Abstract
For the first time ent-labdanes have been synthetised starting from ent-halimic acid, following a route that is the reverse of the biosynthetic one leading to the former compounds.
Introduction
ent-Halimic acid (Figure 1) is a bicyclic diterpene that has been used as a starting material for the synthesis of different bioactive molecules such as the ent-halimanolides [1], chettaphanin I and chettaphanin II [2], (+)-agelasine C [3] and sesterterpenolides with an disydiolane skeleton [4].
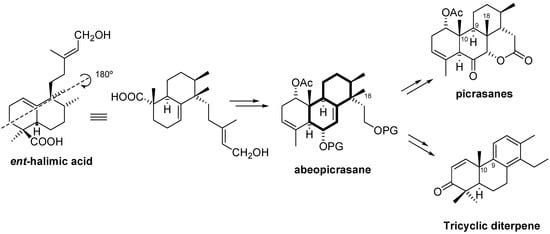
Due to its functionality, ent-halimic acid can be viewed as an excellent synthon for the synthesis of new natural products. For example, the bicyclic system of ent-halimic acid can be visualized as the basis for the BC rings of tricyclic diterpenes or quassinoids with a picrasane skeleton (Scheme 1) whereby the C-4 quaternary carbon and the C-5 carbon of the starting material are incorporated with the correct configuration as C-10 and C-9, respectively, in the targets. However, to the best of our knowledge, a diterpene of the antipodal series has not been used previously as a starting material for the synthesis of diterpenes of the normal series or quassinoids with a picrasane skeleton.
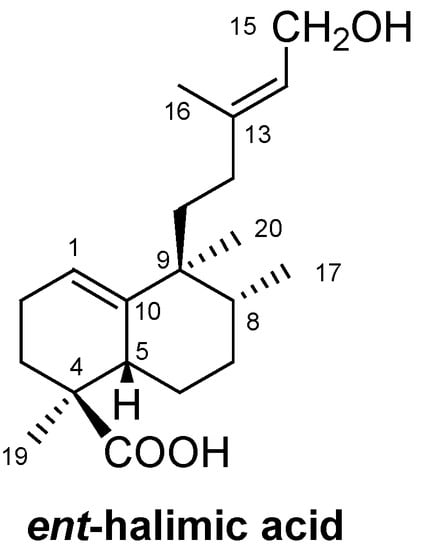
Figure 1.
Scheme 1.
As can be observed in Scheme 1, the synthesis of the picrasane skeleton would require a rearrangement of the abeopicrasane skeleton, namely a 1,2 migration of the Me-18 group, in the same manner as seen in the biosynthetic pathway of euphane and tirucallane, biosynthetic precursors of the quassinoids [5].
Scheme 2.
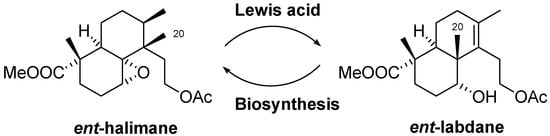
Scheme 2 shows the 1,2-shift of the Me-20 of ent-halimanes that leads to ent-labdanes. A rearrangement of this kind would be required to access the picrasane skeleton quassinoids. This rearrangement would be the opposite of the biosynthetic route, in which the ent-labdanes are the precursors of ent-halimanes [6].
In the present work we have explored the use of ent-halimic acid for the synthesis of biologically active compounds with different carbon skeletons. In particular we have studied the rearrangements with different Lewis acids of epoxides 3 and 4 (Figure 2), derived from ent-halimic acid.
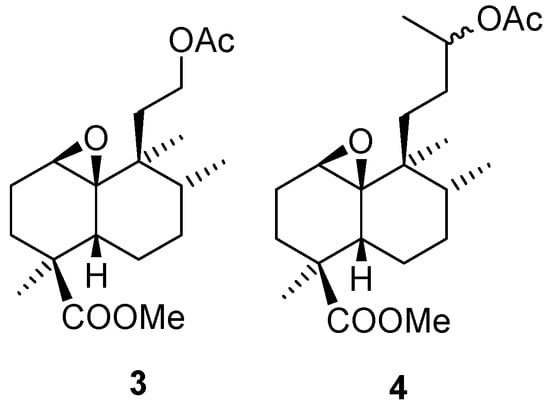
Figure 2.
Starting materials obtained from ent-halimic acid.
Results and Discussion
Synthesis of the starting materials 3 and 4.
The starting materials 3 and 4 were obtained from compound 1, the methyl ester of ent-halimic acid, via two or four carbon atom side chain degradations, respectively, with methylketone 2 being the key intermediate in both syntheses (Scheme 3).
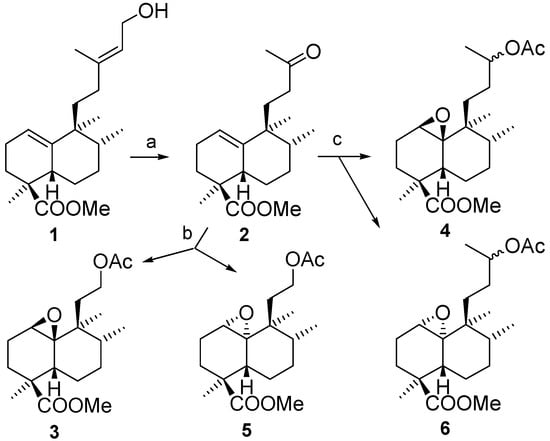
Scheme 3.
