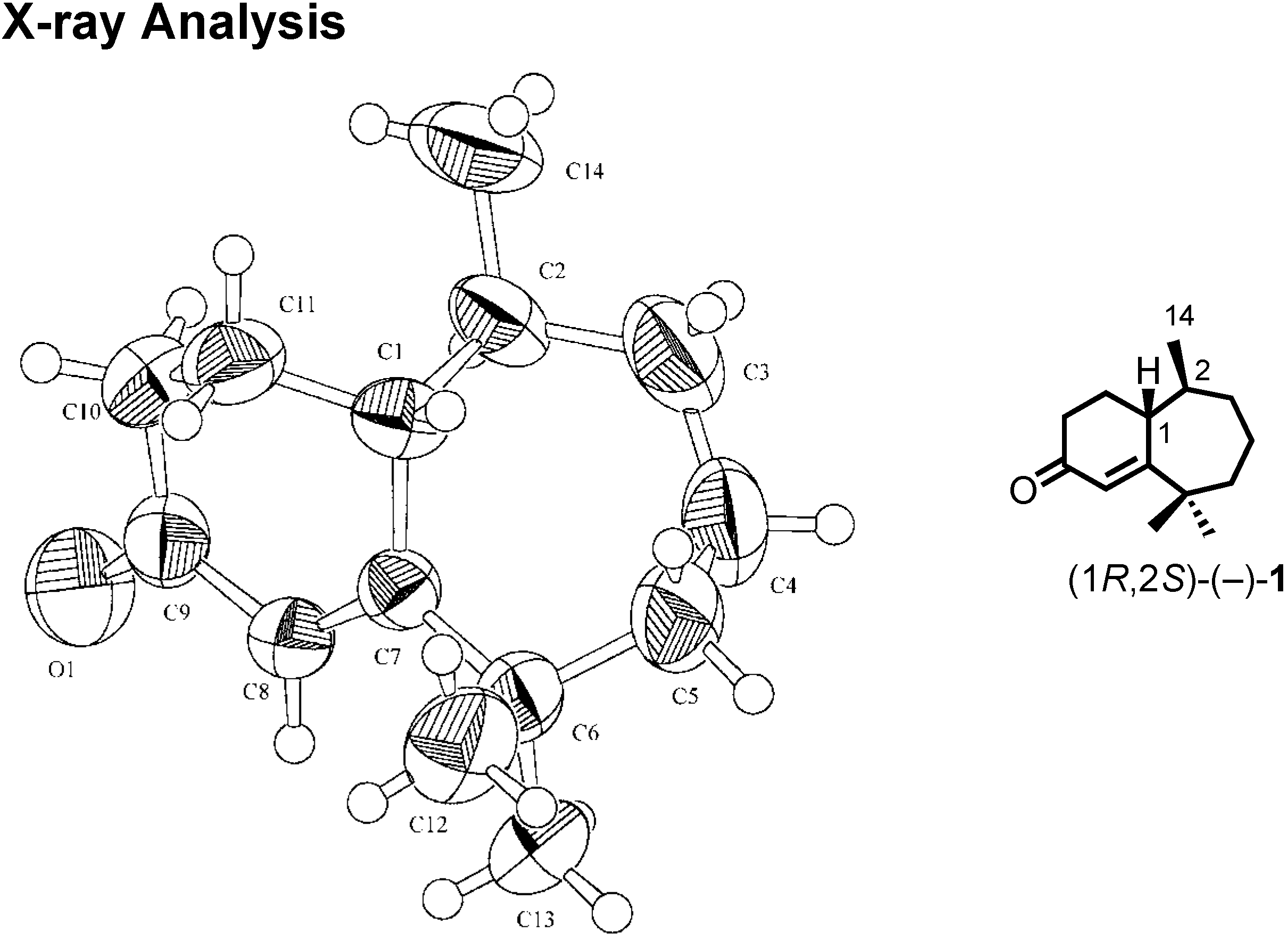
We were confident about our stereochemical assignment, because the stereochemistry of (1
R,2
S)-ketone was unambiguously determined by X-ray analysis (
Figure 18).
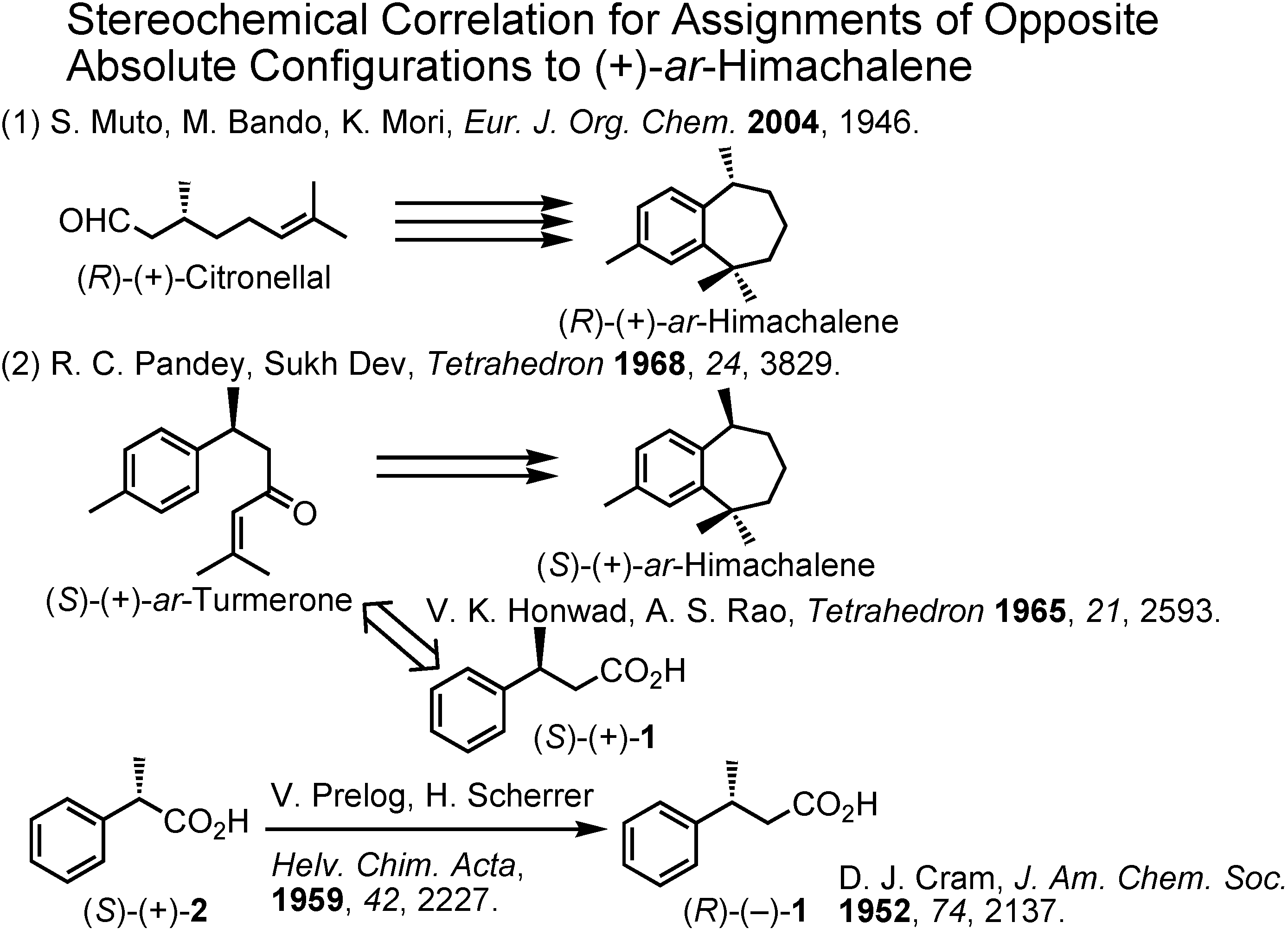
Why were two opposite absolute configurations assigned to (+)-
ar-himachalene? Now, we have to solve this question (
Figure 19). The specific rotations of our final products were measured as hexane solutions according to Bartelt. We assigned the (
R)-configuration to our (+)-
ar-himachalene, because it was derived from (
R)-citronellal. Bartelt assigned the (
S)-configuration to (+)-
ar-himachalene, because Pandey and Sukh Dev gave the (
S)-configuration to (+)-
ar-himachalene by synthesizing it from (
S)-(+)-
ar-turmerone. The (
S)-Configuration was given to (+)-
ar-turmerone by Honwad and Rao on the basis of their correlation of (+)-
ar-turmerone to (
S)-(+)-
1. Its opposite enantiomer (-)-
1 was previously synthesized by Cram, and the (
R)-configuration was assigned to it by Prelog by homologating (
R)-(+)-
2 to (
R)-(-)-
1. Upon seeing such big names as Prelog, Cram and Sukh Dev, I became cautious, and examined their conversion procedures carefully. Then finally I decided to trace the experiment reported by Pandey and Sukh Dev. To do so, I had to synthesize optically active
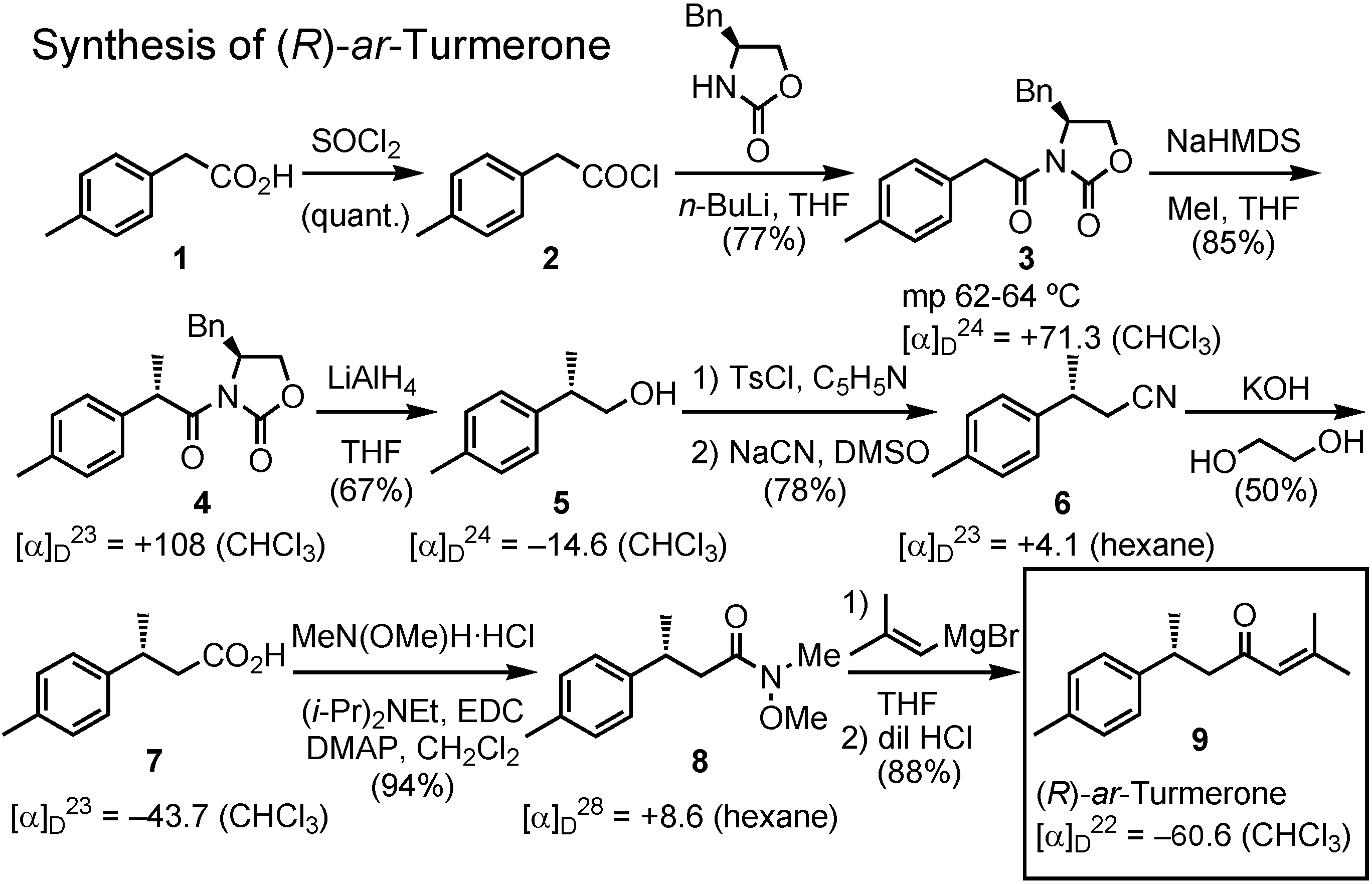
ar-turmerone first, because it was not available in Japan.
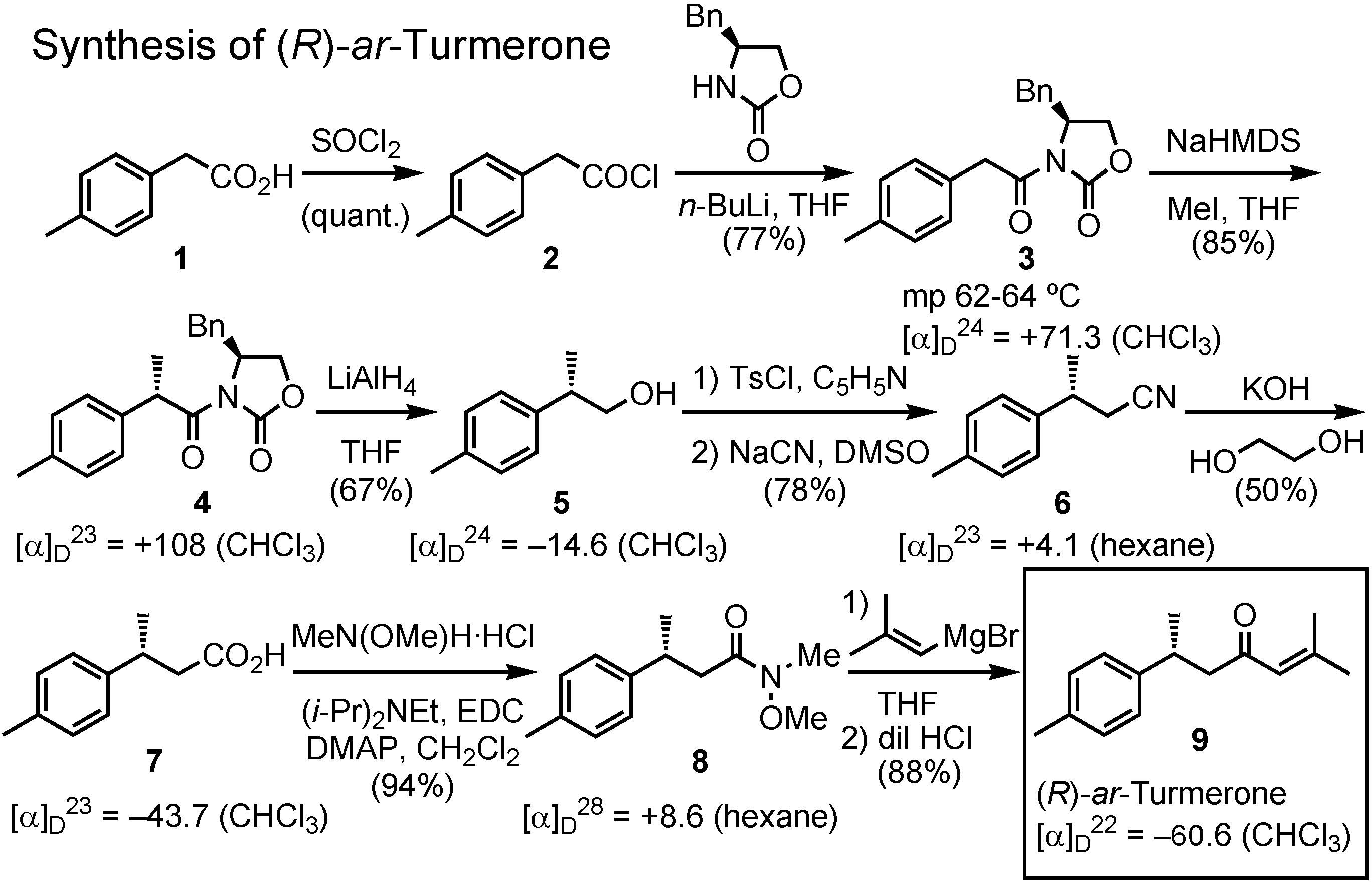
Figure 20 shows my synthesis of (
R)-
ar-turmerone. This experiment was done by me personally in May-July, this year. Commercially available acid
1 was first converted to the corresponding acyl chloride
2, which was coupled with (
S)-4-benzyl-2-oxazolinone, a chiral auxiliary developed by David Evans, to give
3. This was methylated with NaHMDS and methyl iodide to afford
4. Reduction of
4 with lithium aluminum hydride gave
5, which was converted to carboxylic acid
7, the so-called Rupe’s acid, via
6. The acid
7 was derivatized to Weinreb amide
8. Treatment of the amide
8 with 2-methyl- propenylmagnesium bromide afforded (
R)-
ar-turmerone of about 98%
ee, as determined by GC analysis on a chiral stationary phase.
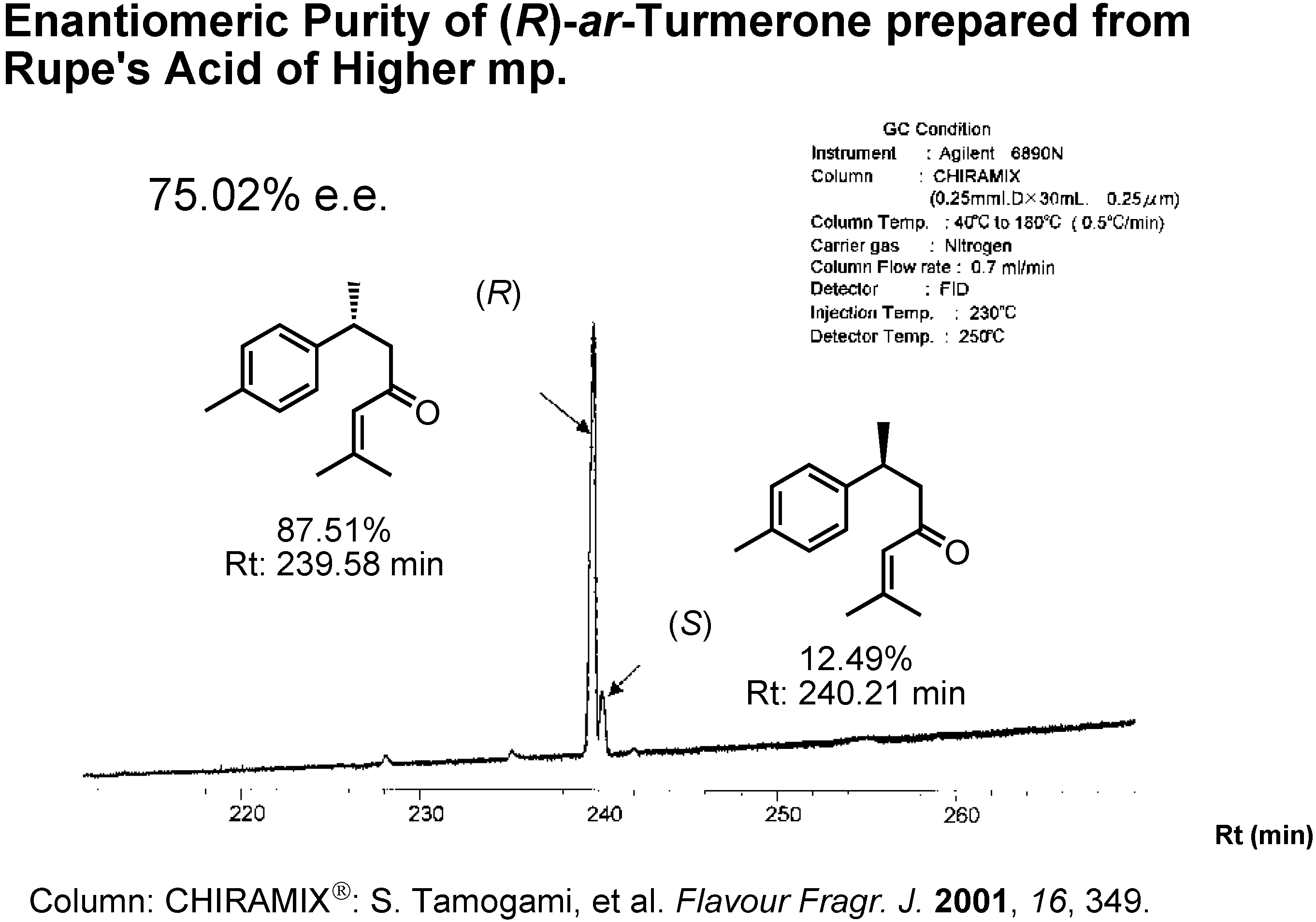
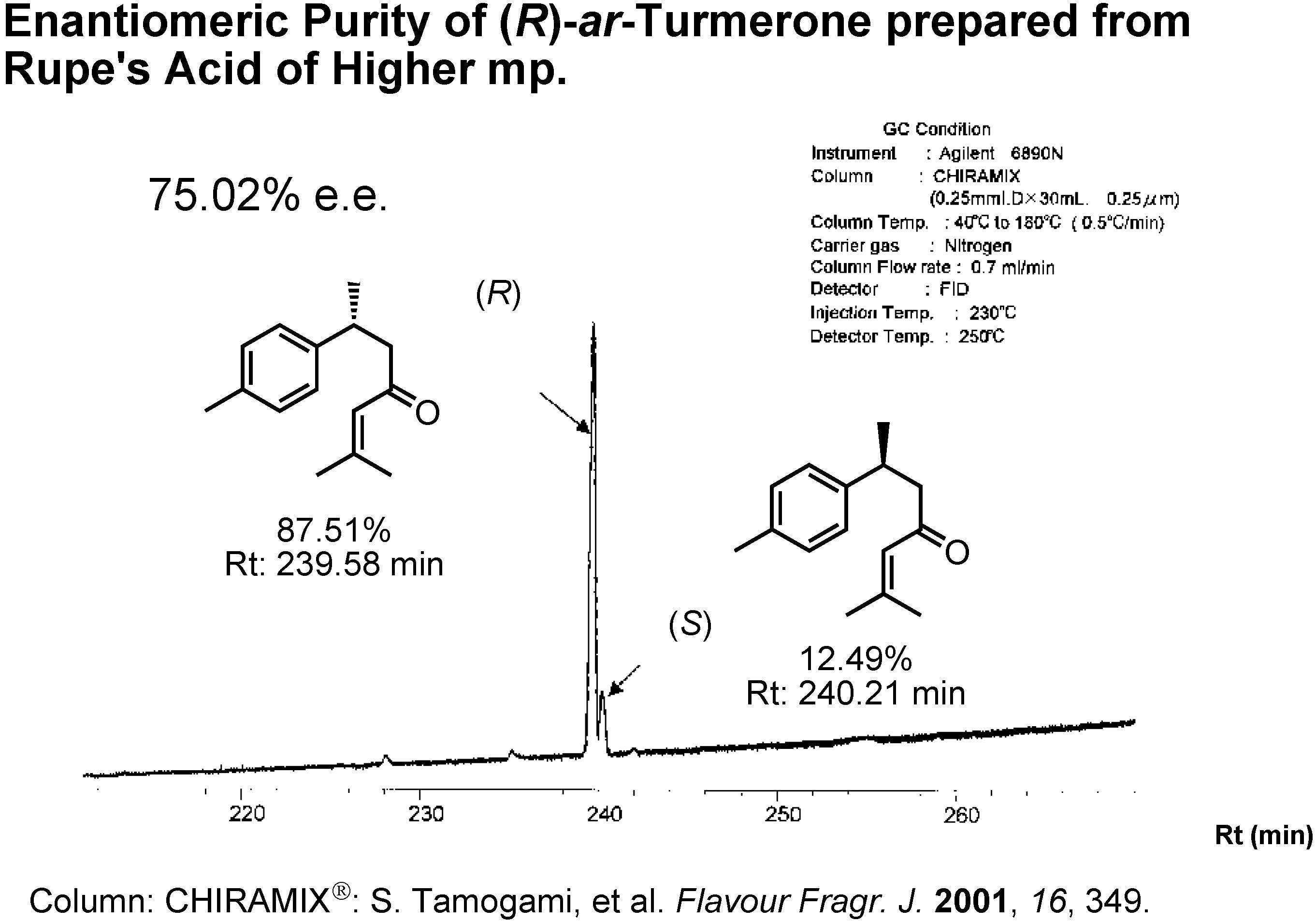
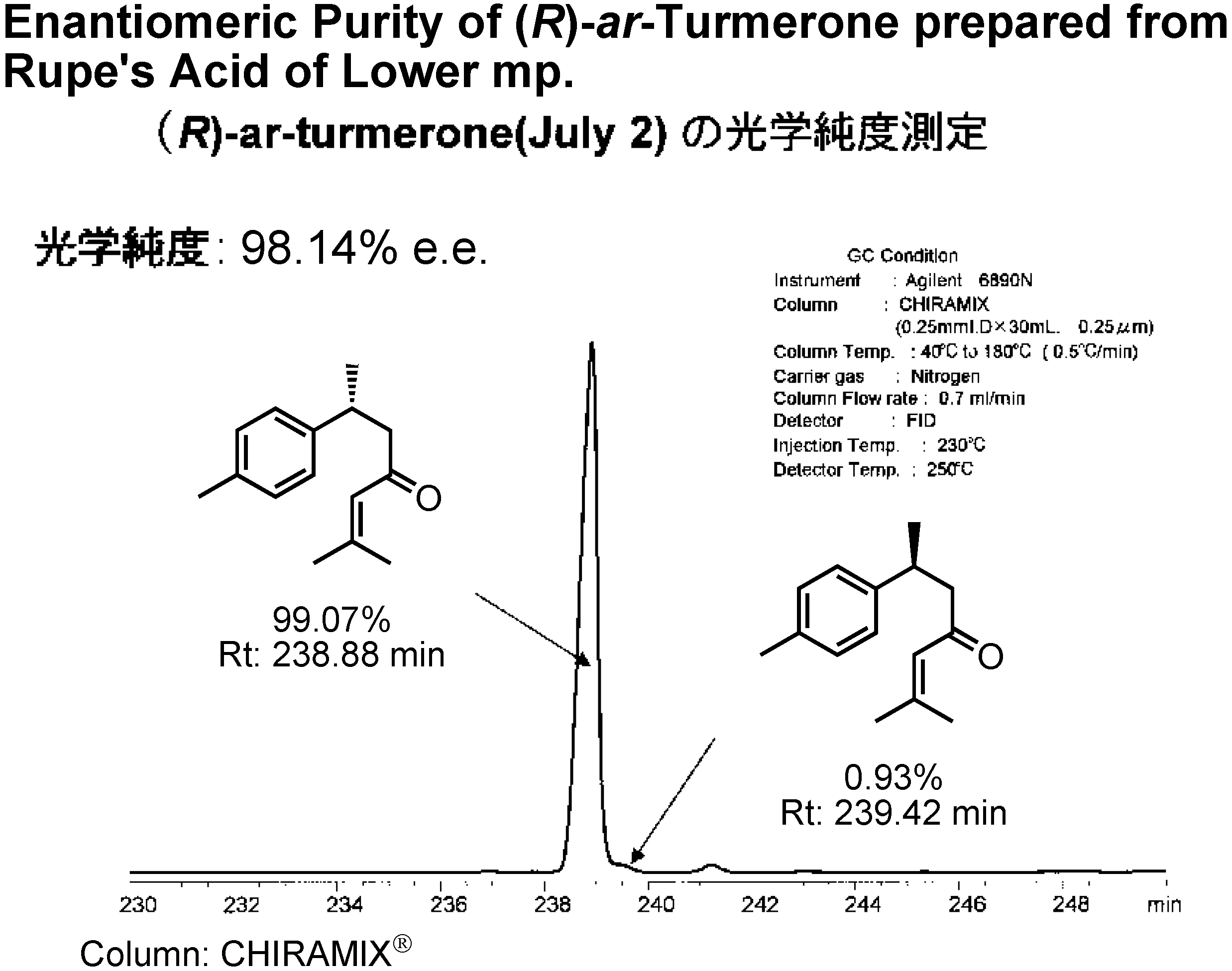
Figure 21 shows the GC analysis of (
R)-
ar-turmerone on a CHIRAMIX chiral stationary phase of Hasegawa Perfume Co. When I used Rupe’s acid of a higher mp as the intermediate, the enantiomeric excess of the resulting
ar-turmerone was 75%
ee.
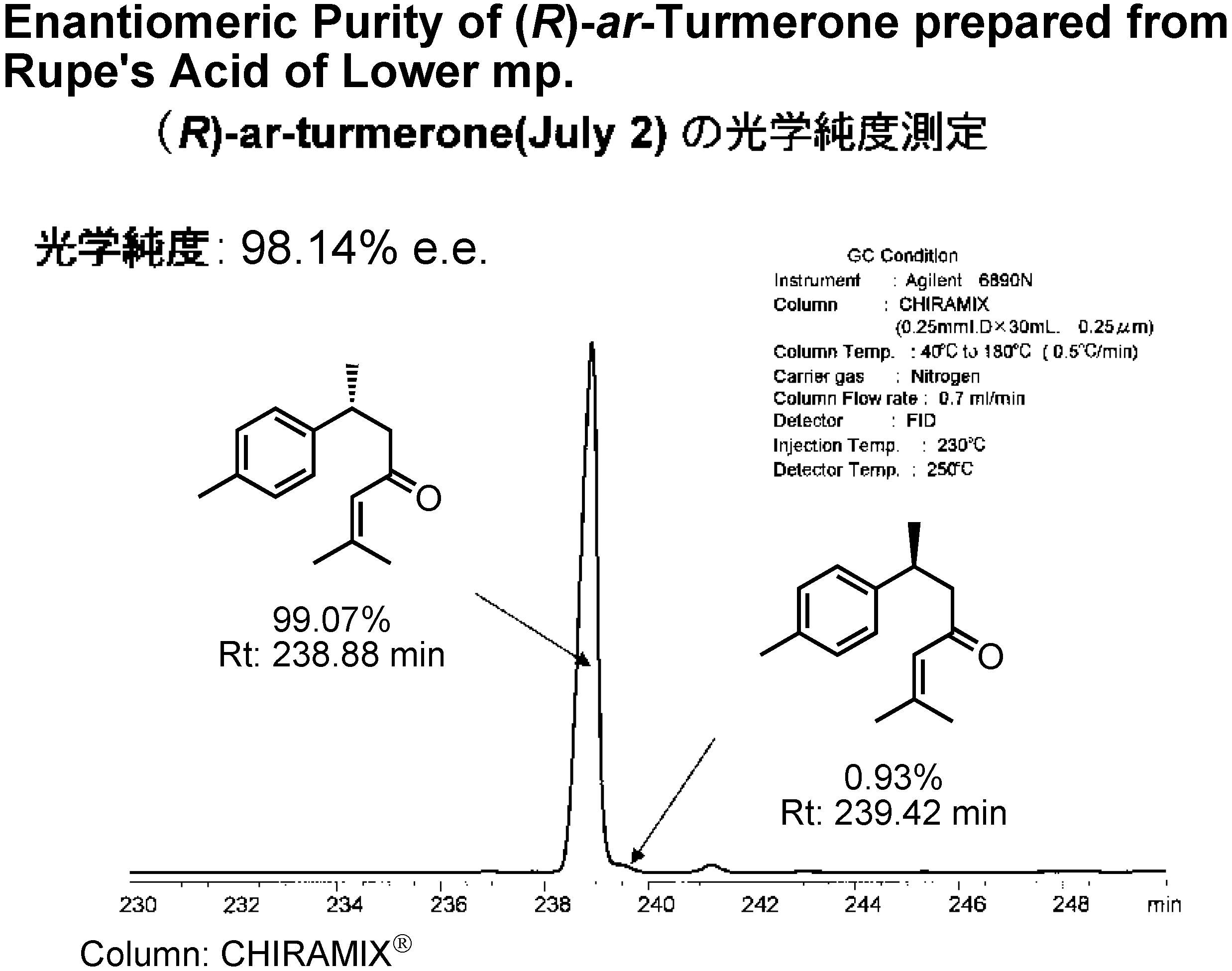
When Rupe’s acid with a lower mp was used as the intermediate, the resulting
ar-turmerone was of 98%
ee (
Fig.22). Lower melting or oily Rupe’s acid was enantiomerically purer than the higher melting material!
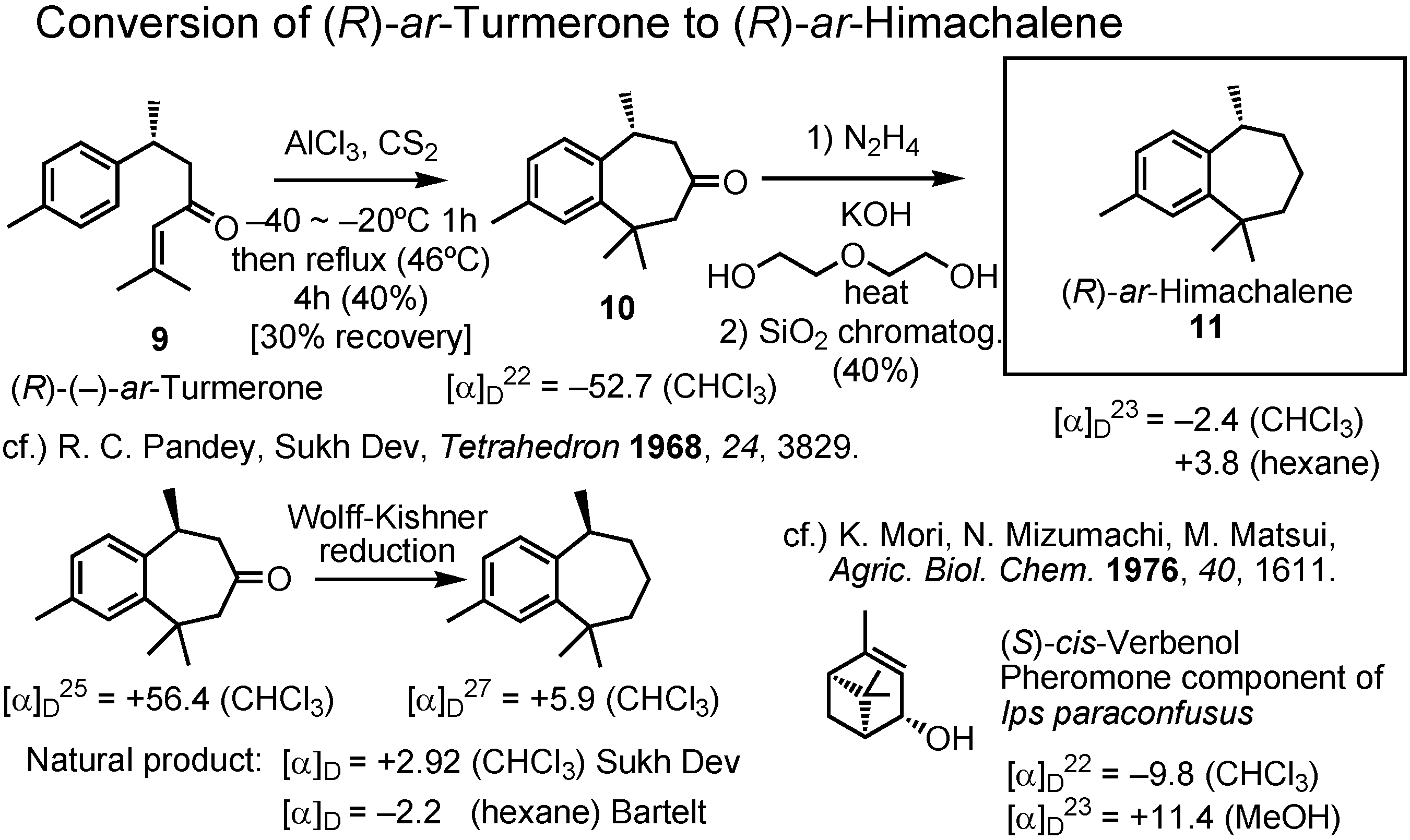
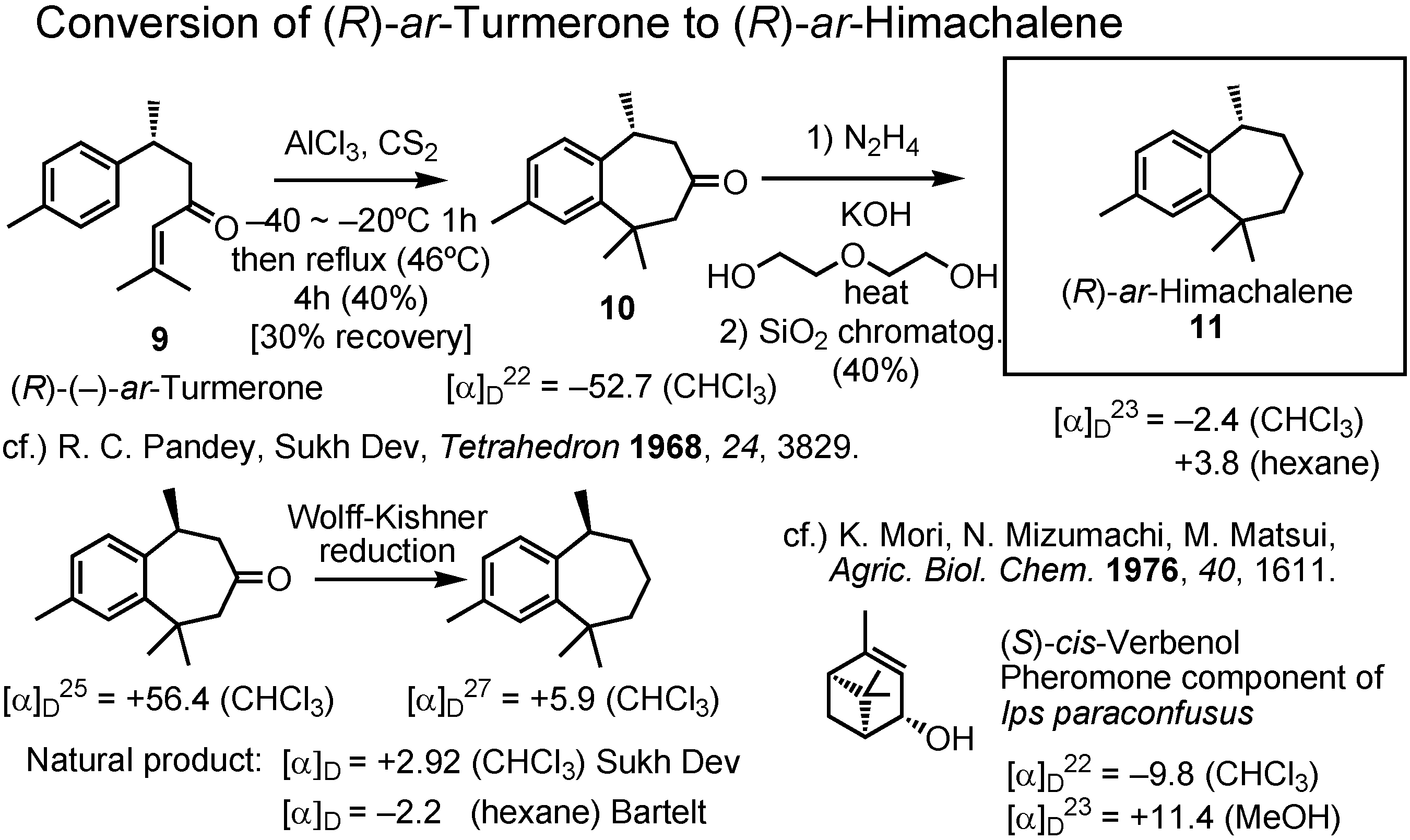
Figure 23 shows the conversion of (
R)-
ar-turmerone to (
R)-
ar-himachalene, and clarifies the reason why Bartelt had made a mistake. The synthetic (
R)-
ar-turmerone was treated with aluminum chloride in carbon disulfide, first at a low temperature then at reflux. The initial low temperature was very important for the success of this cyclization to give
10. Wolff-Kishner reduction of
10 gave (
R)-
ar-himachalene
11. Pandey and Sukh Dev measured the specific rotations of both (
S)-
ar-turmerone and (
S)-
ar-himachalene as chloroform solutions, and they were both dextrorotatory. When the specific rotation of my (
R)-
ar-himachalene was measured, I immediately found the reason why Bartelt made an error. It was levoratory in chloroform, but dextrorotatory in hexane. Although Sukh Dev reported that they used chloroform, Bartelt did not care about the solvent and used hexane, without knowing the fact that a different solvent may change the sign of rotation. A similar phenomenon was reported by us in 1976. (1
S,4
S,5
S)-
cis-Verbenol is a pheromone component of the bark beetle,
Ips paraconfusus. At that time there was confusion. Some people called (
S)-
cis-verbenol as (+)-
cis-verbenol, while others called it (-)-
cis-verbenol. Our study of this compound revealed it to be levorotatory in chloroform, while dextrorotatory in methanol. The important lesson here in this scheme is the fact that the sign of rotation may change by using a different solvent. We should be careful about this fact.
The third topic of my talk is the synthesis of (4
R, 9
Z)-9-octadecen-4-olide, the female sex pheromone of the currant stem girdler,
Janus integer (
Figure 24). This insect is a pest of red currant in North America. This lactone
1 was isolated and identified by Bartelt and his co-workers in the USA. They synthesized the racemate and (
R)-isomer in µg quantities, and proposed the stereochemistry of the pheromone as
R.
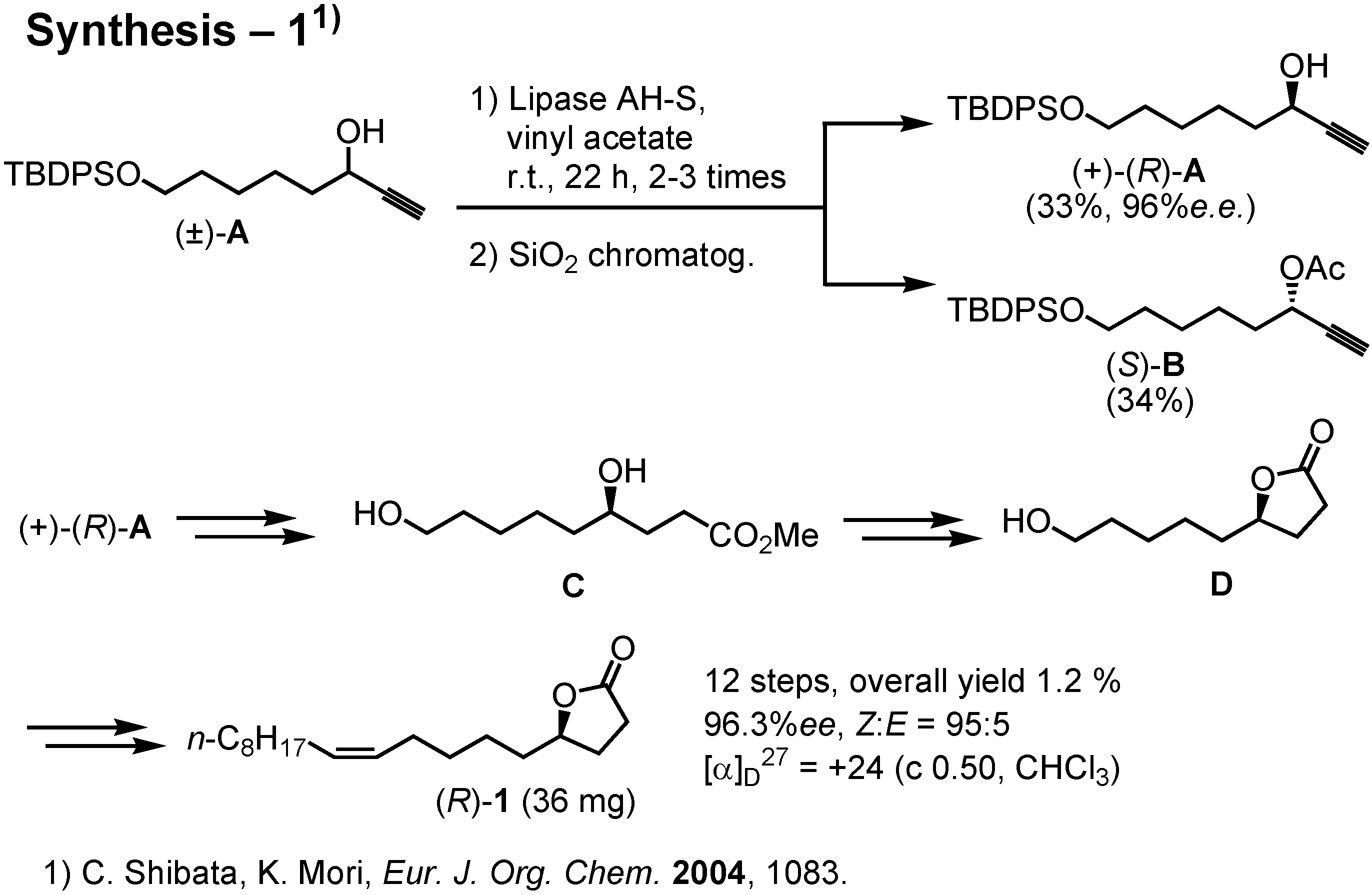
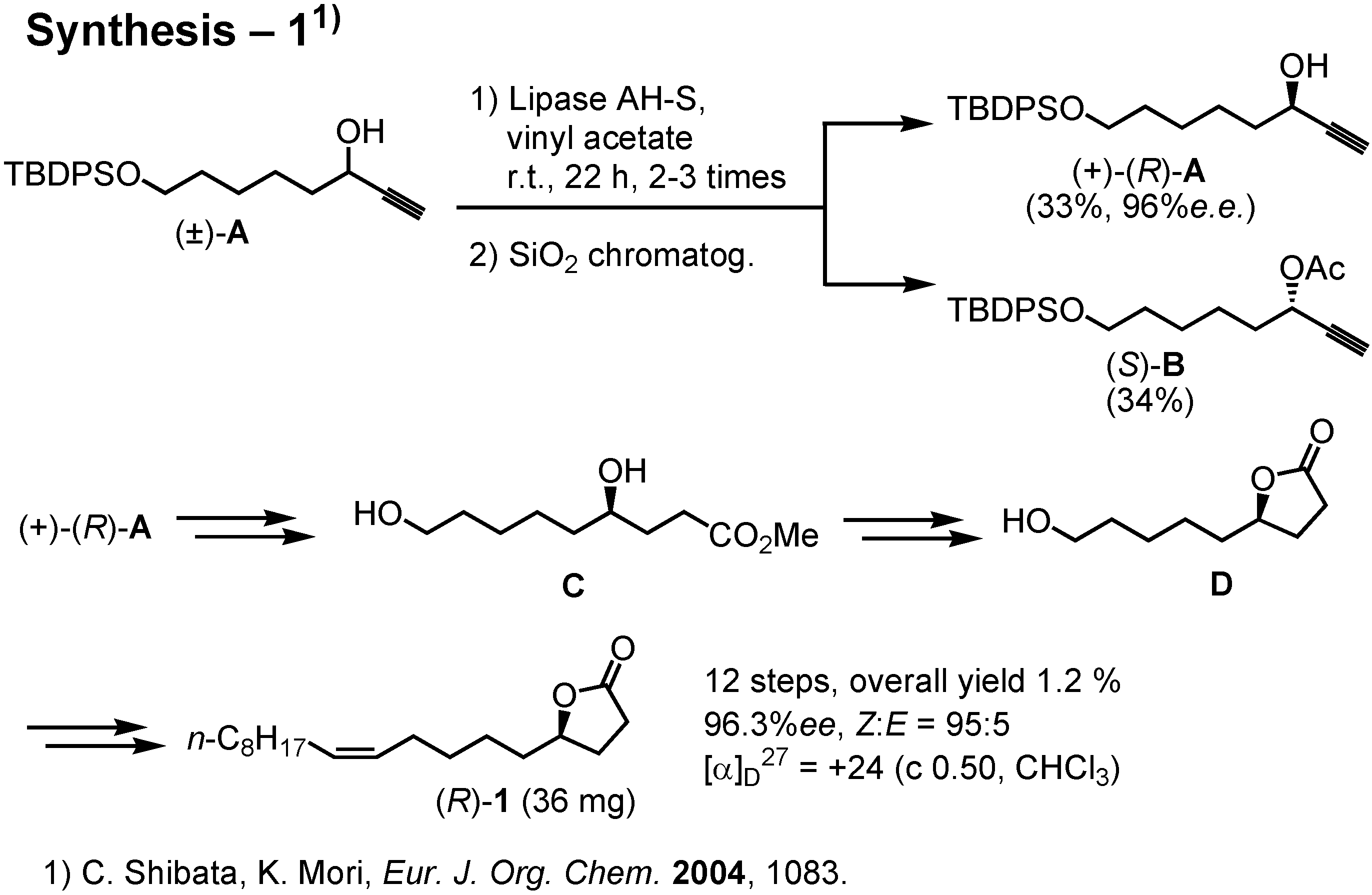
This scheme shows the outline of our recent synthesis, carried out by Ms. Shibata (
Figure 25). We used lipase to achieve kinetic resolution of (±)-
A. Subsequently, (+)-
A was converted to (
R)-
1. The amounts of the enantiomers of
1 secured by this synthesis was only 30-50 mgs. Last year Dr. James at Washington State University in the USA requested me to synthesize gram quantities of (
R)-
1 so that he might be able to carry out a large scale test in the field. I therefore had to develop a new and more efficient synthesis.