Treatment of 2 with urea-hydrogen peroxide and trifluoroacetic acid (UHP-TFAA) [7] for one hour gives the epoxyacetates 3 and 5 with high stereoselectivity as a result of a Baeyer-Villiger reaction and epoxidation of the double bond. By contrast, the reaction of 2 with m-CPBA was slow, gave low yields and is non-stereoselective. The configurations of the epoxide groups were established by nOe experiments, as shown in Figure 3. While in compound 3 H-1 has an nOe with Me-17 and Me-20, in 5 a nOe can only be observed between H-1 and Me-20.
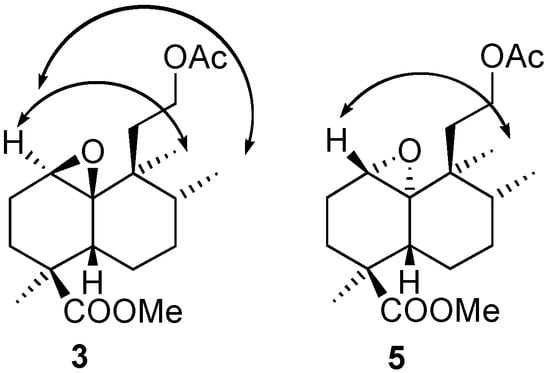
Figure 3.
nOe experiments for determination of the configurations of epoxides 3 and 5.
In order to obtain compound 4 it was necessary to reduce the ketone in 2, followed by acetylation to give the corresponding acetate, which was treated with UHP-TFAA, under the same conditions as before, to give the corresponding epoxides 4 and 6 in excellent yield and with good stereoselectivity.
Treatment of epoxide 4 with Lewis acids
Corey et al. studied the rearrangement of epoxides in their synthesis of gicinoelepin A [8] and after using different Lewis acids and reaction conditions, achieved the rearrangement of an angular methyl with FeCl3-Ac2O. With this precedent in mind we started our study by treatment of epoxide 4 with FeCl3-Ac2O, and after several minutes a mixture of compounds 7 (20%) and 8 (56%) was obtained (Scheme 4).
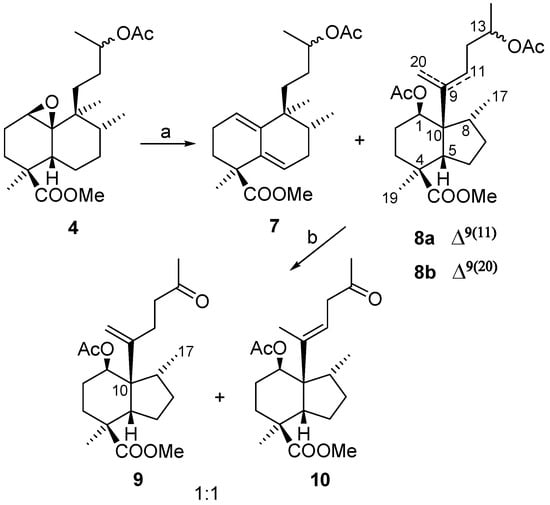
Scheme 4.
The less polar compound, 7, was identified as a heteroannular diene (U.V.: 238 nm) that could arise through the mechanism shown in Figure 4.
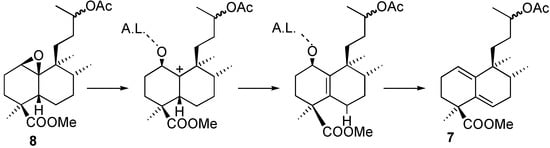
Figure 4.
Proposed mechanism for the formation of 7.
The more polar compound 8 was a mixture of compounds which proved very difficult to separate by chromatography, so it was decided to simplify the analysis by carrying out the hydrolysis of the acetate group, followed by oxidation with TPAP/NMO [9], to thus obtain compounds 9 and 10 in good yield. The structures of these compounds were established by HMQC and HMBC experiments. The presence of an olefin and a methyl on a methine indicates that a skeletal rearrangement has taken place in the reaction of 4 with the Lewis acid. The observed correlation in the NMR experiments between Me-17 and C-10, tell us that B ring of ent-halimane has undergone a contraction to produce a five membered ring, giving a perhydroindene system. The mechanism that explains the formation of these compounds is shown in Figure 5.
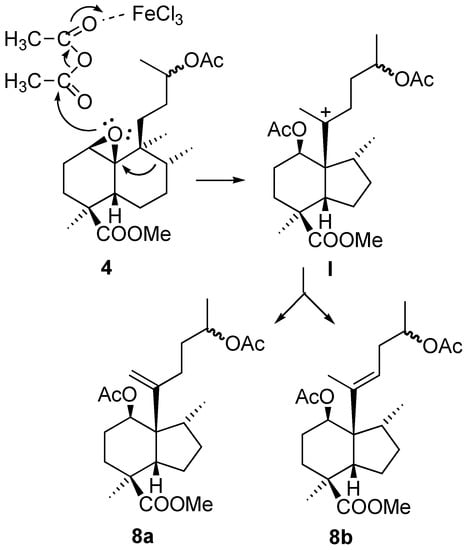
Figure 5.