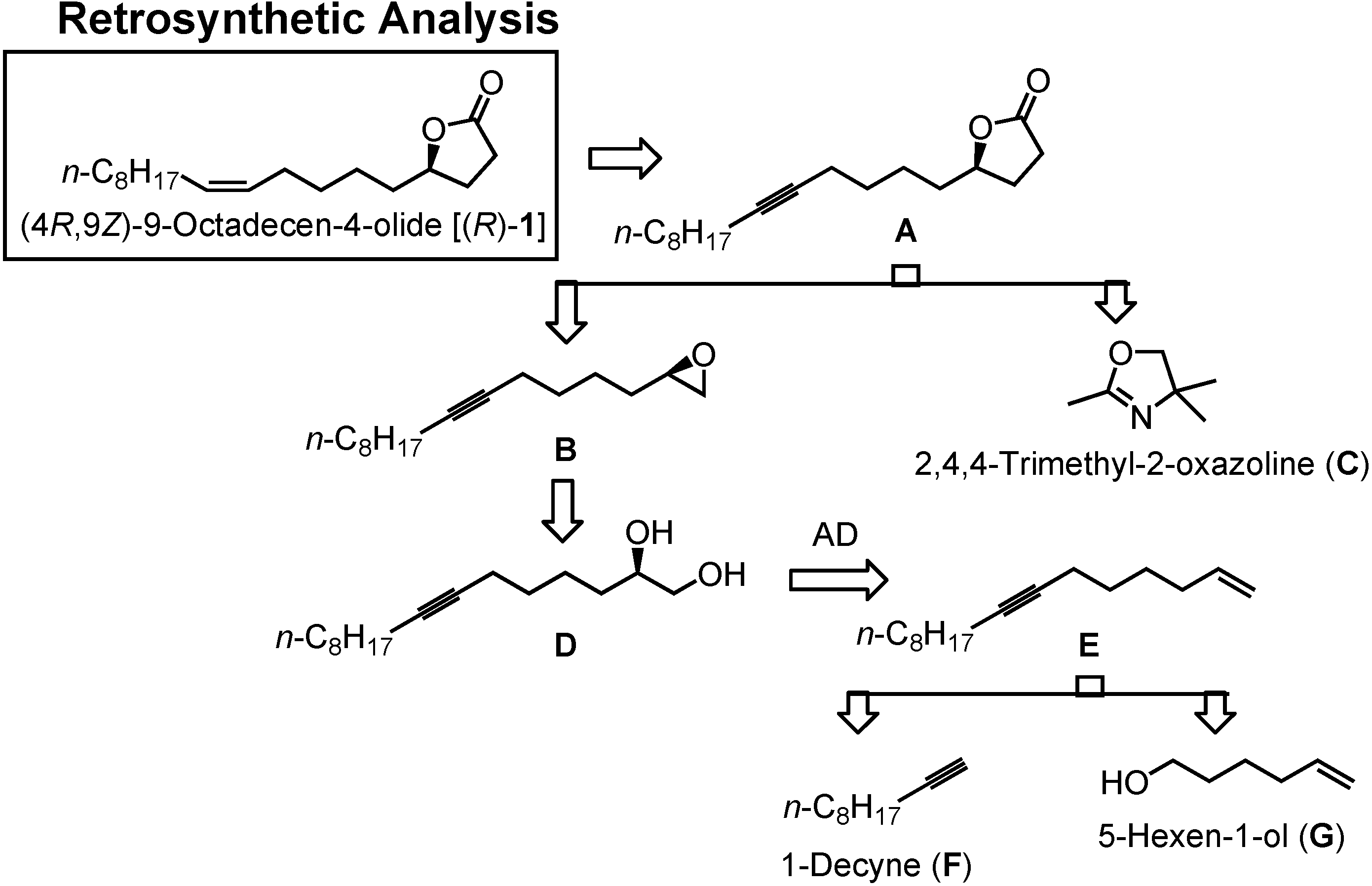
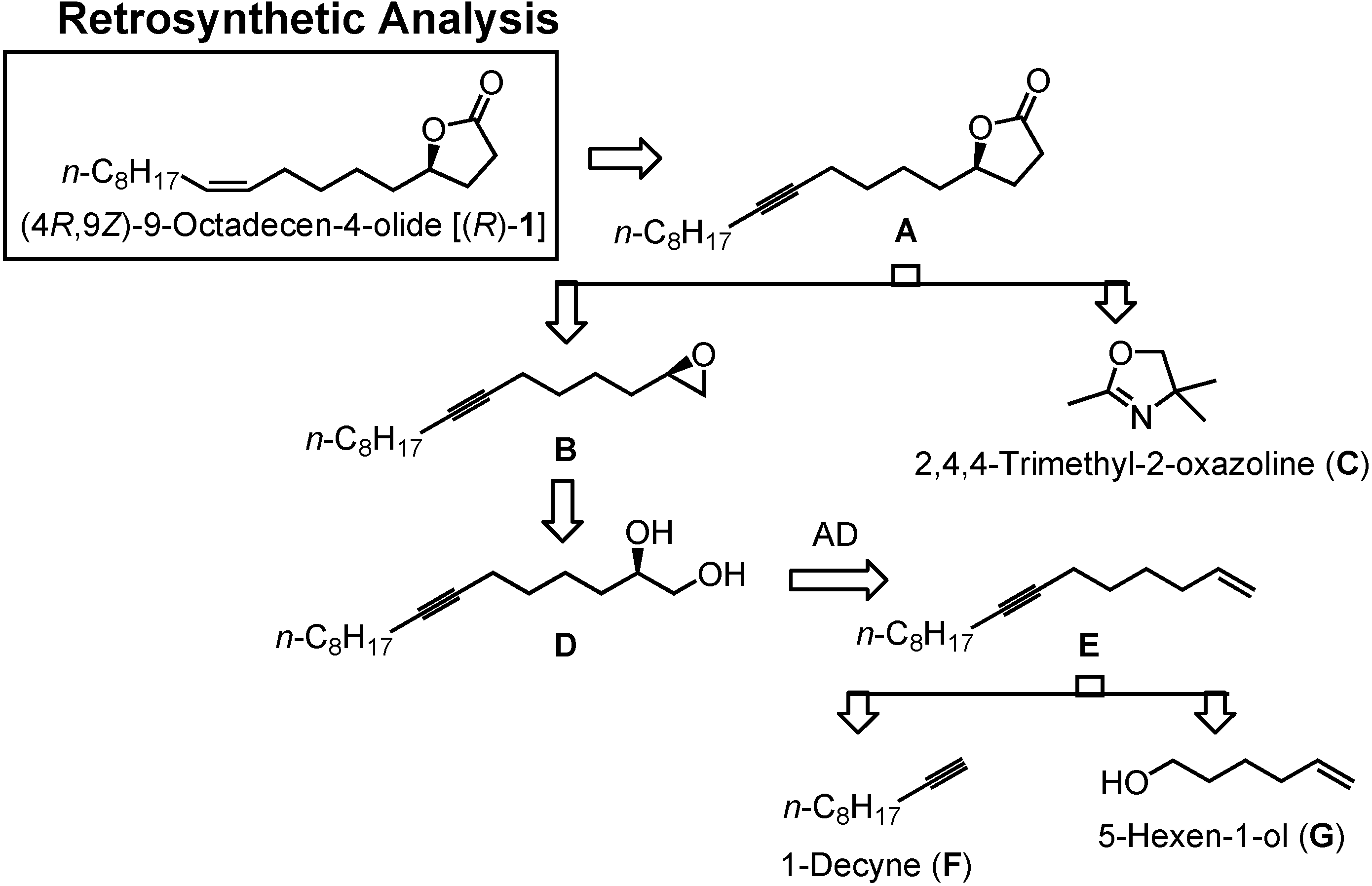
In
Figure 26 you can see my new retrosynthetic analysis. The lactone
1 will be obtained by semi-hydrogenation of the acetylenic lactone
A. Compound
A can be synthesized by alkylating
C with epoxide
B. Epoxide
B is available from glycol
D. Diol
D can be prepared by asymmetric dihydroxylation of
E.
E is to be constructed by combining two commercially available compounds, 1-decyne (
F) and 5-hexen-1-ol (
G).
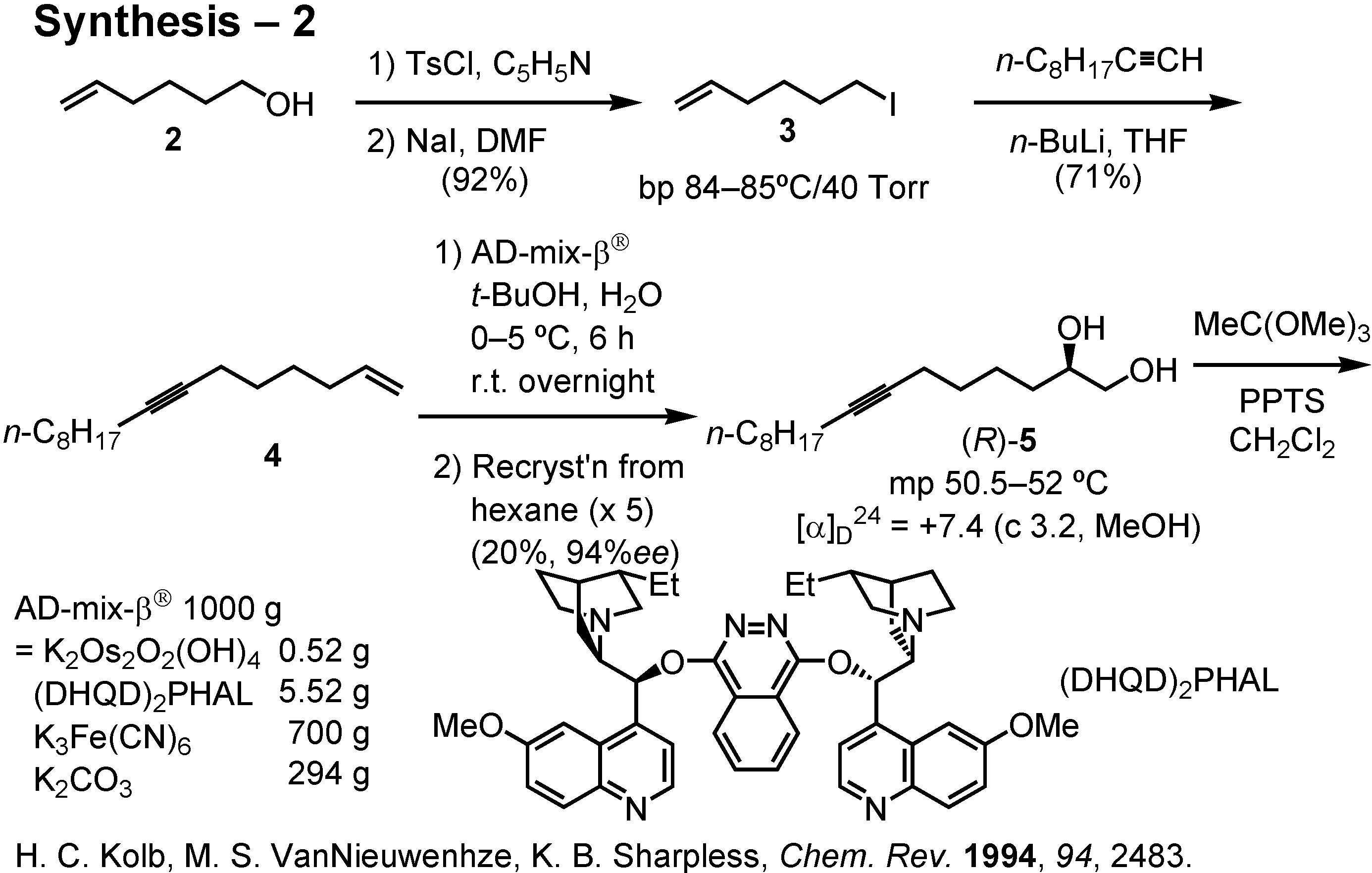
Because I have no one working with me in my pheromone synthesis, I executed the synthesis by myself (
Figure 27). Alcohol
2 was converted to iodide
3 via the corresponding tosylate. Alkylation of octylacetylene with
3 gave enyne
4. This was dihydroxylated by treatment with AD-mix-β
® to give crude diol as a solid. Recrystallization of this solid from hexane gave (
R)-
5 of 87%
ee in 60% yield based on
4. Due to the low melting point and high solubility of (
R)-
5, its purification by recrystallization was difficult. The yield of (
R)-
5 of 94%
ee was only 20% based on
4 after five repeated recrystallizations.
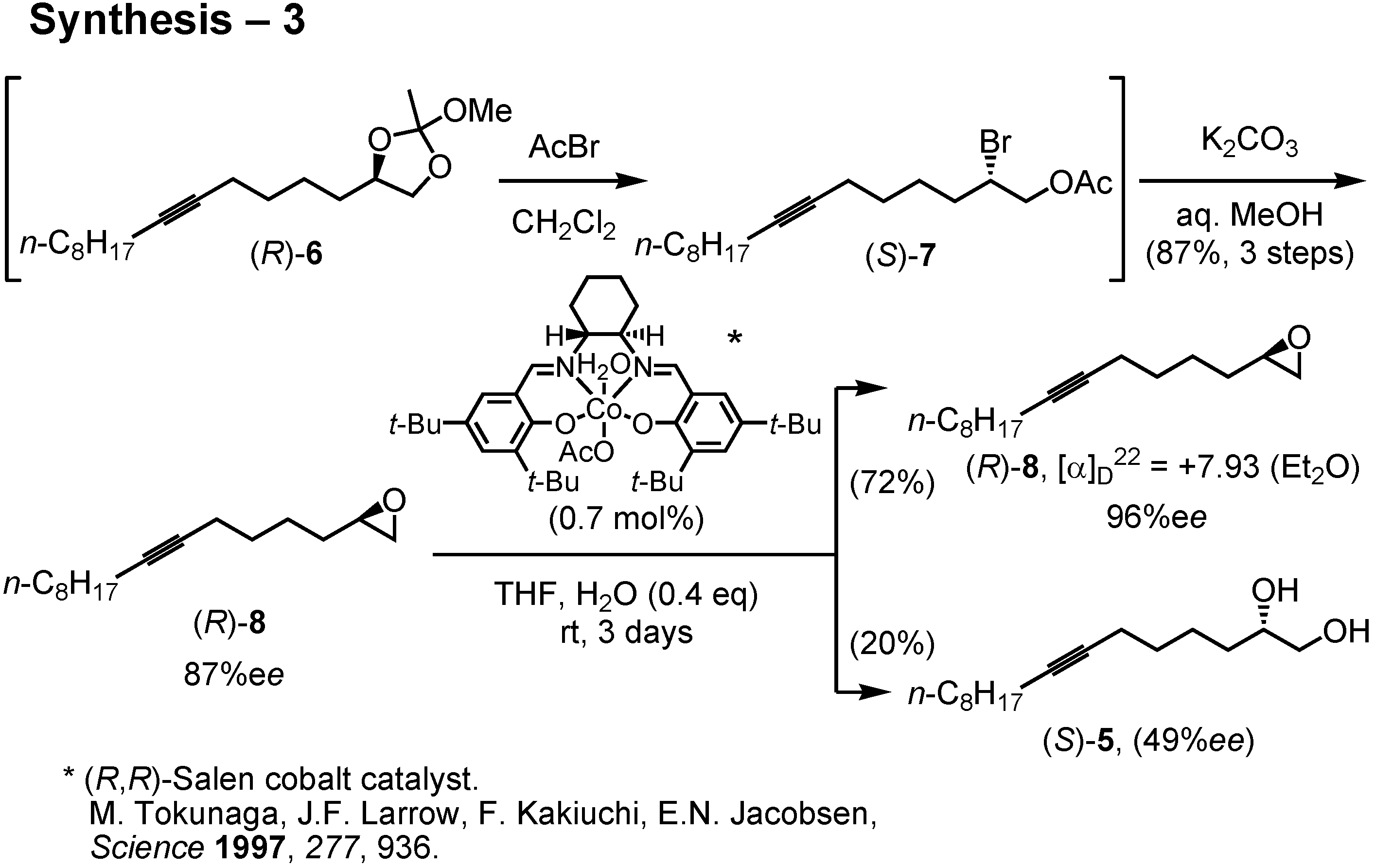
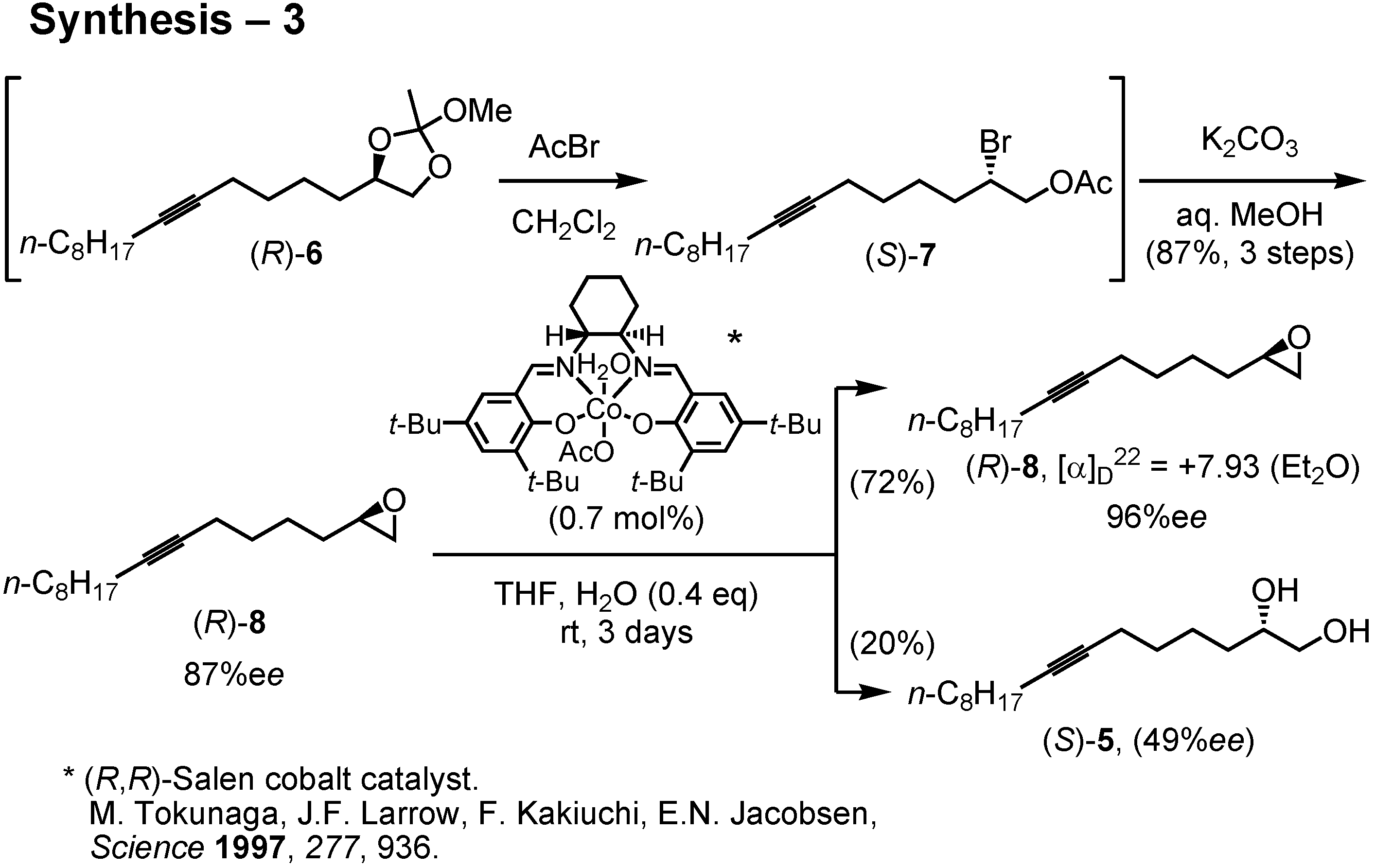
The diol (
R)-
5 of 87%
ee was then converted to epoxide (
R)-
8 by the method of Sharpless. Accordingly, (
R)-
5 was treated with trimethyl orthoacetate and PPTS to give an orthoester. The resulting orthoacetate (
R)-
6 quickly reacted with acetyl bromide to furnish acetoxy bromide (
S)-
7 (
Figure 28). This was treated with potassium carbonate in aqueous methanol to give epoxide (
R)-
8 of 87%
ee. I then purified the epoxide by Jacobsen’s hydrolytic kinetic resolution. Treatment of (
R)-
8 of 87%
ee with Jacobsen’s (
R,
R)-salen cobalt catalyst in the presence of 0.4 equiv. of water in THF afforded in 72% yield epoxide (
R)-
8 of 96%
ee. Purification of (
R)-
8 by Jacobsen’s method was far simpler and more efficient than the recrystallization of diol (
R)-
5. The enantiomeric purity of (
R)-
8 could be determined by chiral GC.
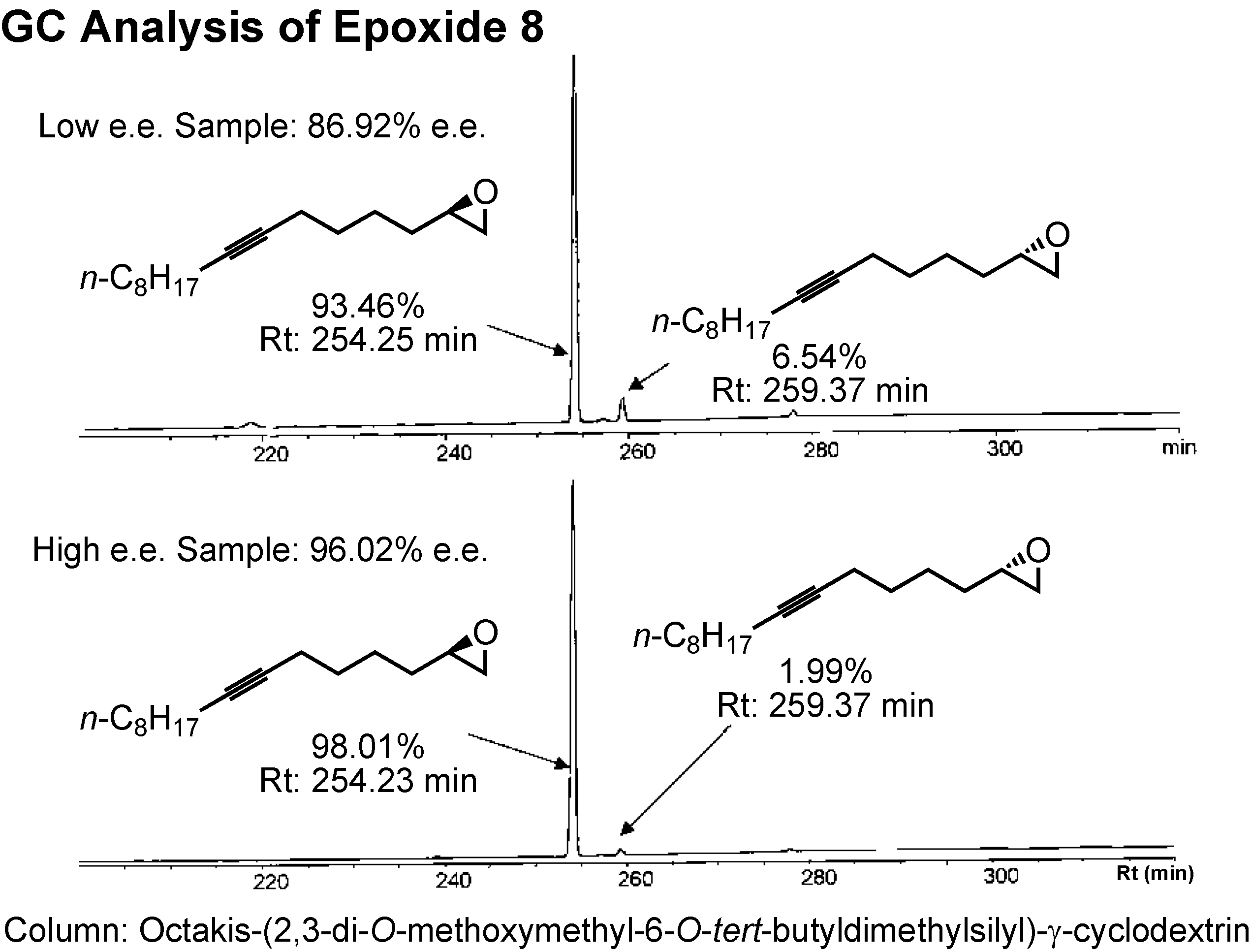
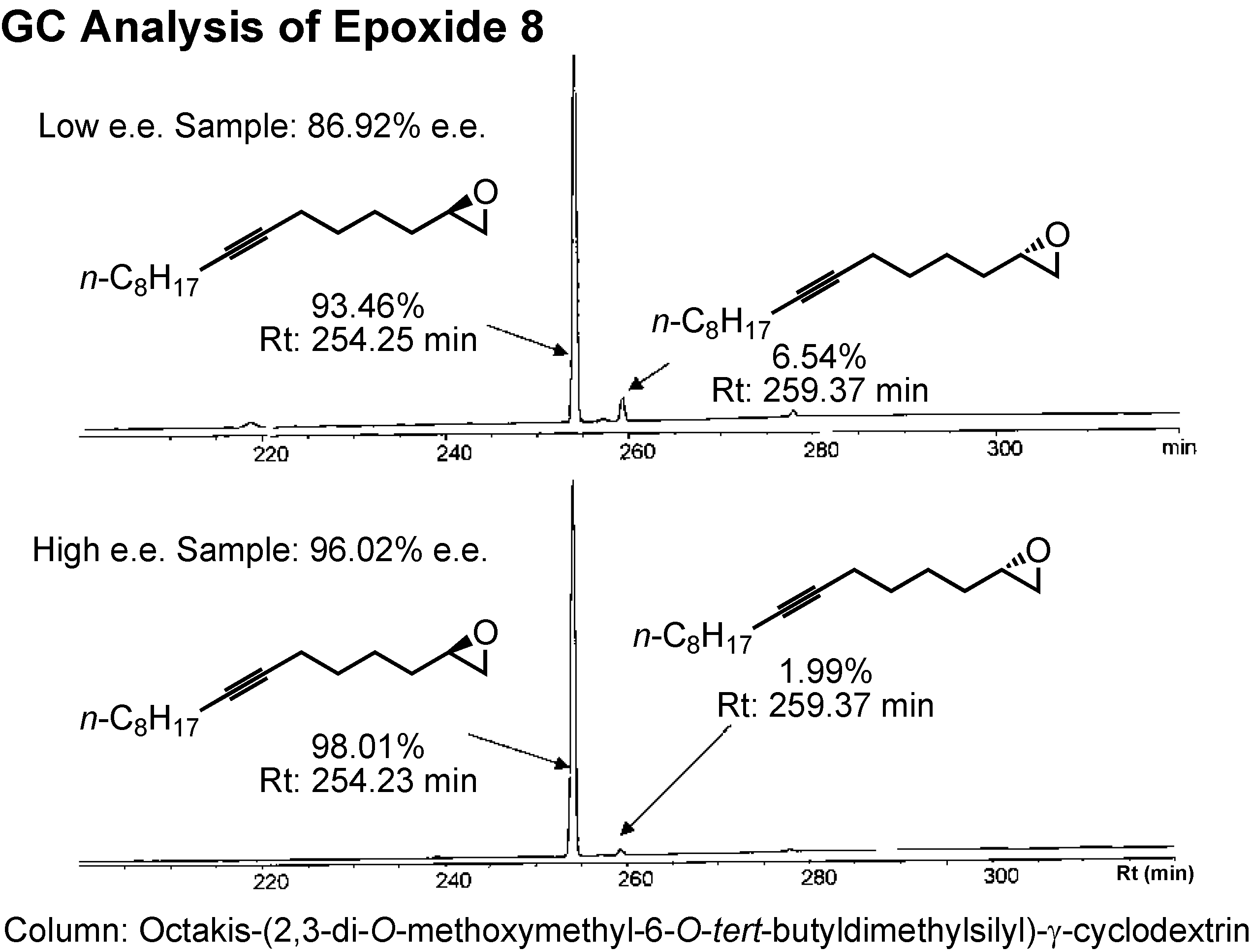
The enantiomers of epoxide
8 could be separated by GC employing a γ-cyclodextrin-based column (
Figure 29). Jacobsen’s HKR afforded the epoxide of 96%
e.e.The epoxide
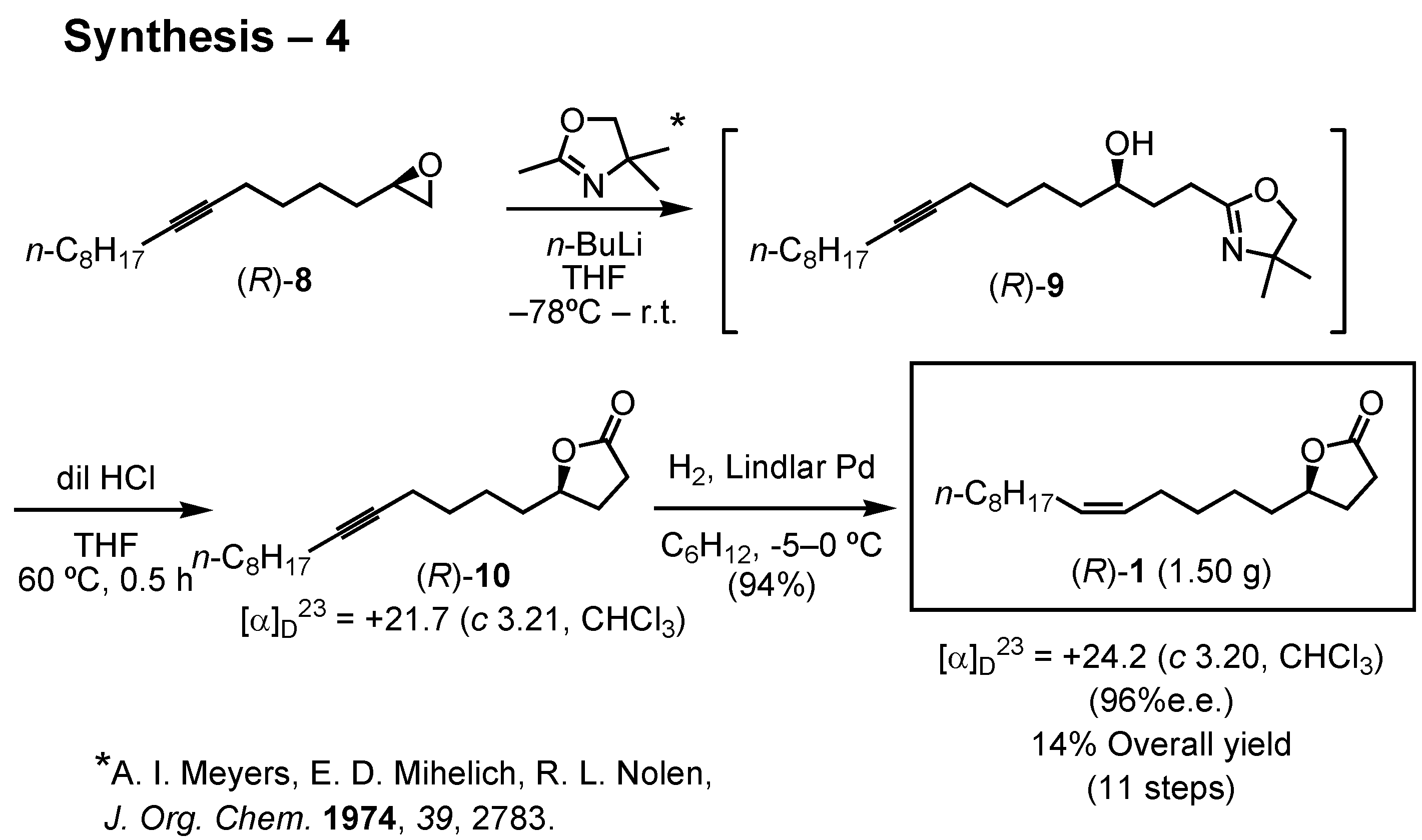
8 furnished the target lactone as shown in this scheme (
Figure 30). Epoxide (
R)-
8 was added to the carbanion derived from 2,4,4-trimethyl-2-oxazoline, and the product was treated with dilute hydrochloric acid to give acetylenic lactone (
R)-
10. Lindlar hydrogenation of the acetylenic lactone (
R)-
10 furnished the target lactone (
R)-
1 of 96%
ee. The overall yield of (
R)-
1 was 14% through 11 steps. I prepared 4 g of (
R)-
1 and sent it to Dr. James. In the next flight season in 2005, he will carry out a large-scale field bioassay in the USA. When gram quantities of a pheromone are needed, we have to develop an efficient synthetic route to achieve the goal.
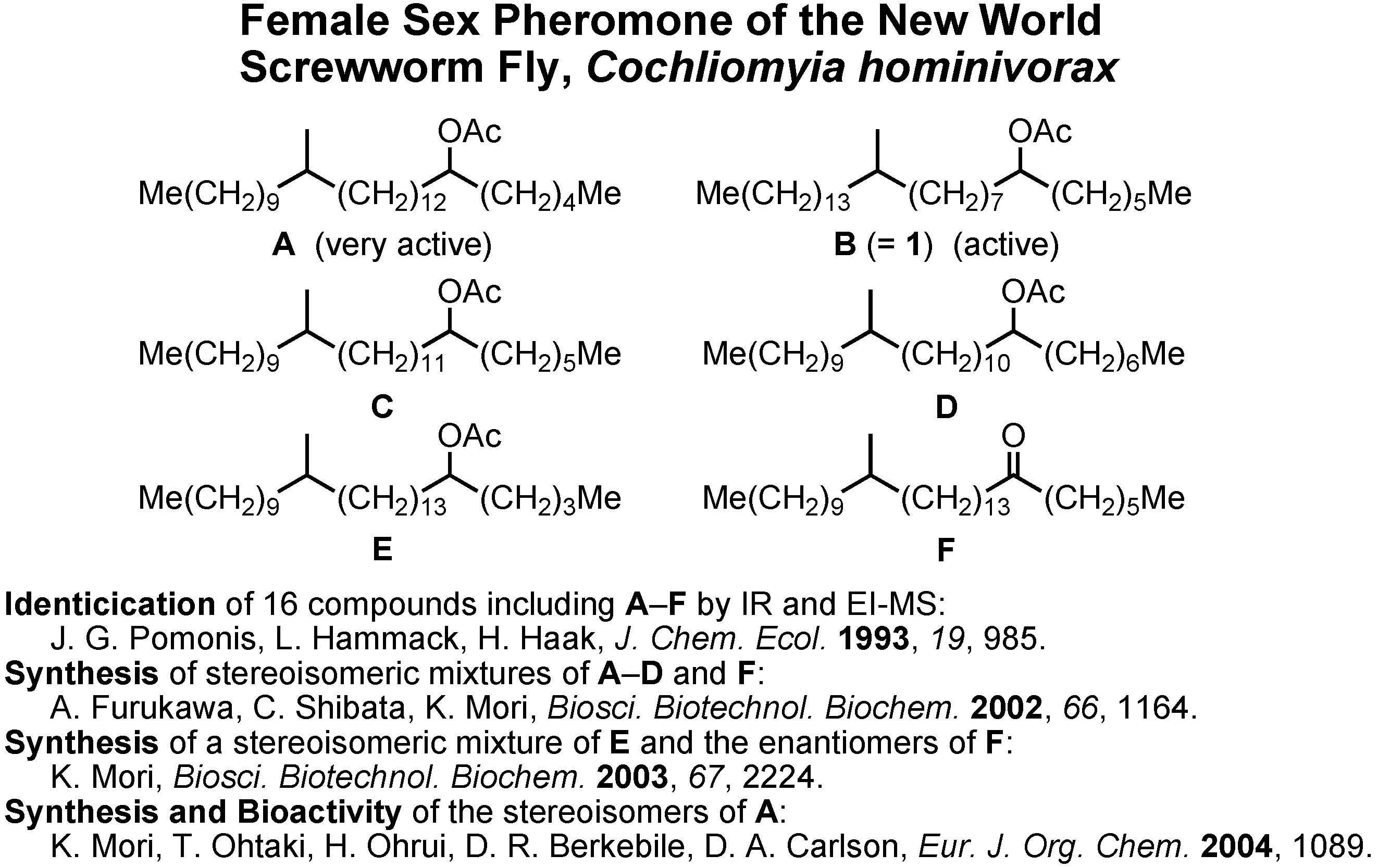
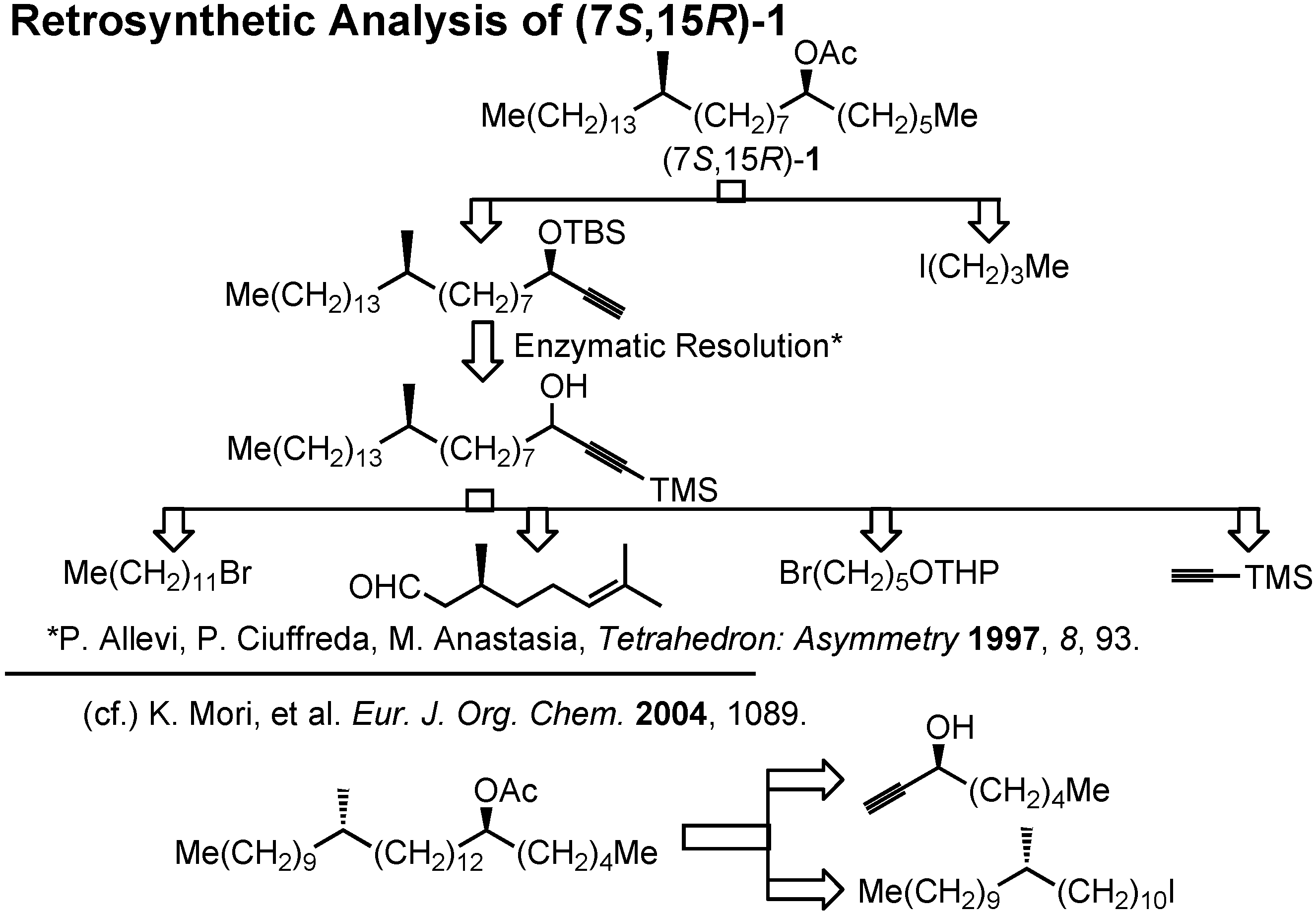
Let us turn to the next and fourth topic, the female sex pheromone of the new world screwworm fly,
Cochliomyia hominivorax (
Figure 31). The screwworm fly is a serious pest to livestock in Central and South America. Its female-produced sex pheromone was first studied by Pomonis at U.S. Department of Agriculture in 1993. They isolated 16 pheromone candidates from females, but they were unable to identify the pheromonally active compounds. In cooperation with Dr. D.A. Carlson at U.S. Department of Agriculture, I began the cooperative work on this pheromone in 2001. By his intuition Dr. Carlson chose these six compounds as real pheromone candidates. We synthesized them as racemic and diastereomeric mixtures, and Dr. Carlson found
A and
B to be pheromonally active.
Because four stereoisomers are possible for
A and
B, it is necessary to synthesize them in order to clarify the stereochemistry-pheromone activity relationship. Very recently, I synthesized four stereoisomers of
A. Ohtaki and Ohrui analyzed their enantiomeric purities, and Berkebile and Carlson found all of them to be pheromonally active.
Figure 32 picture shows the bioassay of these pheromone components The male fly can perceive these pheromone molecules. If a µg quantity of
A or
B was put on the surface of the dead body of the fellow male fly, the living male fly reacted, and embraced the dead body as if it were a living female fly.
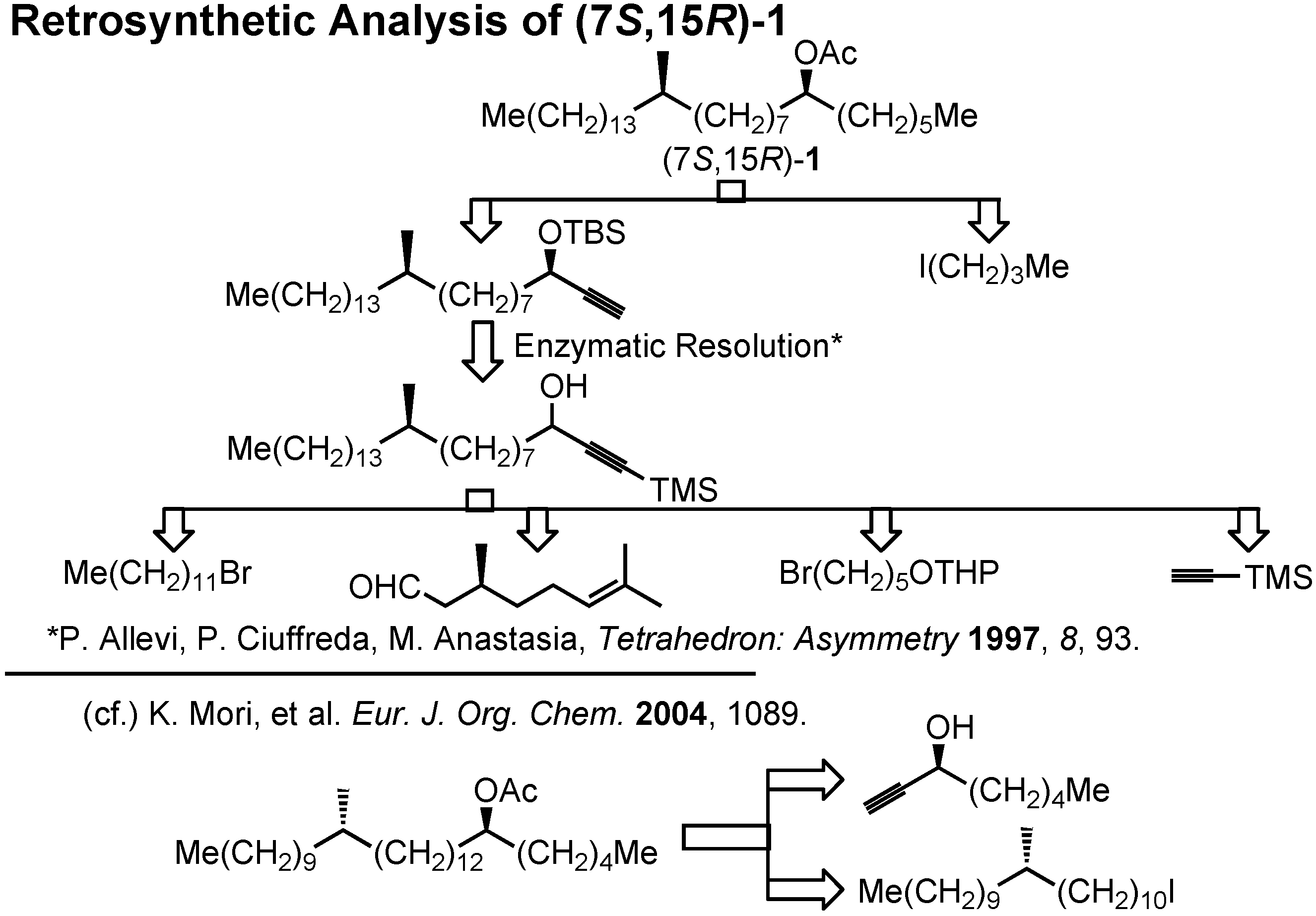
Here is our retrosynthetic analysis (
Figure 33). In the case of our recent synthesis of this acetate, we could employ commercially available enantiomers of 1-octyn-3-ol. But in the present case of
1, we could not depend on commercially available starting materials. Therefore, enzymatic resolution of an acetylenic intermediate was chosen as the key step.