Proposed mechanism for the formation of 8.
The Lewis acid coordination with acetic anhydride promotes a concerted rearrangement to produce olefins 8a and 8b. The stereochemistry for C-1, C-8 and C-10 is proposed in accordance with the shown mechanism. In a ROESY experiment of 10 (Figure 6) nOes can be observed between H-1 and Me-17, H-5 and Me-20, H-11 and H-2β and finally H-12 and Me-20, which corroborate the proposed configurations.
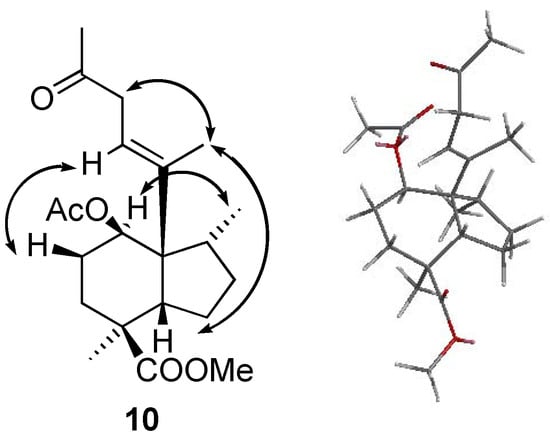
Figure 6.
nOe experiments for determination of the configuration of 10 and a Chem 3D model to better understand the nOes.
To test other Lewis acids compound 4 was treated with BF3.Et2O and TiCl4, The best results are shown in Scheme 5. Treatment of 4 with BF3.Et2O in ether gave a mixture of compounds 7, 11, 12 and 13, with ketone 11 as the major product. The synthesis of this ketone can be rationalized by the opening of the oxirane by the Lewis acid and a subsequent α-face C-1→C10 hydride rearrangement. Compounds 12 and 13 are perhydroindenes, analogues of 9 and 10, with the hydroxy group of C-1 forming a δ-lactone with the methoxycarbonyl in C-18.
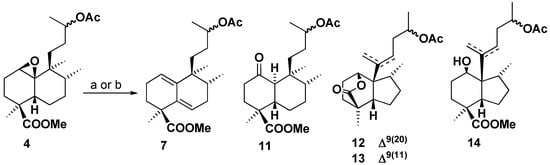
Scheme 5.
Treatment of 4 with TiCl4 gave similar results as those obtained with BF3.Et2O, but in this case the corresponding hydroxyester 14 was obtained as well. As can be seen, the rearrangement of compound 4 with different Lewis acid did not give us the required results, so we decided to change the starting material to compound 3 which possesses a less demanding side chain.
Treatment of epoxide 3 with Lewis acids
Treatment of epoxide 3 with FeCl3/Ac2O takes place to give compounds 15 and 16 that were separated by column chromatography (Scheme 6). The less polar compound 15 corresponds to a diene similar to 7 obtained by the analogous treatment of 4. The more polar compound is 16, which is produced by a skeletal rearrangement similar to that of compounds 8.
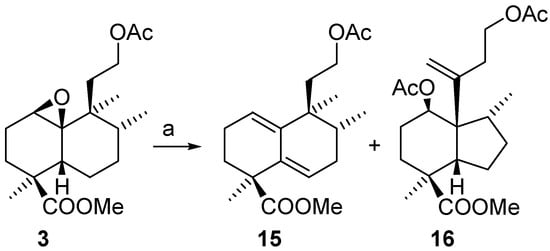
Scheme 6.
The perhydroindene structure for 16 was established spectroscopically by 2D-NMR experiments. Treatment of epoxide 3 with BF3.Et2O (Scheme 7) produced a mixture of compounds that could be separated by column chromatography.
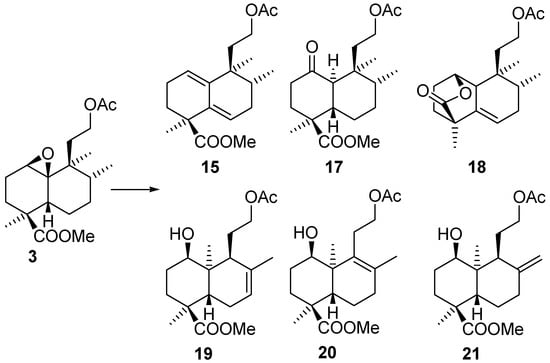
Scheme 7.
Besides diene 15 obtained in the previous treatment (Scheme 6), we obtained ketone 17 (analogous to 11), lactone 18, produced by transesterification of the hydroxy group in C-1 with the methoxycarbonyl at C-18 and, as the main component, a more polar mixture constituted by three compounds − 19, 20 and 21 − all of them ent-labdanes, as desired. In the 1H-NMR spectra of 19 and 20 signals corresponding to a methyl group on a double bond can be observed in ring B instead of the methyl on a methine. In the minor compound, 21 a terminal double bond in ring B is observed instead.