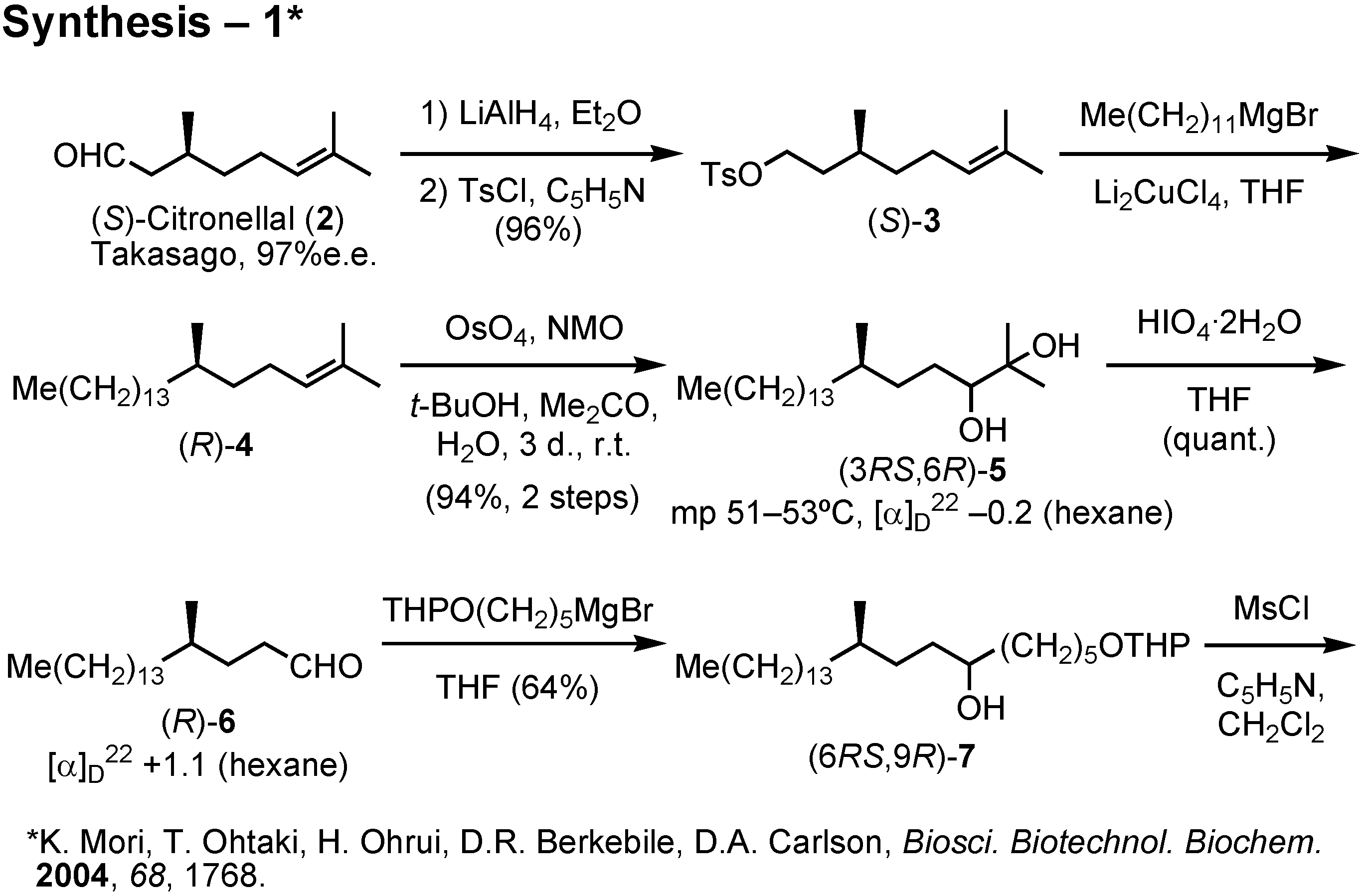
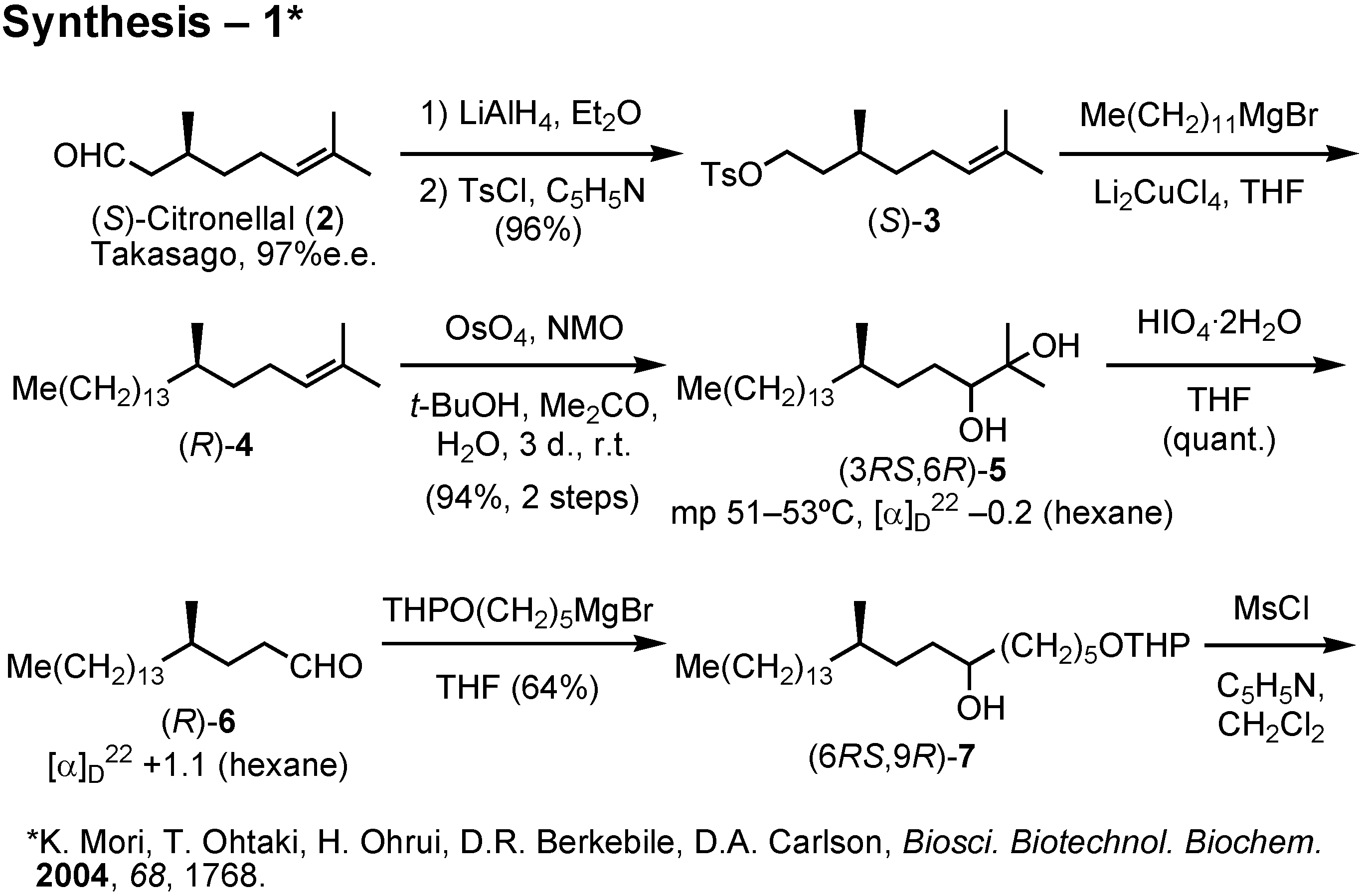
Lipase-catalyzed resolution of acetylenic alcohols is a known process as studied by Anastasia. The key acetylenic alcohol could be prepared from 1-bromododecane, citronellal, 5-tetrahydropyranyloxy- pentyl bromide and trimethylsilylacetylene. This synthesis (
Figure 34) was also carried out by me. (
S)-Citronellal was reduced with lithium aluminum hydride, and the resulting alcohol was tosylated to give
3. Chain-elongation of
3 with dodecylmagnesium bromide in the presence of dilithium tetrachlorocuprate under the Schlosser conditions afforded (
R)-
4. This was dihydroxylated with osmium tetroxide to give
5 as crystals. Glycol
5 was cleaved with periodic acid to give aldehyde (
R)-
6. Further chain-elongation of
6 with tetrahydropyranyloxypentylmagnesium bromide furnished
7, whose secondary hydroxy group was removed by reduction of the corresponding mesylate.
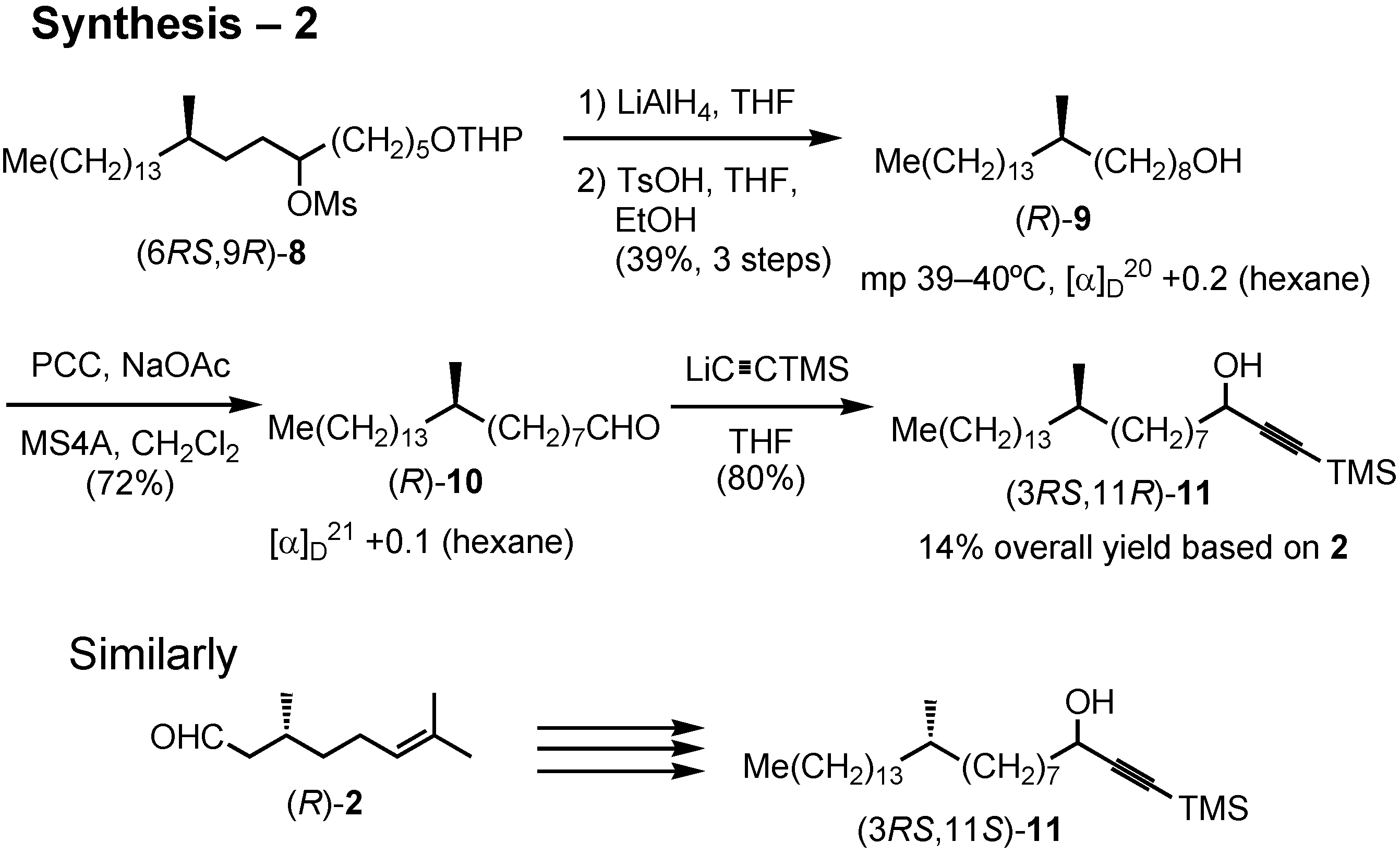
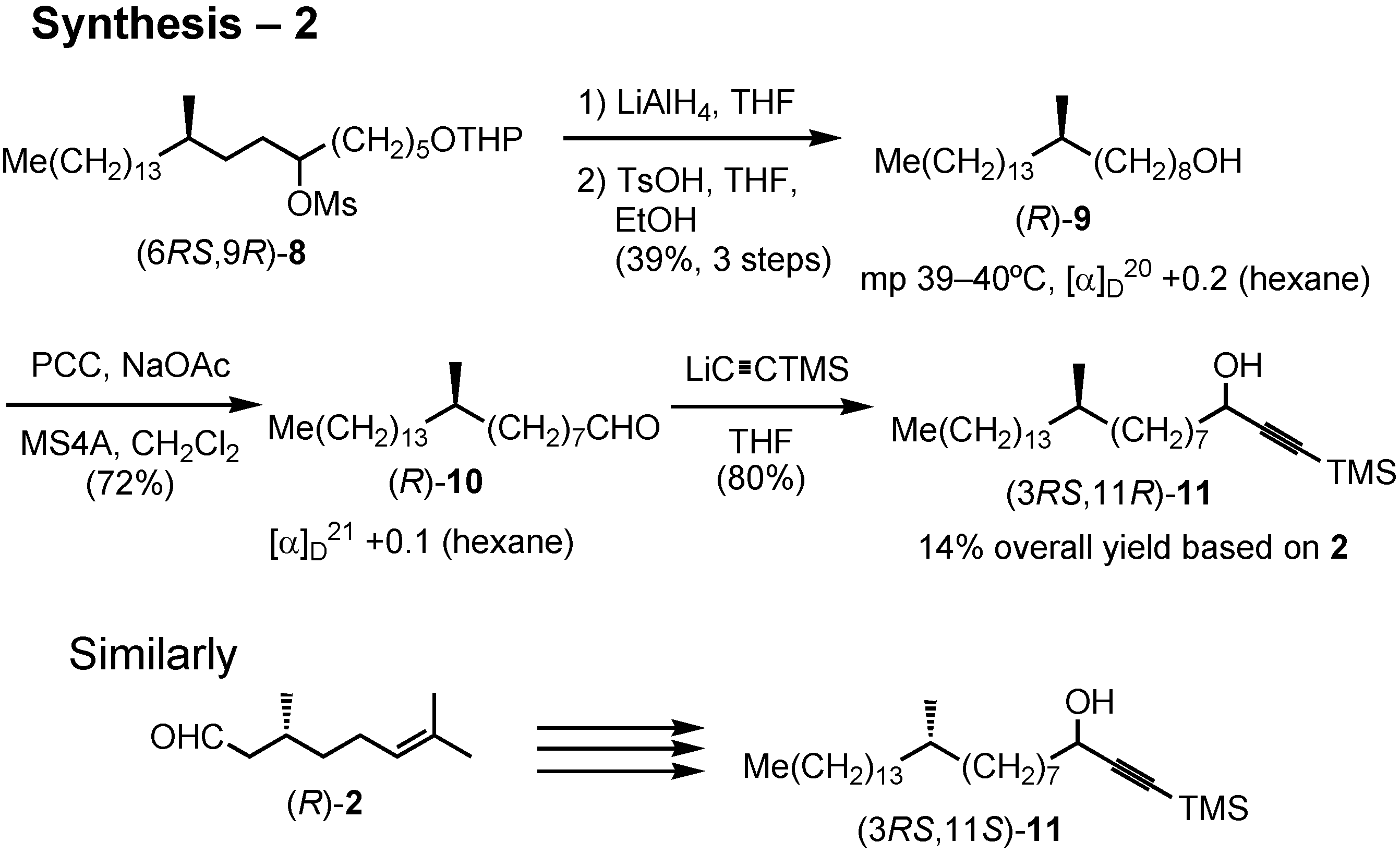
After removal of the THP protective group, crystalline alcohol (
R)-
9 was obtained (
Figure 35). PCC oxidation of (
R)-
9 gave aldehyde
10. Addition of lithium trimethylsilylacetylide to
10 gave acetylenic alcohol
11, the substrate for the enzymatic kinetic resolution. Similarly, its (3
RS,11
S)-isomer was synthesized from (
R)-citronellal.
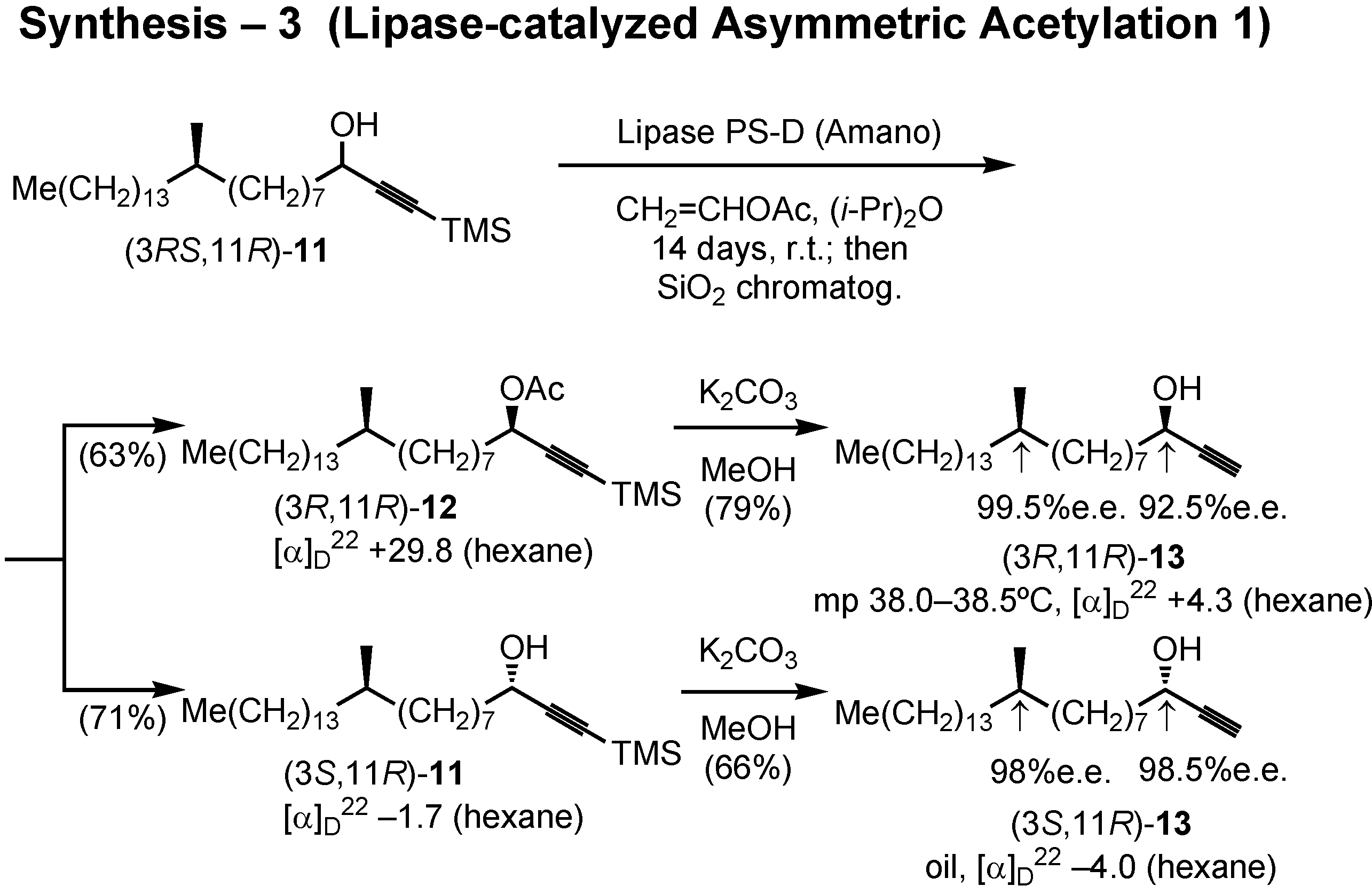
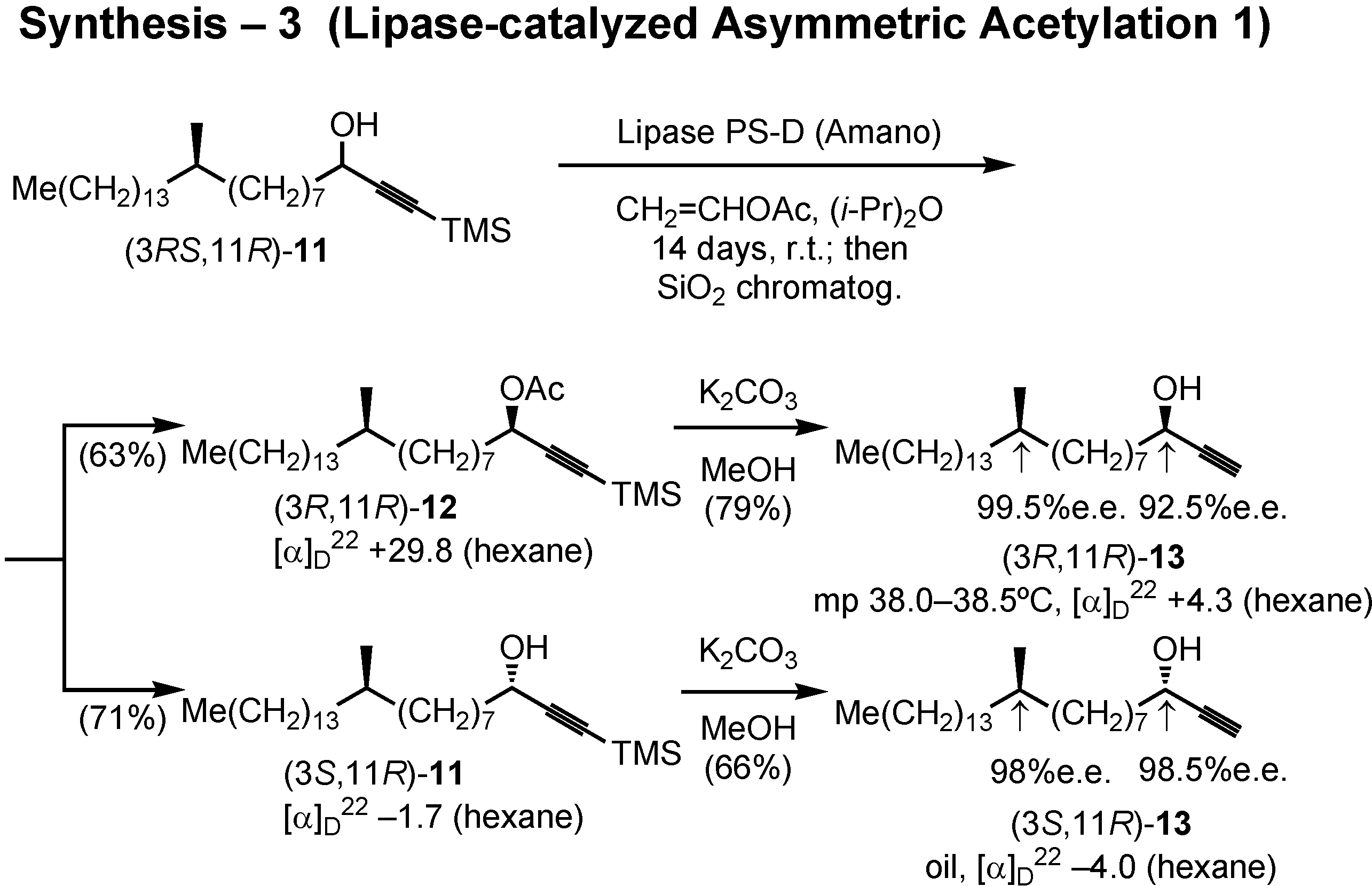
After some preliminary screening experiment, lipase PS-D (Amano) was chosen to achieve asymmetric acetylation of
11 (
Figure 36). Accordingly, lipase PS-D and vinyl acetate were added to a solution of (3
RS,11
R)-
11 in diisopropyl ether, and the mixture was stirred for 2 weeks at room temperature. The product was separated by silica gel chromatography to give the acetylated (
R)-acetate
12 and non-acetylated (
S)-alcohol
11. Treatment of
12 with potassium carbonate in methanol removed both TMS and acetyl protective groups to give alcohol (3
R,11
R)-
13. Similarly, (3
S,11
R)-
11 yielded (3
S,11
R)-
13. The stereochemical purity of these alcohols was satisfactory.
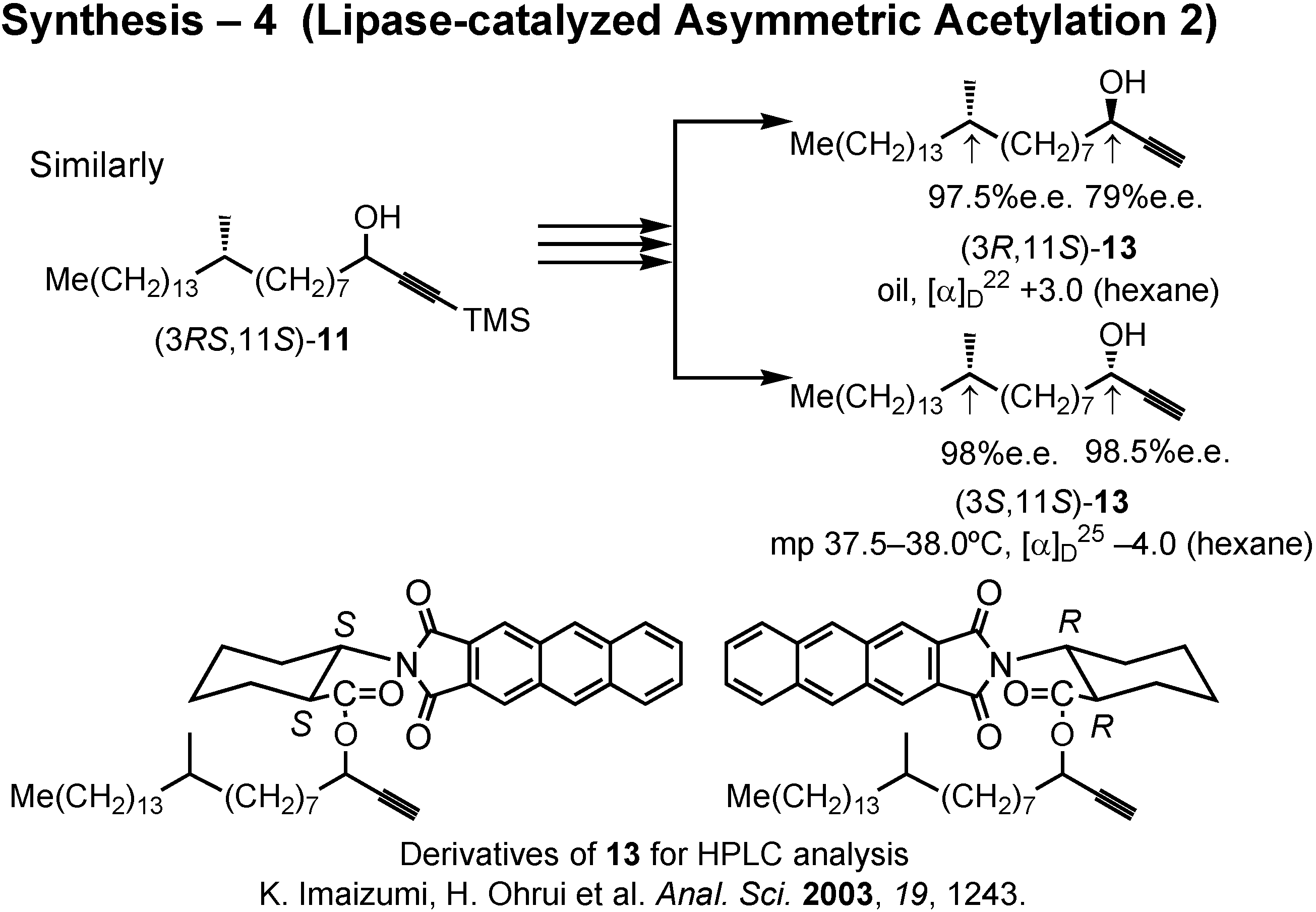
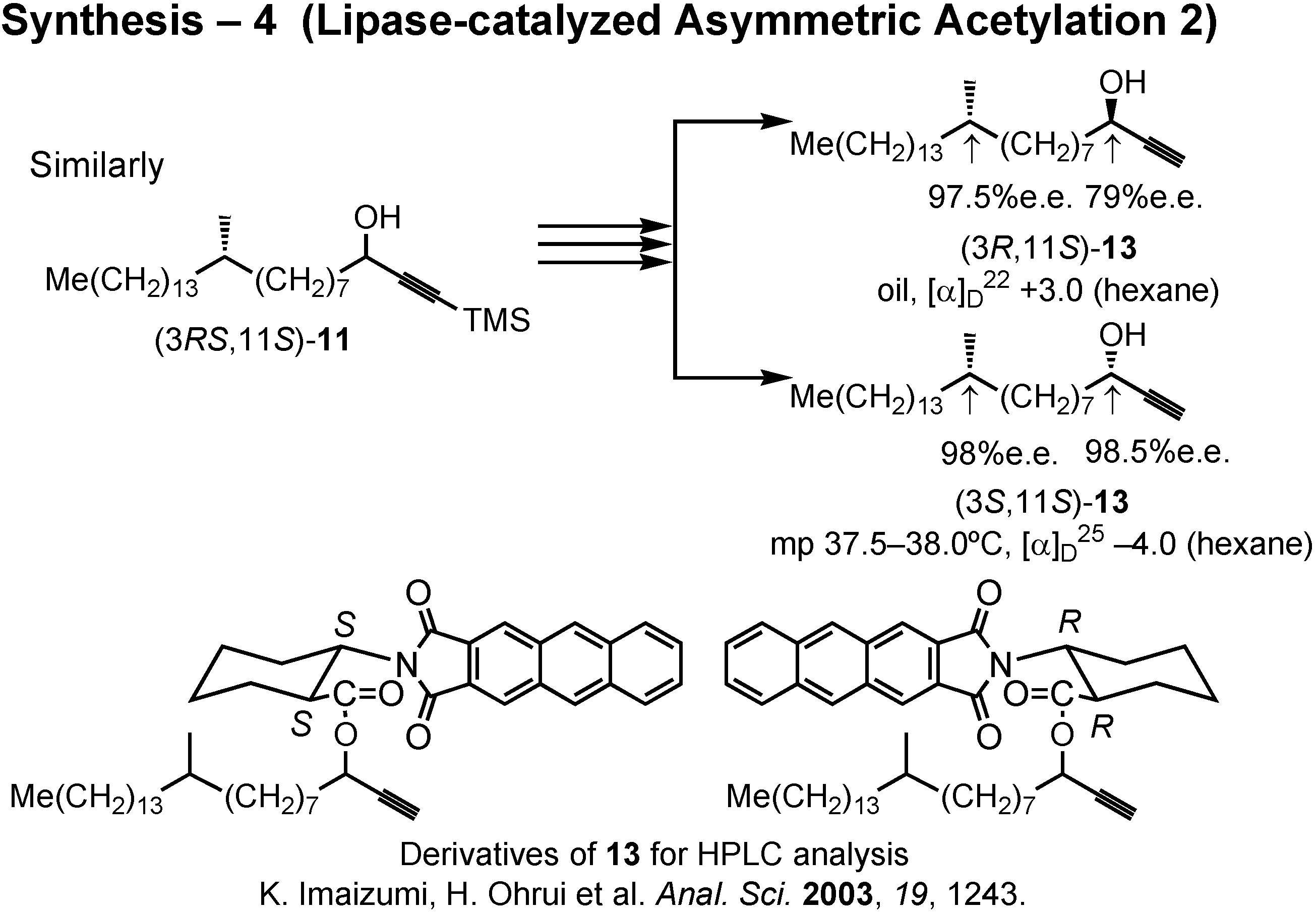
In the same manner, (3
RS,11
S)-
11 furnished (3
R,11
S)-
13 and (3
S,11
S)-
13 (
Fig.37). The stereochemical purity of these alcohols were determined by the HPLC analysis of these derivatives prepared by the method of Ohrui.
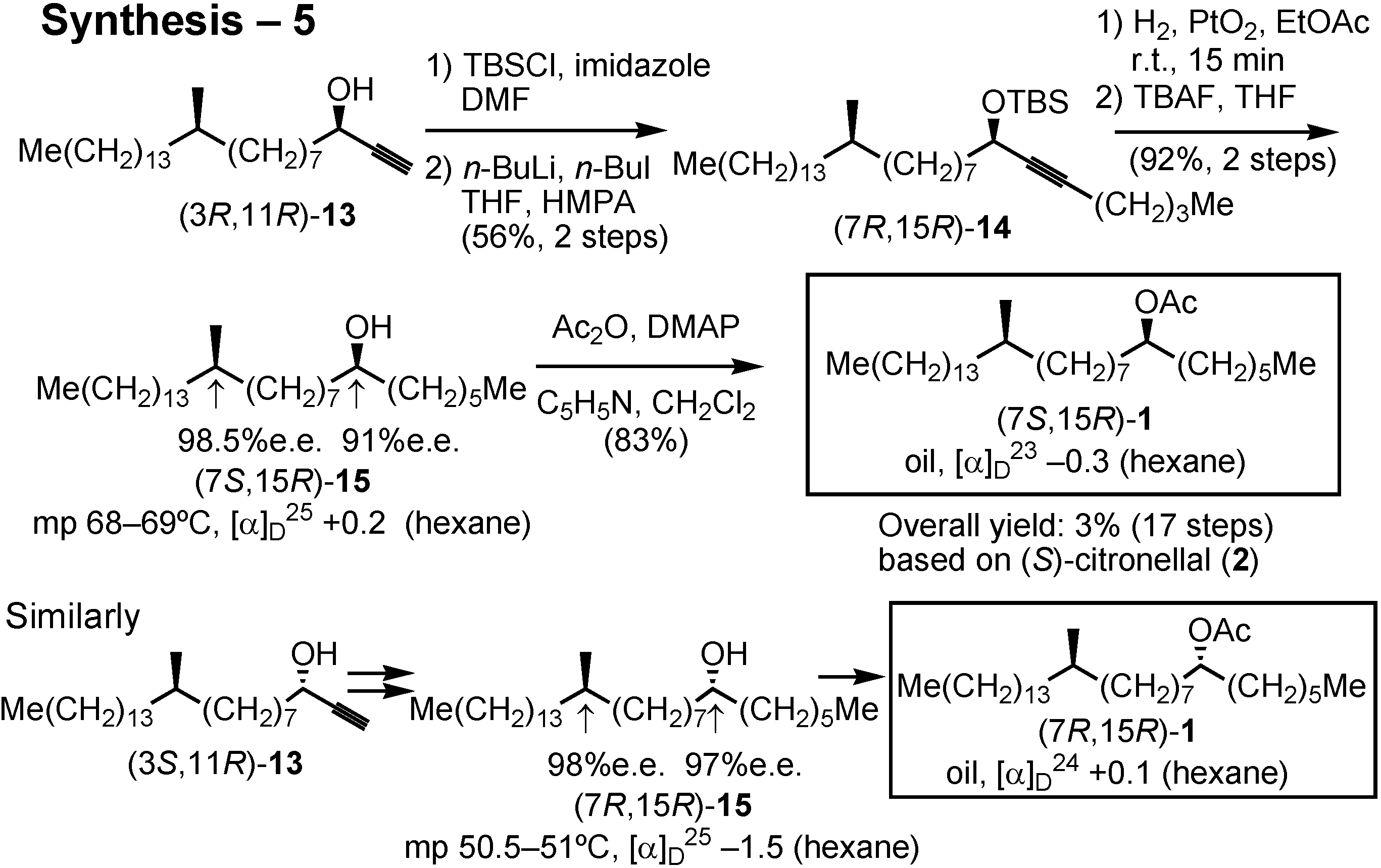
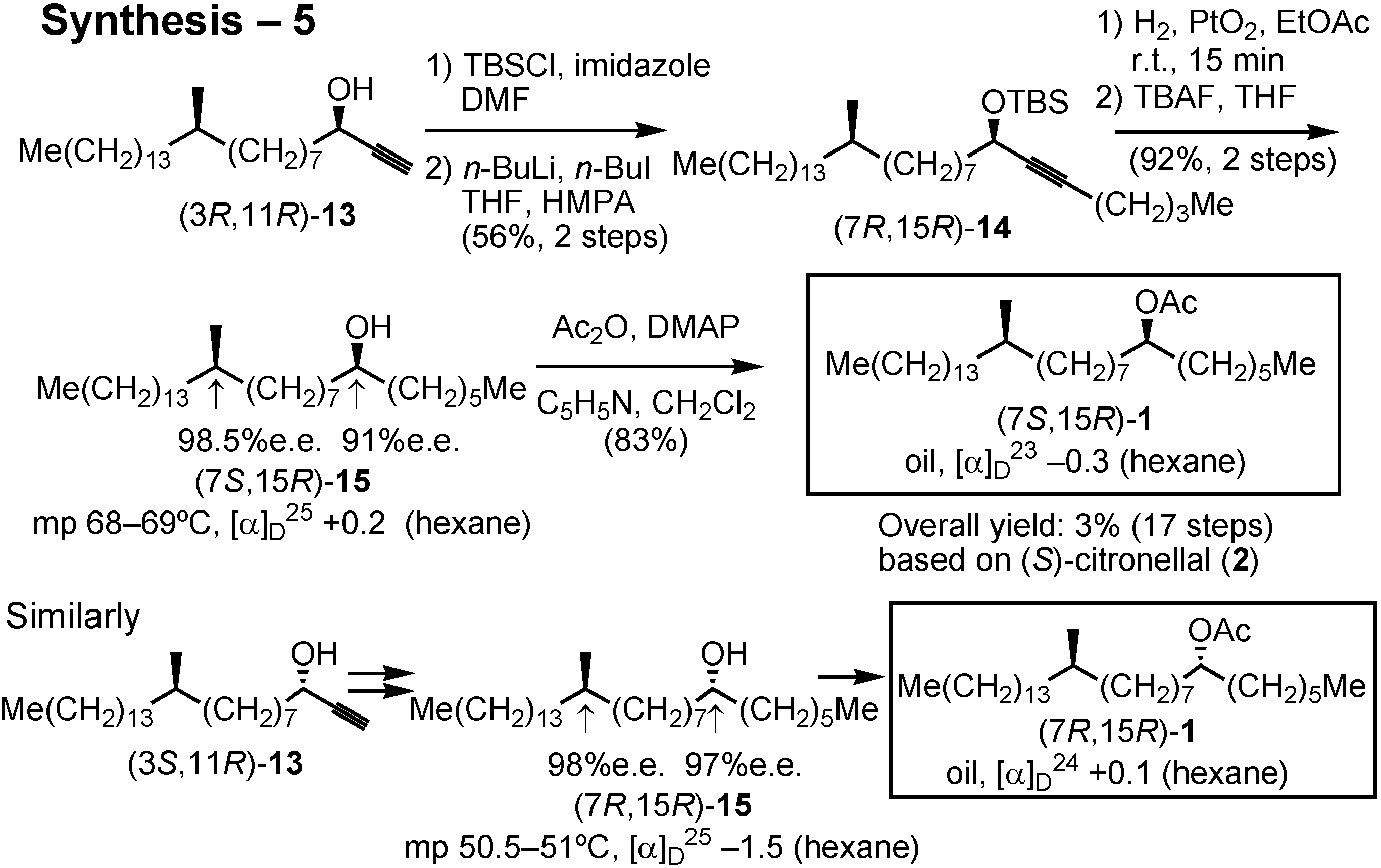
Conversion of acetylenic alcohol
13 to the final product
1 is shown in this scheme (
Figure 38). Chain-elongation of
13 was executed after converting it to the corresponding TBS ether by employing butyllithium and butyl iodide. The resulting
14 was hydrogenated over Adams’ platinum oxide, and the TBS protective group was removed to give alcohol
15 as crystals. Its stereochemical purity was also determined. Finally, acetylation of
15 gave the final product, acetate
1. Similarly, three other isomers were also synthesized.
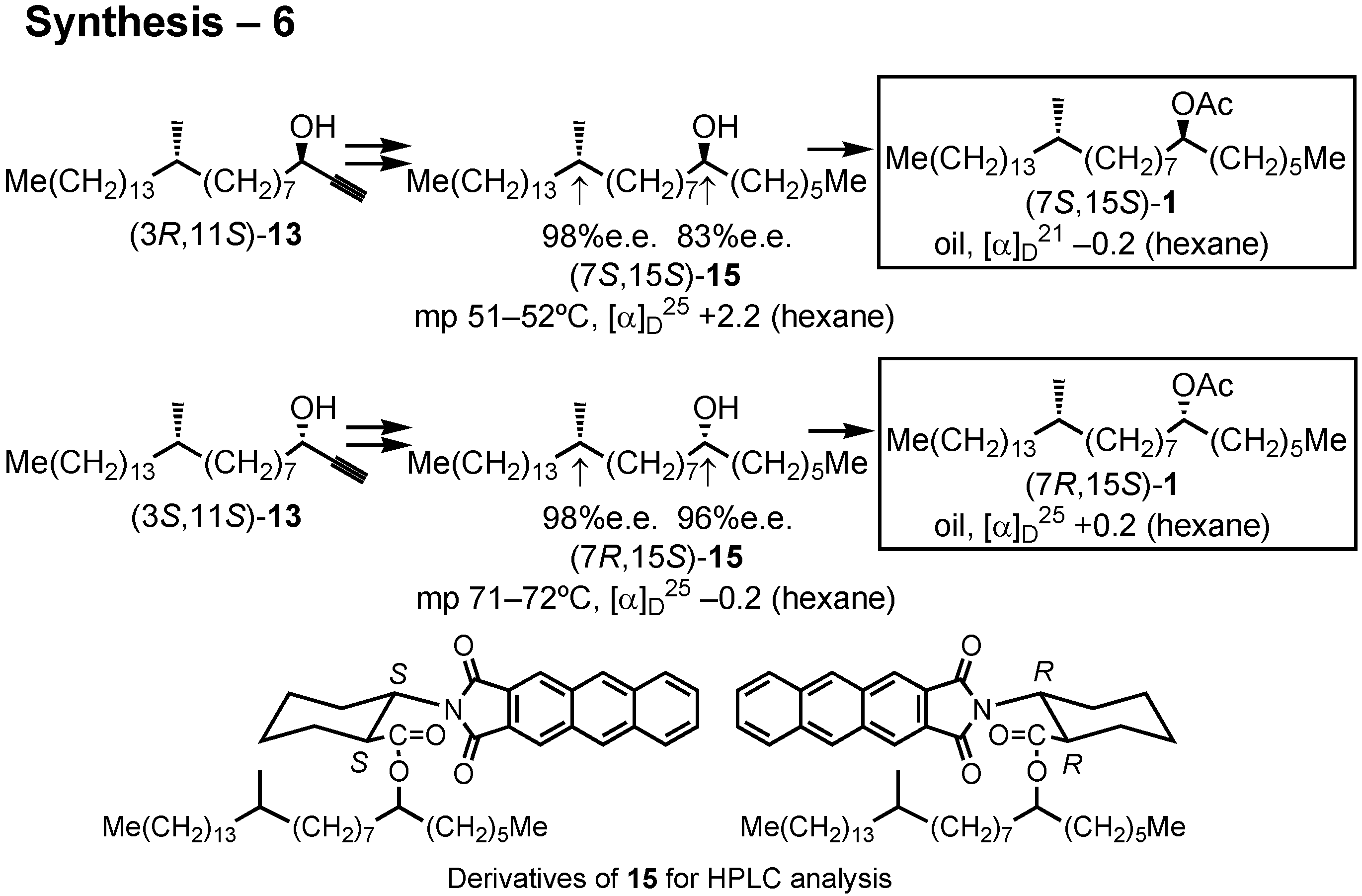
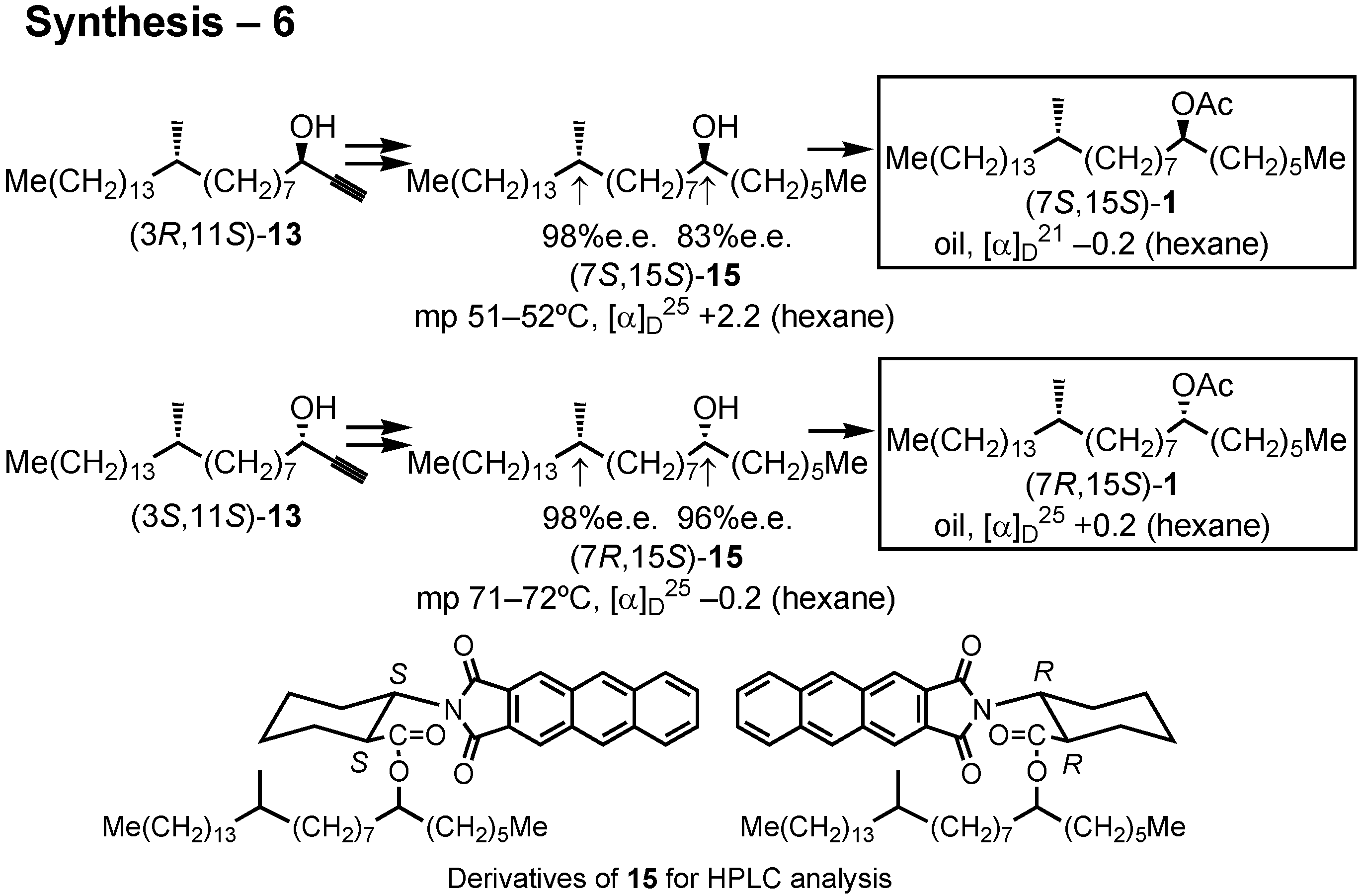
The stereochemical purity of these alcohols
15 were also determined by the HPLC analysis of their derivatives (
Figure 39). I thus synthesized all the four stereoisomers of the screwworm fly pheromone, and their bioassay was carried out in the USA. In this particular case, all of the four stereoisomers showed pheromone activity.