Conclusions
The transformation of ent-halimanes to ent-labdanes has been achieved for the first time. This methodology open the way for the use of the available ent-halimanes for the synthesis of tri- and tetracyclic compounds of the normal series such as tricyclic diterpenes and degraded triterpenes like quassinoids with a picrasane skeleton. Future work will involve using this kind of rearrangement on tricyclic compounds with similar system to the tirucalanes and euphanes that are the biological precursors in which this rearrangement takes place.
Experimental
General
Unless otherwise stated, all chemicals were purchased as the highest purity commercially available and were used without further purification. IR spectra were recorded on a BOMEM 100 FT-IR or an AVATAR 370 FT-IR Thermo Nicolet spectrophotometers. 1H- and 13C-NMR spectra were performed in CDCl3 and referenced to the residual peak of CHCl3 at δ 7.26 ppm and δ 77.0 ppm, for 1H- and 13C-, respectively, using Varian 200 VX and Bruker DRX 400 instruments. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. MS were performed at a VG-TS 250 spectrometer at 70 eV ionising voltage. Mass spectra are presented as m/z (% rel. int.). HRMS were recorded on a VG Platform (Fisons) spectrometer using chemical ionization (ammonia as gas) or Fast Atom Bombardment (FAB) technique. For some of the samples, a QSTAR XL spectrometer was employed for electrospray ionization (ESI). Optical rotations were determined on a Perkin-Elmer 241 polarimeter in 1 dm cells. Diethyl ether and THF were distilled from sodium, and dichloromethane was distilled from calcium hydride under an Ar atmosphere.
Methyl 12-acetoxy-1β,10β-epoxy-13,14,15,16-tetranor-ent-haliman-18-oate (3) and methyl 12-acetoxy-1α,10α-epoxy-13,14,15,16-tetranor-ent-haliman-18-oate (5).
An ice cooled solution of 2 (1.41 g, 4.59 mmol) and UHP (5.49 mg, 58.35 mmol) in CH2Cl2 (55 mL) was treated with TFAA (4.6 mL, 32.57 mmol) under argon. The reaction mixture was stirred at room temperature for 1 h, then a 40% aqueous solution of NaHSO3 was added and the stirring was continued for an additional 30 min. The mixture was extracted with Et2O and washed successively with a 6% aqueous solution of NaHCO3, water and brine. The organic layer was dried (Na2SO4), evaporated and chromatographed on silica gel (95:5→9:1 hexane/EtOAc) giving 3 (1.34 g, 95%) and 5 (54 mg, 2%).
Compound 3: a colourless solid; +52.1 (c 0.4, CHCl3); mp 55 °C; IR (film) ν ( cm-1) 1736, 1464, 1373, 1246, 1125, 1030; 1H-NMR (400 MHz;) 4.21 and 4.08 (1H, two dt, JAB = 11.4, 5.7 Hz, H-12), 3.62 (3H, s, -COOMe), 2.96 (1H, s, H-1), 2.78-2.72 (1H, m, H-5), 2.40-2.30 (1H, m), 2.22-2.00 (2H, m), 2.03 (3H, s, MeCOO-), 1.82-1.73 (1H, m), 1.71-1.55 (3H, m), 1.45-1.38 (2H, m), 1.23-1.12 (2H, m), 0.95 (3H, s, Me-19), 0.91 (3H, d, J = 6.8 Hz, Me-17), 0.72 (3H, s, Me-20); 13C-NMR (100 MHz) 53.6 (C-1), 20.1 (C-2), 23.2 (C-3), 42.4 (C-4), 38.6 (C-5), 21.0 (C-6), 28.3 (C-7), 38.7 (C-8), 39.3 (C-9), 64.4 (C-10), 36.5 (C-11), 61.6 (C-12), 14.9 (C-17), 177.4 (C-18), 25.4 (C-19), 15.5 (C-20), 51.3 (-COOMe), 21.1 (MeCOO-), 170.4 (MeCOO-); EIMS m/z (%) 279 (M+ - 59, 12), 263 (10), 219 (21), 203 (14), 192 (51), 173 (61), 163 (38), 119 (38), 105 (63), 91 (58), 55 (100).
Compound 5: a colourless oil; -8.94 (c 0.3, CHCl3); IR (film) ν ( cm-1) 1738, 1462, 1375, 1236, 1125, 1034; 1H-NMR (400 MHz) 4.12 and 4.07 (1H, two dt, JAB = 10.7, 5.7 Hz, H-12), 3.64 (3H, s, -COOMe), 2.92 (1H, s, H-1), 2.50-2.55 (1H, m, H-5), 2.10-1.55 (8H, m), 2.04 (3H, s, MeCOO), 1.52-1.42 (2H, m), 1.38-1.30 (1H, m), 1.19 (3H, s, Me-19), 0.98 (3H, d, J = 6.8 Hz, Me-17), 0.71 (3H, s, Me-20); 13C-NMR (100 MHz) 51.3 (C-1), 20.5 (C-2), 31.5 (C-3), 45.3 (C-4), 36.8 (C-5), 21.0 (C-6), 28.9 (C-7), 38.4 (C-8), 39.3 (C-9), 64.1 (C-10), 36.7 (C-11), 61.5 (C-12), 15.5 (C-17), 178.6 (C-18), 17.5 (C-19), 15.9 (C-20), 52.0 (-COOMe), 21.1 (MeCOO-), 171.0 (MeCOO-); EIMS m/z (%) 279 (M+ - 59, 9), 220 (18), 203 (12), 193 (54), 173 (48), 163 (38), 105 (63), 91 (100).