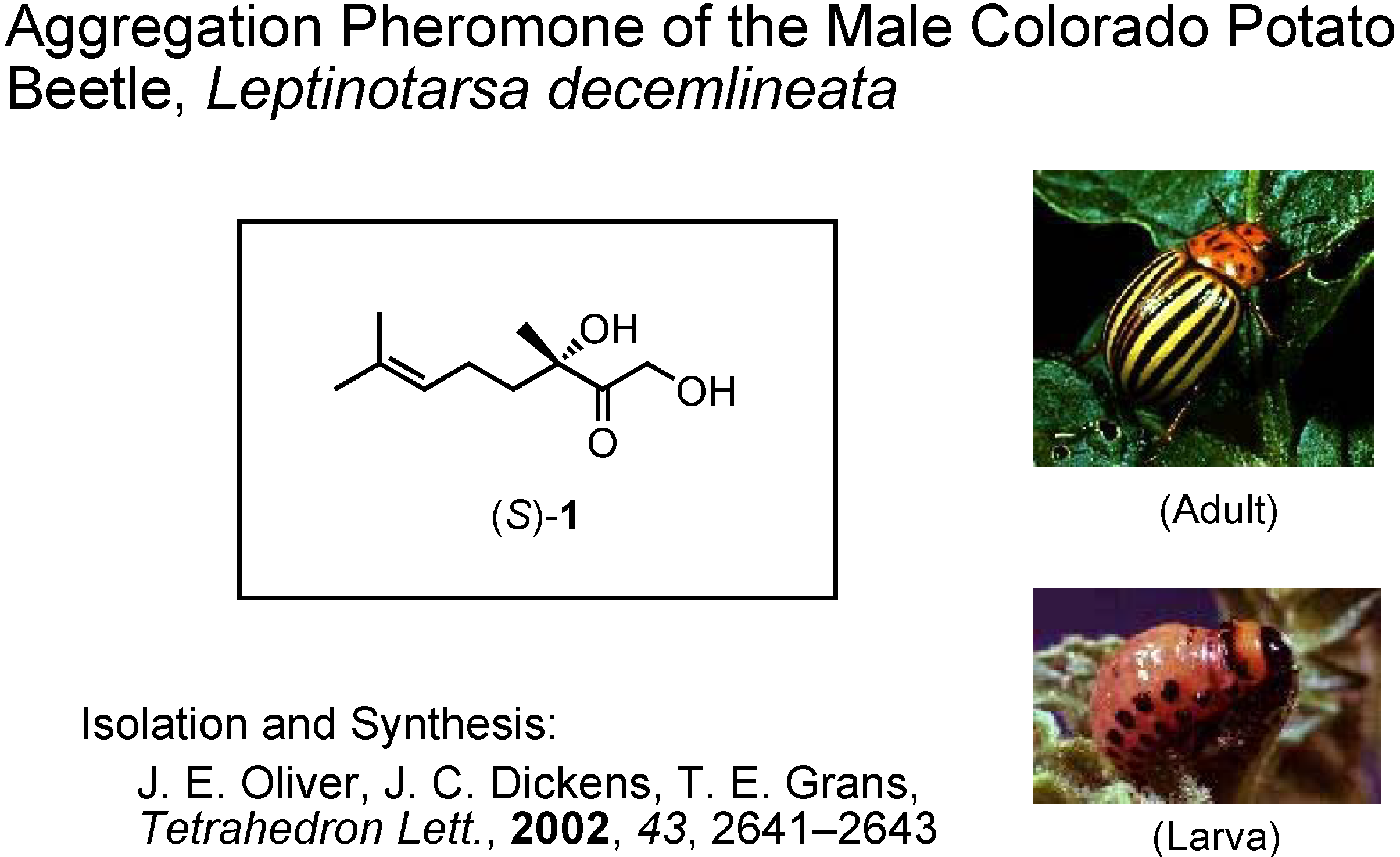
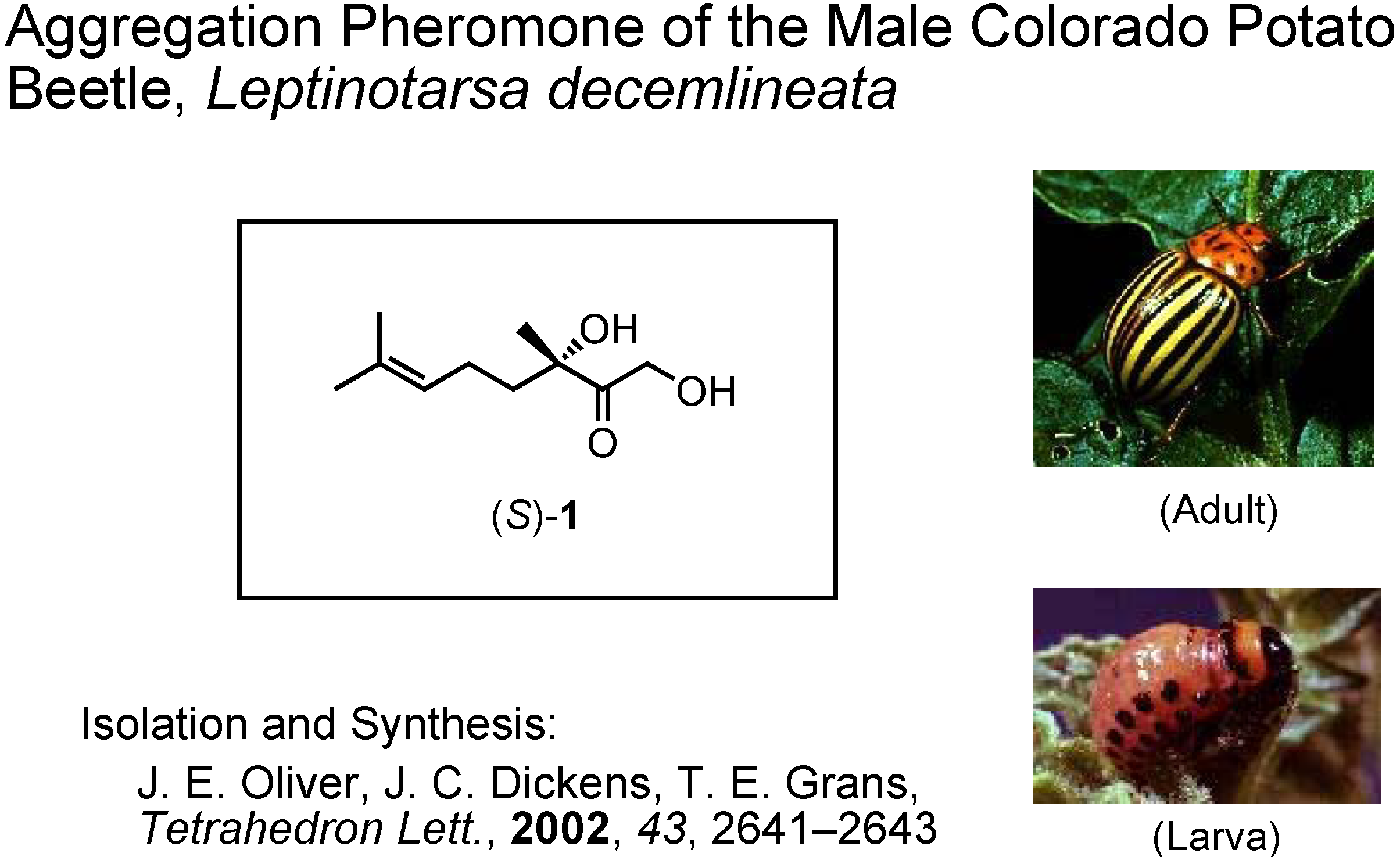
My fifth and last topic is the male-produced aggregation pheromone of the Colorado potato beetle,
Leptinotarsa decemlineata (
Figure 40). This is a pest insect of potatoes in the USA. Its isolation and synthesis were reported by Oliver at U.S. Department of Agriculture in 2002. They synthesized this unique monoterpene from (
S)-linalool, and established its absolute configuration as
S. Dr. Dickens at U.S. Department of Agriculture requested me to synthesize gram quantities of (
S)-
1, because their synthesis provided only mg quantities of it. The problem was how to make
S-stereocenter in a simple manner. We planned to use an enzymatic method. Dr. Tashiro mainly did the experimental work.
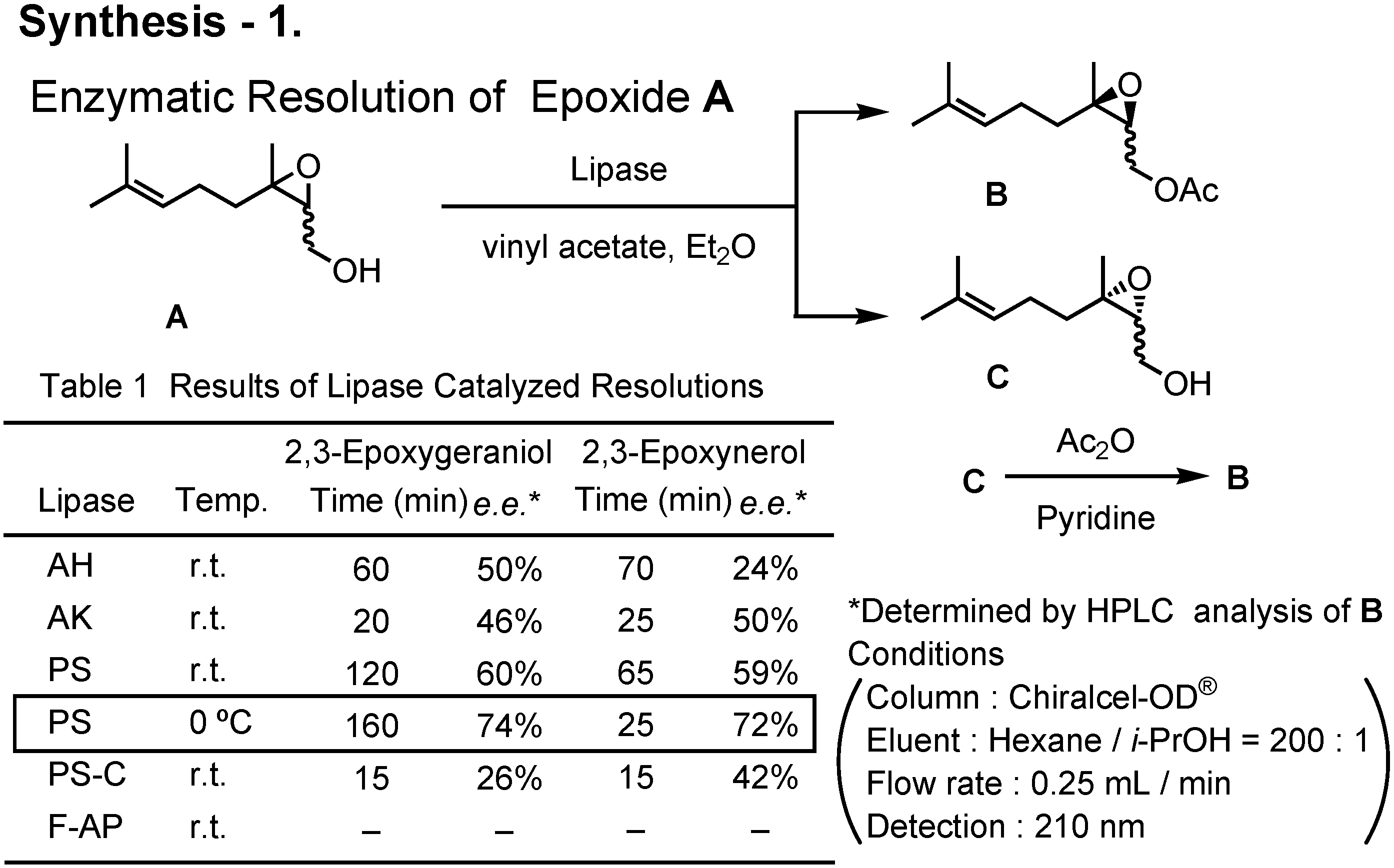
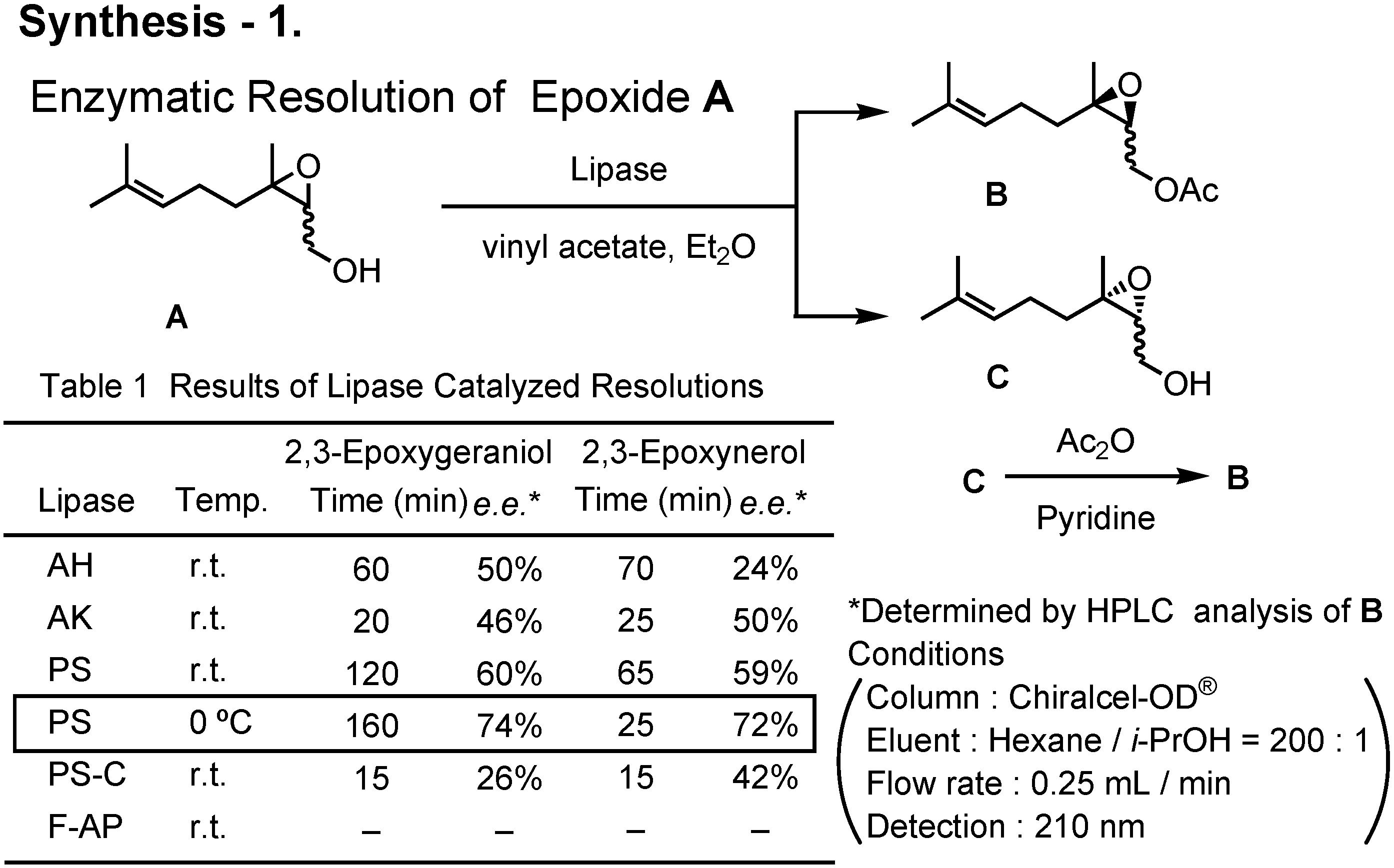
Various lipases were screened to achieve enzymatic kinetic acetylation of 2,3-epoxygeraniol and 2,3-epoxynerol (
Figure 41). When used at 0°C, lipase PS was found to be the best.
By repeating the lipase-catalyzed kinetic resolution three times, (2
S,3
R)-2,3-epoxyneryl acetate (
5) was prepared (
Figure 42). Its enantiomeric purity was about 97%
ee. Treatment of
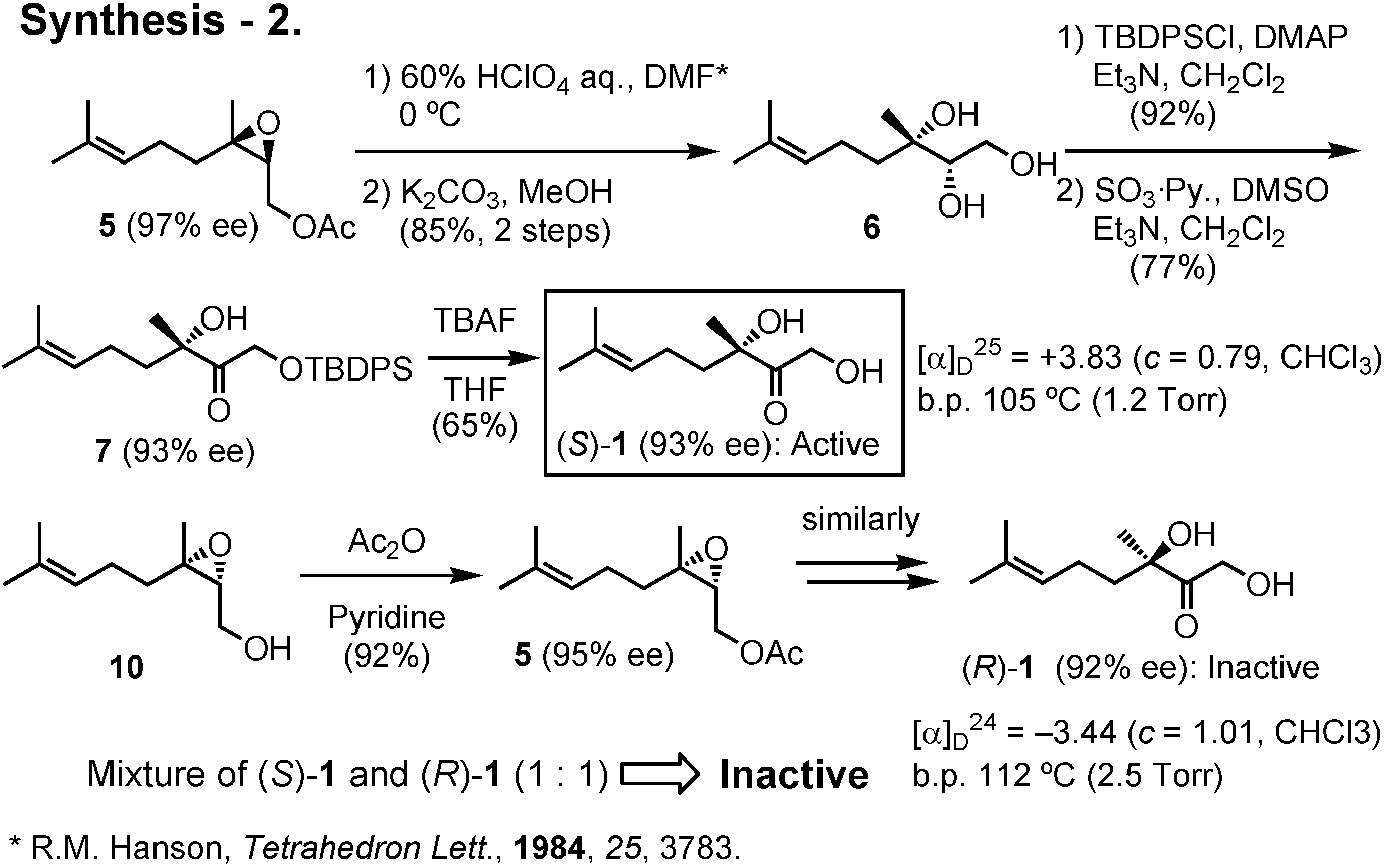
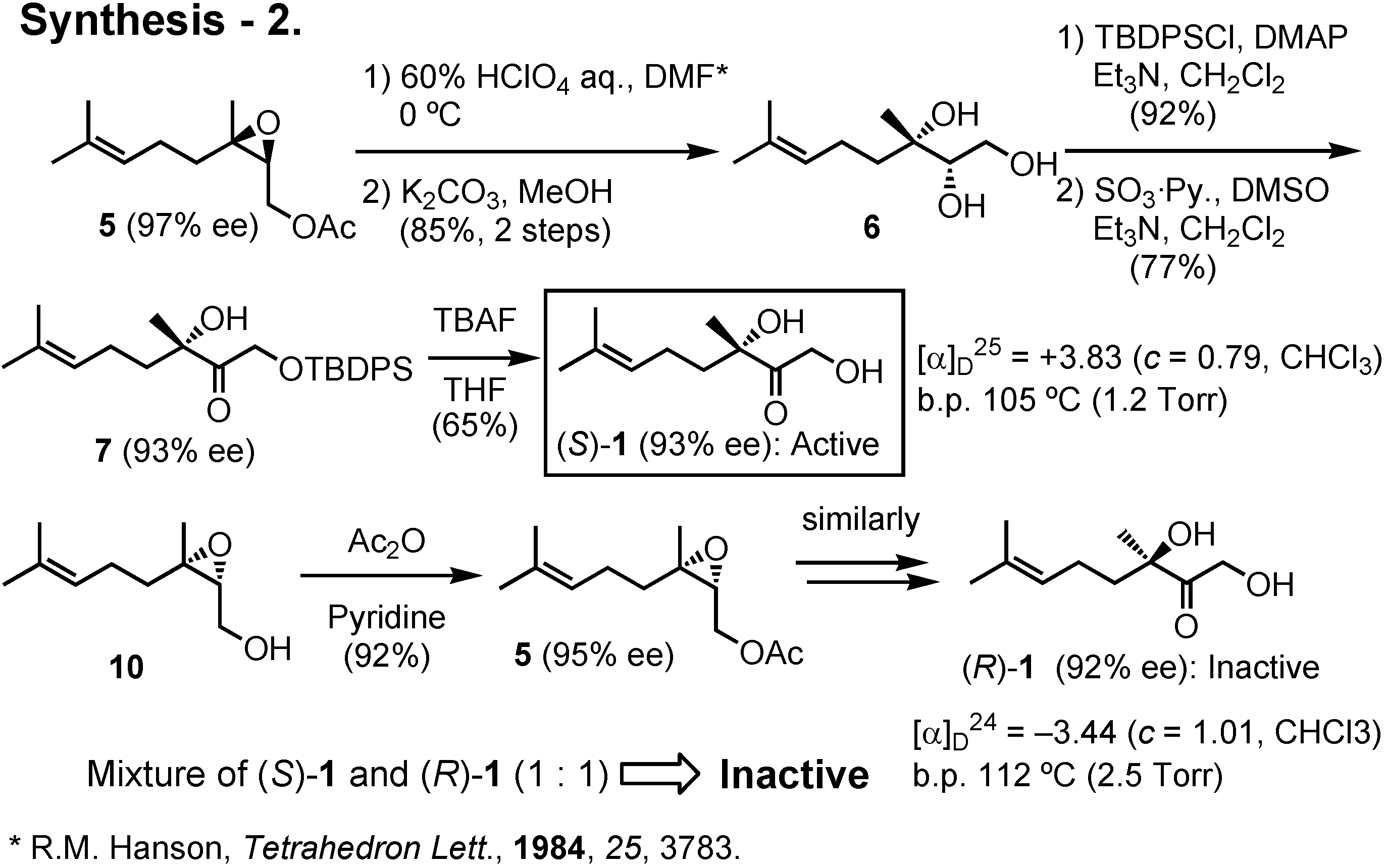
5 with 60% perchloric acid in DMF at 0°C gave (2
S,3
S)-
6 with Walden inversion at C-3. The primary hydroxy group of
6 was then protected as TBDPS ether, and the secondary hydroxy group was oxidized to give
7. Removal of the TBDPS protective group of
7 afforded the Colorado potato beetle pheromone (
S)-
1 in gram quantities. Its enantiomeric purity was 93%
ee. Similarly, (2
R,3
S)-
10 afforded (
R)-
1. Bioassay of these samples in the USA revealed that (
S)-
1 was active, while a 1:1 mixture of (
S)-
1 and (
R)-
1 was inactive. Thus (
R)-
1 seems to inhibit the activity of the natural pheromone with
S-configuration.