Methyl 13-acetoxy-1β,10β-epoxy-14,15-dinor-ent-haliman-18-oate (4) and methyl 13-acetoxy-1α,10α-epoxy-14,15-dinor-ent-haliman-18-oate (6).
To an ice cooled solution of 2 (0.37 g, 1.21 mmol) in MeOH (12 mL) was added NaBH4 (47 mg, 1.25 mmol). After being stirred at room temperature for 20 min the reaction mixture was diluted with Et2O and water, acidified with a few drops of a 2M aqueous solution of HCl, and extracted with Et2O. The organic layer was washed with H2O, dried (Na2SO4) and evaporated to give a hydroxyester (0.31 g, 83 %), which was used in the next step without further purification. To a solution of this hydroxyester (3.65 g, 11.85 mmol) in dry pyridine (5.5 mL) was added acetic anhydride (5.5 mL). The mixture was stirred at room temperature for 20 h, and then it was poured into ice-water and extracted with EtOAc. The organic layer was washed successively with aqueous 2M HCl, aqueous 6% NaHCO3, water and brine. The resulting solution was then dried (Na2SO4) and evaporated to obtain the desired acetate (3.98 g, 96%). An ice cooled solution of acetate (375 mg, 1.07 mmol), UHP (1.85 g, 19.66 mmol) in CH2Cl2 (12 mL) was treated with TFAA (0.8 mL, 5.67 mmol) under argon. The reaction mixture was stirred at room temperature for 30 min. Following the same work-up described above, the residue obtained was purified by column chromatography and 4 (322 mg, 90%) and 6 (54 mg, 6%) were thus obtained.
Compound 4: a colourless oil; IR (film) ν ( cm-1) 1736, 1464, 1373, 1246, 1125 1030; 1H-NMR (400 MHz) 4.91-4.78 (2H, m, H-13 and H-13’), 3.63 (6H, s, -COOMe), 2.94-2.92 (2H, m, H-1 and H-1’), 2.80-2.68 (2H, m, H-5 and H-5’), 2.30-1.10 (26H, m), 2.03 and 2.01 (3H, two s, MeCOO-), 1.23 and 1.21 (3H, d each, J = 6.2 Hz, Me-16 and Me-16’), 0.96 (6H, s, Me-19 and Me-19’), 0.91 (6H, d, J = 7.0 Hz, Me-17 and Me-17’), 0.65 (6H, s, Me-20 and Me-20’); 13C-NMR (100 MHz) 52.8/52.7 (C-1), 20.0 (C-2), 23.1 (C-3), 42.4 (C-4), 38.1/37.8 (C-5), 20.9 (C-6), 28.1 (C-7), 38.6 (C-8), 39.9 (C-9), 64.7 (C-10), 30.2/30.1 (C-11), 33.8/33.2 (C-12), 71.8/71.7 (C-13), 19.9 (C-16), 15.4 (C-17), 177.3 (C-18), 25.4 (C-19), 17.0 (C-20), 51.1/51.0 (-COOMe), 21.2 (MeCOO-), 170.6 (MeCOO-); HRMS (EI): m/z calcd for C21H34O5 (M+ + Na): 389.2298, found 389.2304.
Compound 6: as a colourless oil; IR (film) ν ( cm-1) 2948 1732, 1474, 1375, 1249, 1112 1025; 1H-NMR (400 MHz) 4.91-4.78 (1H, m, H-13), 3.65 (3H, s,-COOMe), 2.93 (1H, d, J = 3.8 Hz, H-1), 2.56-2.44 (1H, m, H-5), 2.05 (3H, s, MeCOO-), 2.02-0.90 (13H, m), 1.23 (3H, d, J = 6.4 Hz, Me-16), 1.00 (3H, d, J = 7.0 Hz, Me-17), 1.20 and 0.65 (3H, s each, Me-19 and Me-20); HRMS (EI) m/z calcd for C21H34O5 (M+ + Na): 389.2298, found: 389.2290.
Methyl 13-acetoxy-14,15-dinor-ent-halima-1(10),5-dien-18-oate (7), methyl 1β,13R and S-diacetoxy-14,15-dinor-8(9→10)abeo-ent-halim-9(11)E-en-18-oate (8a) and methyl 1β,13R and S-diacetoxy-14,15-dinor-8(9→10)abeo-ent-halim-9(20)-en-18-oate (8b).
a) To a solution of 4 (86 mg, 0.23 mmol) in EtOAc (0.5 mL) and Ac2O (0.5 mL), FeCl3 (9 mg, 0.055 mmol) was added and the mixture was stirred under argon at room temperature for 30 min. The reaction mixture was poured into ice-water and extracted with n-Hexane. The organic layer was washed successively with aqueous 6% NaHCO3, and water. The resulting solution was then dried (Na2SO4) and evaporated, a residue was obtained and purified by column chromatography (95:5→8:2 hexane/EtOAc), yielding 7 (16 mg, 20%) and 8a/8b (54 mg, 56%).
b) To a solution of 4 (86 mg, 0.23 mmol) in 10:1 Ac2O/CH2Cl2 (2.3 mL), FeCl3 (42 mg, 0.26 mmol) in Ac2O was added. The mixture was stirred under argon at -78°C for 24 h. Following the same procedure described above, column chromatography afforded 7 (15 mg, 19%) and 8a/8b (56 mg, 58%).