Now let me summarize my pheromone work (
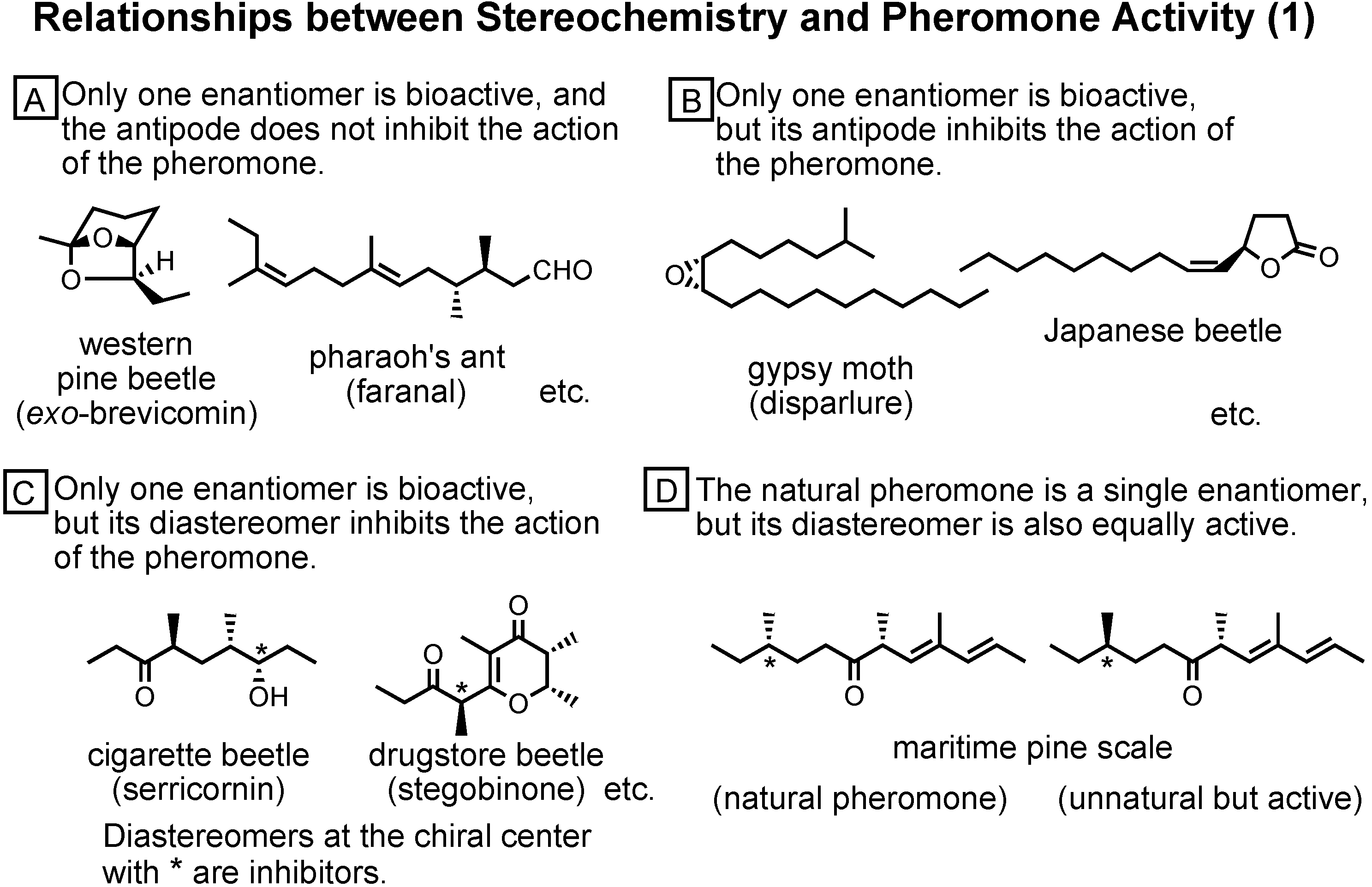
Figure 43). By synthesizing and biotesting the stereoisomers of pheromones, we could clarify the relationship between stereochemistry and pheromone activity. The relationship is not simple but complicated.
In group A, only one enantiomer is bioactive, and the antipode does not inhibit the action of the pheromone. This is common to other bioactive molecules.
In group B, only one enantiomer is bioactive, but its antipode inhibits the action of the pheromone. In the case of the Japanese beetle pheromone, its racemate is inactive.
In group C, only one enantiomer is bioactive, but its diastereomer inhibits the action of the pheromone. We must prepare diastereomerically pure pheromones.
In group D, the natural pheromone is a single enantiomer, but its diastereomer is also equally active. The stereochemical recognition at the receptor site seems to be more flexible.
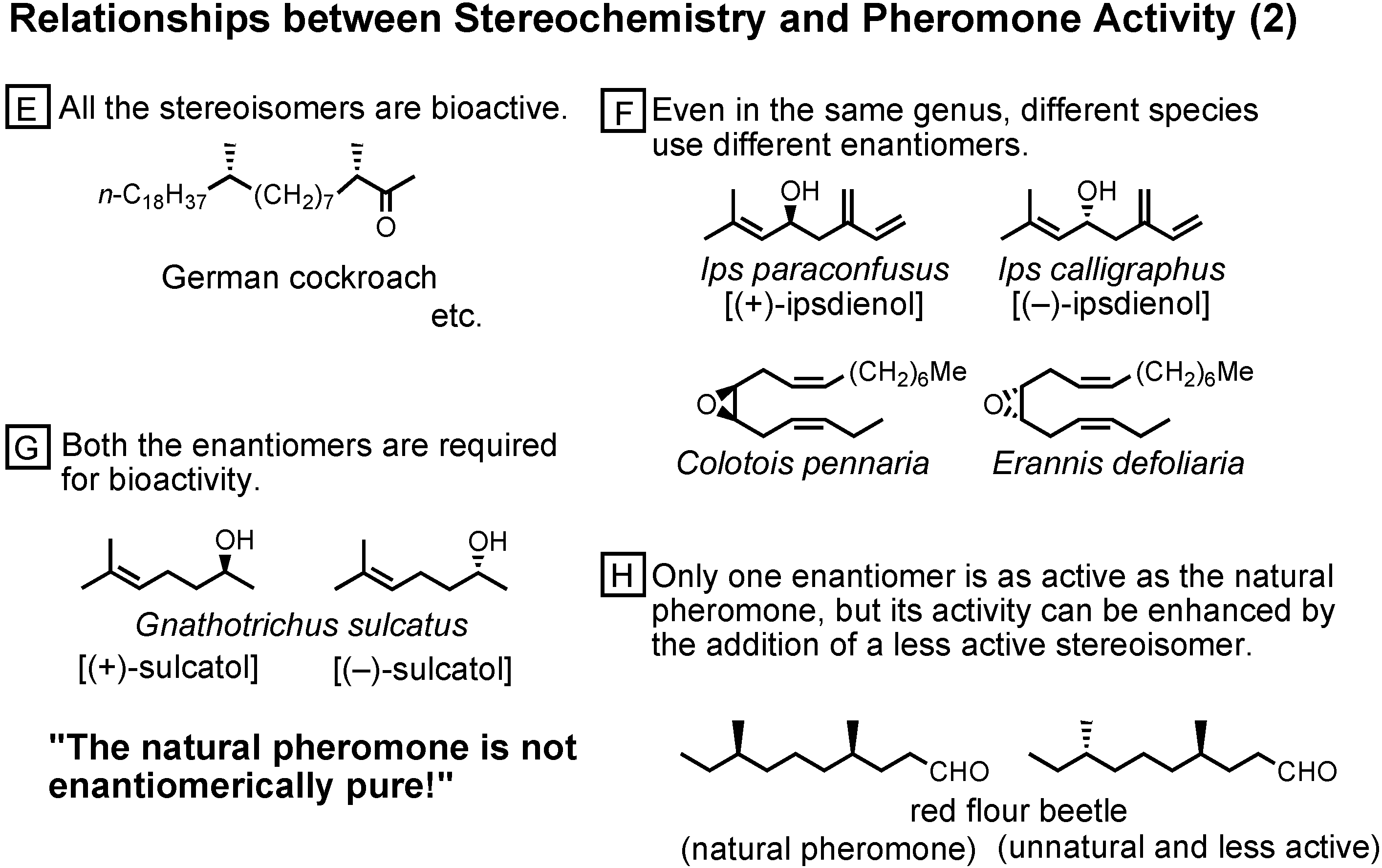
In group E, all the stereoisomers are bioactive (
Figure 44). The pheromone receptor must be very flexible.
In group F, even in the same genus, different species use different enantiomers. Enantiomerism is employed to avoid mixed marriages.
In group G, both the enantiomers are required for bioactivity. To show bioactivity, both the enantiomers must exist.
In group H, only one enantiomer is as active as the natural pheromone, but its activity can be enhanced by the addition of a less active stereoisomer. I cannot understand the reason why.
In group I, one enantomer is active on male insects, while the other is active on females (
Figure 45). In this particular case of olive fruit fly, the female-produced sex pheromone is a racemate.
In group J, only the meso-isomer is active, while the enantiomers are inactive.
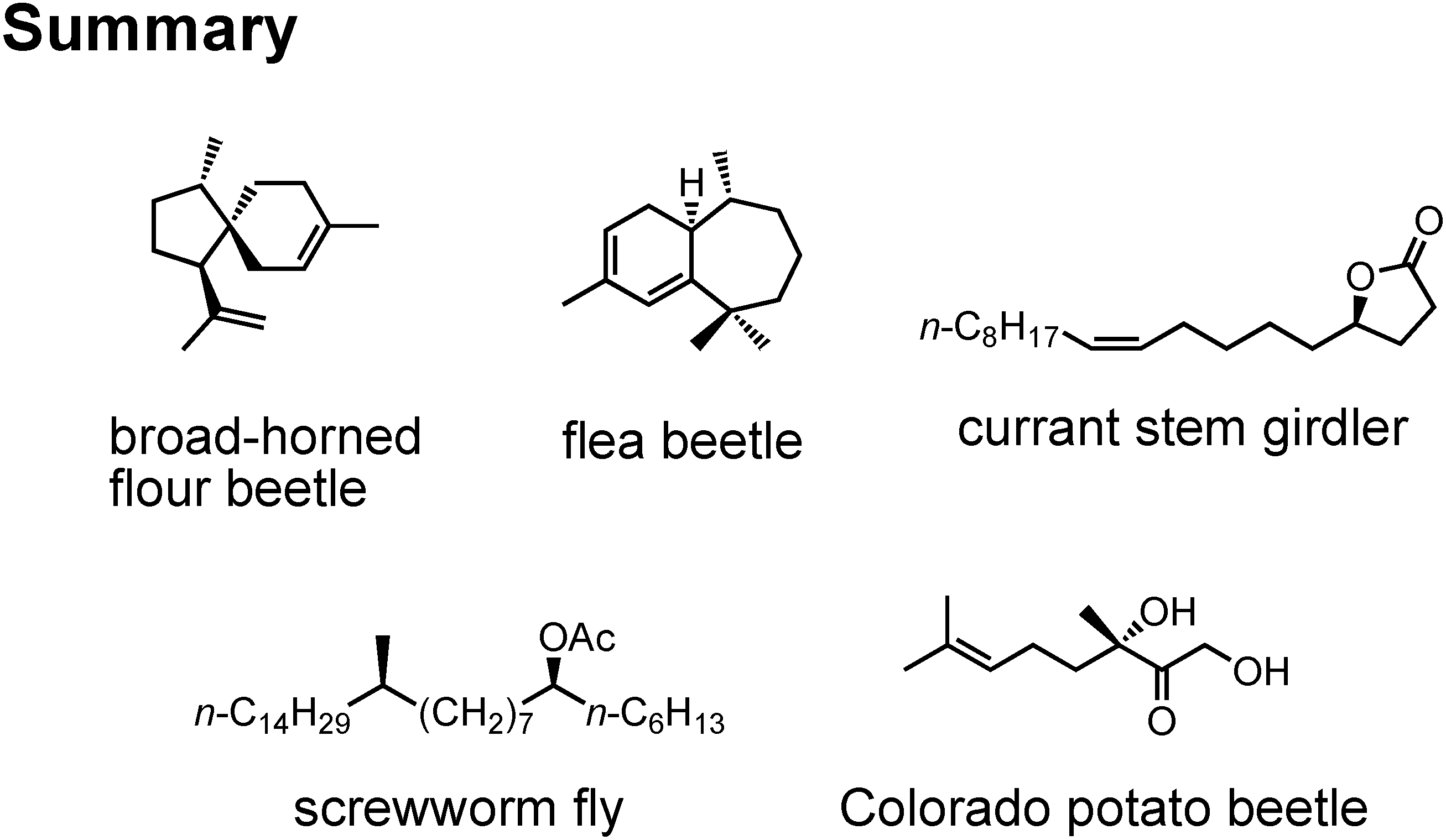
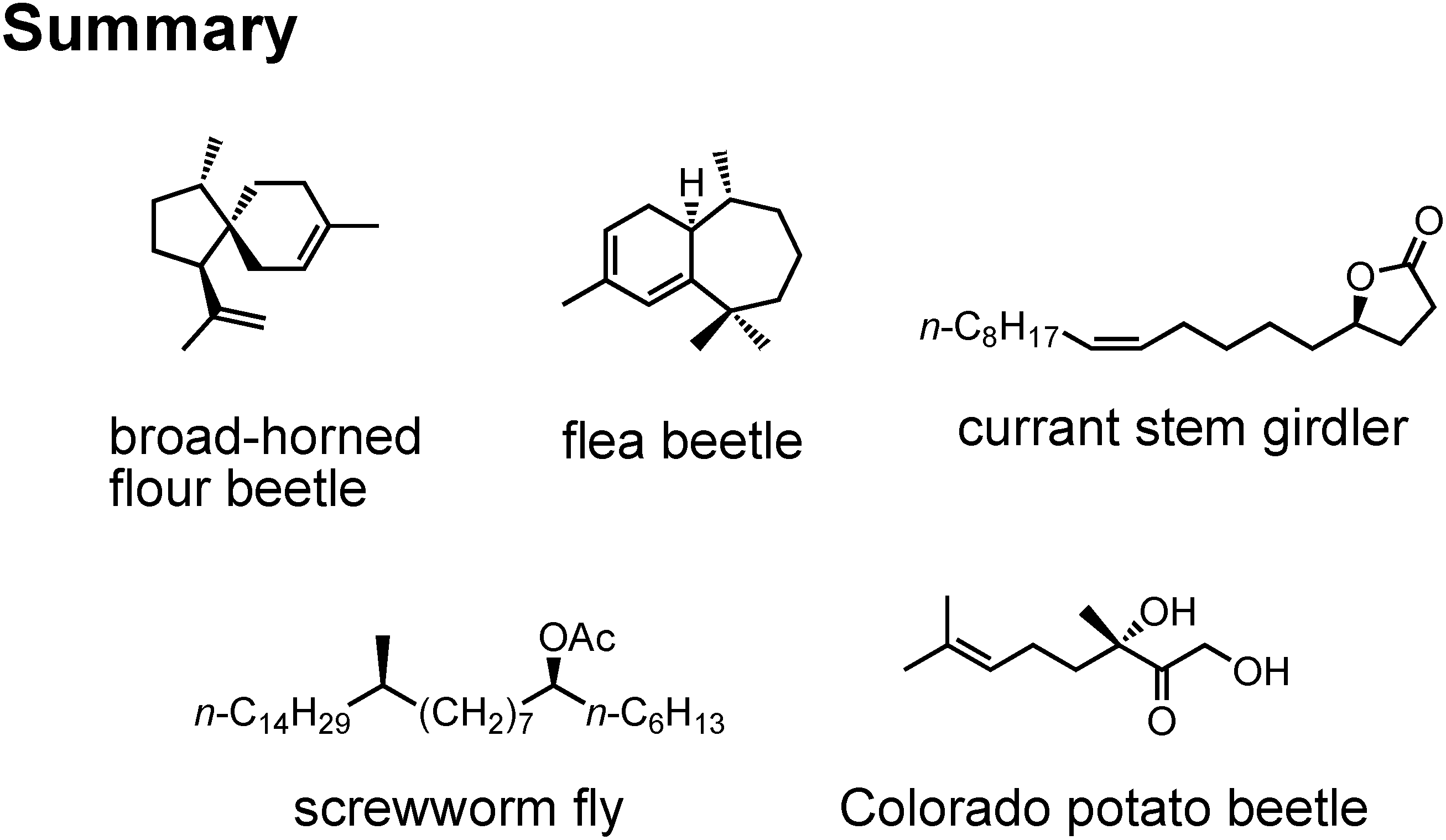
I have talked on the synthesis of these five new pheromones (
Figure 46).
Enantioselective synthesis is possible by employing (1) enantiopure starting materials, (2) chemical asymmetric reactions and (3) enzymatic asymmetric reactions (
Figure 47). Criteria for choosing one of them are (1) selectivity, (2) efficiency, and (3) simplicity. Our goal is to provide useful materials to others. In summary, diversity is the keyword of pheromone response. In the case of chiral pheromones, we must synthesize and bioassay stereoisomers to know the stereochemistry-bioactivity relationships. Only after that, we may be able to think about the practical use of pheromones. The diversity in pheromone response could be clarified only through experimental efforts of chemists and biologists. I thank all of my co-workers to provide synthetic samples and then to bioassay them.
Thank you for your attention.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
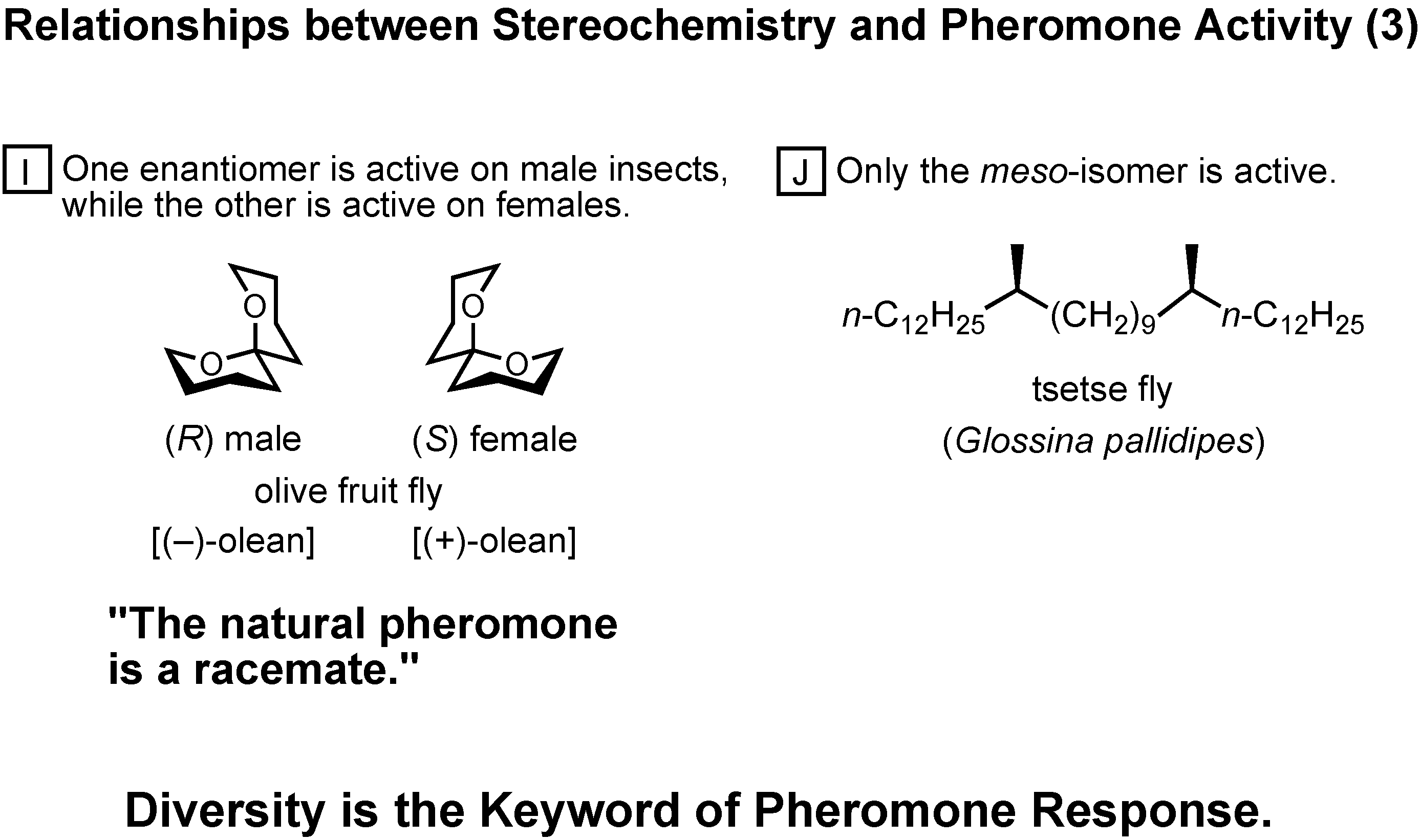{kind=link}
{kind=link}
{kind=link}