Compound 7: a colourless oil; UV: 238 nm (EtOH); IR (film) ν ( cm-1) 3046, 1738, 1240, 1100, 1032; 1H-NMR (400 MHz) 5.51-5.42 (4H, m, H-1, H-6, H-1’ and H-6’), 4.87-4.78 (2H, m, H-13 and H-13’), 3.60 (6H, s, -COOMe), 2.02 and 2.01 (3H, s each, MeCOO-), 1.31 (6H, s, Me-19 and Me-19’), 1.12 and 1.18 (3H, d each, J = 6.0 Hz, Me-16 and Me-16’), 0.92 (6H, s, Me-20 and Me-20’), 0.78 and 0.77 (3H, d each, J = 7.0 Hz, Me-17 and Me-17’); HRMS (EI) m/z calcd for C21H32O4 (M+ + Na): 371.2193, found 371.2182.
Compound 8a: a colourless oil; IR (film) ν ( cm-1) 2954, 1732, 1458, 1374, 1243, 1135, 1039; 1H- NMR (400 MHz) 5.79-5.72 (1H, m, H-11), 5.04-4.92 (2H, m, H-1 and H-13), 3.70 (3H, s, -COOMe), 2.79 (1H, t, J = 10.0 Hz, H-5), 2.38-1.08 (11H, m), 1.97 (6H, s, MeCOO-), 1.51 (3H, s, Me-20), 1.24 (3H, d, J = 6.8 Hz, Me-16), 1.03 (3H, s, Me-19), 0.72 (3H, d, J = 6.5 Hz, Me-17); HRMS (EI) m/z calcd for C23H36O6 (M+ +H): 409.2590, found 409.2534.
Compound 8b: a colourless oil; IR (film) ν ( cm-1) 2954, 1732, 1458, 1374, 1243, 1135, 1039; 1H-NMR (400 MHz) 5.40 and 4.97 (1H, two s, H-20), 5.04-4.92 (2H, m, H-1 and H-13), 3.54 (3H, s, ‑COOMe), 2.80 (1H, t, J = 10.0 Hz, H-5), 2.36-1.08 (13H, m), 2.04 y 2.03 (3H, two s, MeCOO-), 1.23 (3H, d, J =6.8 Hz, Me-16), 1.03 (3H, s, Me-19), 0.75 (3H, d, J = 6.5 Hz, Me-17); HRMS (EI) m/z calcd forC26H36O6 (M+ +H): 409.2590, found 409.2566.
Methyl 1β-acetoxy-13-oxo-14,15-dinor-8(9→10)abeo-ent-halim-9(20)-en-18-oate (9) and methyl 1β-acetoxy-13-oxo-14,15-dinor-8(9→10)abeo-ent-halim-9(11)E-en-18-oate (10)
The 8a/8b mixture (51 mg, 0.13 mmol) was treated with potassium carbonate in MeOH (3%, 4 mL) for 8 h at room temperature. The MeOH was removed and the residue was diluted with water and extracted with Et2O. The organic fractions were combined and washed with 2M aqueous HCl solution and water. Evaporation of solvent after drying over Na2SO4 gave a residue which was chromatographed on silica gel (95:5 hexane/EtOAc ) affording the desired hydroxy ester (34 mg, 74%), which was directly used in the next step. To a mixture of hydroxy ester (34 mg, 0.092 mmol), N-methylmorpholine N-oxide (NMO) (17 mg, 0.13 mmol) and molecular sieves (41 mg, 500 mg/mmol) in dry CH2Cl2 (1 mL) under argon, at room temperature was added TPAP (2 mg, 0.004 mmol). The reaction mixture was stirred for 30 min and then was filtered through a short pad of silica gel eluting with EtOAc. The solvent was evaporated and the residue was purified by chromatography on silica gel (99:1 CHCl3/Me2CO) yielding 9 (14 mg, 42%) and 10 (15 mg, 45%).
Compound 9: a colourless oil; +16.4 (c 0.5, CHCl3); IR (film) ν ( cm-1) 2942, 1737, 1458, 1359, 1255, 1211, 1145, 1025; 1H-NMR (400 MHz) 5.41 (1H, s, H-20), 5.03 (1H, s, H-20), 4.98 (1H, dd, J = 14.0, 6.0 Hz, H-1), 3.57 (3H, s, -COOMe), 2.85 (1H, t, J = 10.0 Hz, H-5), 2.56 (2H, ddd, J = 12.0, 8.0, 4.0 Hz, H-12), 2.32-2.28 (1H, m, H-2), 2.32-2.22 ( 1H, m, H-11), 2.21-2.11 (1H, m, H-8), 2.18 (3H, s, Me-16), 2.10-1.95 (1H, m, H-3), 1.99 (3H, s, MeCOO-), 1.88-1.81 (1H, m, H-7), 1.78-1.72 (1H, m, H-11), 1.75-1.60 (2H, m, H-6), 1.70-1.60 (1H, m, H-2), 1.62-1.48 (1H, m, H-3), 1.47-1.39 (1H, m, H-7), 1.03 (3H, s, Me-19), 0.77 (3H, d, J = 7.0 Hz, Me-17); 13C-NMR (100 MHz) 71.1 (C-1), 26.3 (C-2), 28.6 (C-3), 43.3 (C-4), 49.6 (C-5), 23.3 (C-6), 29.0 (C-7), 42.1 (C-8), 145.4 (C-9), 55.4 (C-10), 25.5 (C-11), 44.3 (C-12), 208.6 (C-13), 29.9 (C-16), 15.0 (C-17), 177.1 (C-18), 26.2 (C-19), 115.8 (C-20), 51.3 (-COOMe), 21.7 (MeCOO-), 170.6 (MeCOO-); EIMS m/z (%) 387 (M+ + Na, 360), 382 (173), 365 (50), 305 (90), 245 (50), 227 (57); HRMS (EI) m/z calcd for C21H32O5 (M+ + Na): 387.2142, found 387.2137.
Compound 10: a colourless oil; +27.7 (c 0.4, CHCl3); IR (film) ν ( cm-1) 2964, 1721, 1452, 1370, 1255, 1195, 1129, 1030; 1H-NMR (400 MHz) 6.05 (1H, t, J = 6 Hz, H-11), 5.01 (1H, dd, J = 14.0, 6.0 Hz, H-1), 3.52 (3H, s, -COOMe), 3.19 (1H, dd, J = 16.0, 8.0 Hz, H-12), 3.10 (1H, dd, J = 16.0, 8.0 Hz, H-12), 2.80 (1H, t, J = 10.0 Hz, H-5), 2.35-2.25 (1H, dd, J = 12.0, 4.0 Hz, H-2), 2.24-2.11 (1H, m, H-8), 2.16 (3H, s, Me-16), 2.10-1.40 (4H, m, H-7, H-3), 2.0 (3H, s, MeCOO-), 1.75-1.60 (2H, m, H-6), 1.70-1.62 (1H, m, H-2), 1.55 (3H, s, Me-20), 1.03 (3H, s, Me-19), 0.73 (3H, d, J = 7.0 Hz, Me-17); 13C NMR (100 MHz) 71.2 (C-1), 26.4 (C-2), 28.4 (C-3), 43.4 (C-4), 48.9 (C-5), 23.2 (C-6), 28.5 (C-7), 42.3 (C-8), 136.2 (C-9), 55.5 (C-10), 121.9 (C-11), 44.7 (C-12), 206.9 (C-13), 29.2 (C-16), 14.7 (C-17), 176.9 (C-18), 26.2 (C-19), 13.3 (C-20), 51.3 (-COOMe), 21.7 (MeCOO-), 170.5 (MeCOO-); EIMS m/z (%) 387 (M+ + Na, 166), 382 (80), 375 (58), 305 (43), 293 (32), 277 (35), 227 (22), 181 (20); HRMS (EI): m/z calcd for C21H32O5 (M+ + Na): 387.2142, found 387.2144.
Compounds 11, 12 and 13
To a solution of 4 (83 mg, 0.23 mmol) in dry C6H6 (5 mL), BF3·Et2O (0.1 mL, 0.87 mmol) was added under argon at 0°C. After being stirred at room temperature for 30 min, the mixture was poured into ice-water, and extracted with Et2O. The organic layer was washed successively with aqueous 6% NaHCO3 and water. The resulting solution was then dried (Na2SO4) and evaporated, giving a residue that was purified by column chromatography (95:5→9:1 hexane/EtOAc), yielding 7 (9 mg, 11%), 11 (39 mg, 47%), 12 (10 mg, 13%) and 13 (12 mg, 15%).
Methyl (10R)-14,15-dinor-13-acetoxy-1-oxo-ent-haliman-18-oate (11): a colourless oil; IR (film) ν (cm-1) 1732, 1717, 1456, 1373, 1258, 1138, 1071, 955; 1H-NMR (400 MHz) 4.87-4.78 (2H, m, H-13 and H-13’), 3.69 (6H, s, -COOMe), 2.04 and 2.02 (3H, two s, MeCOO-), 1.36 (6H, s, Me-19 and Me-19’), 1.22 and 1.21 (3H, two d, J = 6.3 Hz, Me-16 and Me-16’), 1.04 (6H, s, Me-20 and Me-20’), 0.86 (6H, d, J = 7.1 Hz, Me-17 and Me-17’); HRMS (EI) m/z calcd for C21H34O5 (M+ + Na): 389.2298, found 389.2312.
13R and S-acetoxy-14,15-dinor-8(9→10)abeo-ent-halim-9(20)-en-18(1)-olide (12): a colourless oil; IR (film) ν ( cm-1) 2926, 1748, 1463, 1375, 1249, 1101, 1025; 1H-NMR (400 MHz) 5.08 (1H, s, H-20), 5.02-4.86 (1H, m, H-13), 4.97 (1H, s, H-20), 4.82-4.78 (1H, m, H-1), 2.36-1.02 (14H, m), 2.03 (3H, s, MeCOO-), 1.24-1.21 (3H, d, J = 6.8 Hz, Me-16), 1.16 (3H, s, Me-19), 0.98-0.95 (3H, d, J = 6.8 Hz, Me-17); HRMS (EI) m/z calcd for C20H30O4 (M+ +Na): 357.2036, found 357.2025.
13R and S-acetoxy-14,15-dinor-8(9→10)abeo-ent-halim-9(11)E-en-18(1)-olide (13): a colourless oil; +0.1 (c 0.86, CHCl3); IR (film) ν ( cm-1) 2942, 1737, 1463, 1370, 1255, 1107, 1030; 1H-NMR (400 MHz) 5.38-5.30 (1H, m, H-11), 4.98-4.93 (1H, m, H-13), 4.80 (1H, t, J = 3.8 Hz, H-1), 2.40-2.30 (1H, m, H-12), 2.22-2.10 (1H, m, H-5), 2.10 (3H, s, MeCOO-), 1.98-1.96 (1H, m, H-8), 1.84-1.80 (2H, m, H-6), 1.80-1.78 (2H, m, H-2), 1.62 (3H, s, Me-20), 1.45-1.38 (1H, m, H-12), 1.44-1.34 (2H, m, H-7), 1.42-1.40 (2H, m, H-3), 1.23 (3H, d, J = 6.8 Hz, Me-16), 1.15 (3H, s, Me-19), 0.95 (3H, s, Me-17); 13C-NMR (100 MHz) 76.5 (C-1), 24.1 (C-2), 23.4 (C-3), 40.6 (C-4), 47.8 (C-5), 26.4 (C-6), 34.6 (C-7), 43.3 (C-8), 138.6 (C-9), 56.5 (C-10), 120.0 (C-11), 34.7 (C-12), 70.6 (C-13), 19.6 (C-16), 12.7 (C-17), 178.1 (C-18), 20.2 (C-19), 14.0 (C-20), 170.7 (MeCOO-), 21.3 (MeCOO-); EIMS m/z (%) 335 (M+ + H, 335), 275 (140), 229 (55); HRMS (EI) m/z calcd for C20H30O4 (M+ +H): 335.2217, found 335.2223.
Methyl 13R and S-acetoxy-1-hydroxy-14,15-dinor-8(9→10)abeo-ent-halim-9(11)E-en-18-oate and methyl 13R and S-acetoxy-1-hydroxy-14,15-dinor-8(9→10)abeo-ent-halim-9(20)-en-18-oate (14)
At -44°C and under argon, a solution of 4 (114 mg, 0.31 mmol) in CH2Cl2 (7 mL), was treated with TiCl4 (0.35 mL, 2.52 mmol) and stirred at room temperature for 1 h. The reaction was quenched by addition of Et2O/H2O and extracted with Et2O. The organic layer was washed successively with aqueous 6% NaHCO3, and water. The resulting solution was then dried (Na2SO4) and evaporated, a residue was obtained and purified by column chromatography (95:5 hexane/EtOAc), yielding 14 (29 mg, 22%), 7 (26 mg, 25%), 11 (30 mg, 27%) and 12/13 (8 mg, 8%).
Compound 14: a colourless oil; IR (film) ν ( cm-1) 3458, 2926, 1732, 1463, 1375, 1244, 1134, 1063, 992; 1H-NMR (400 MHz) 5.38-5.30 (1H, m, H-11), 5.07 and 4.98 (1H, two s, H-20), 5.00-4.96 (1H, m, H-13), 3.98 (1H, br s, H-1), 3.63 (3H, s, -COOMe), 2.01 (3H, s, MeCOO-), 1.56 (3H, s, Me-20), 1.20 (3H, d, J = 6.8 Hz, Me-13), 1.14 (3H, s, Me-19), 0.85 (3H, d, J = 6.5 Hz, Me-17); HRMS (EI) m/z calcd for C21H34O5 (M+ +H): 367.2406, found 367.2395.
Methyl 12-acetoxy-13,14,15,16-tetranor-ent-halima-1(10),5-dien-18-oate (15) and methyl 1β,12-diacetoxy-13,14,15,16-tetranor-8(9→10)abeo-ent-halim-9(20)E-en-18-oate (16).
To a solution of 3 (137 mg, 0.41 mmol) in EtOAc (0.85 mL) and Ac2O (0.85 mL) was added FeCl3 (16 mg, 0.096 mmol). The reaction mixture was stirred under argon at room temperature for 1 h. Then ice-water was added and extracted with n-hexane. The organic layer was washed successively with aqueous 6% NaHCO3, and water. The resulting solution was dried (Na2SO4) and evaporated, a residue was obtained and purified by column chromatography (95:5→9:1→8:2 hexane/EtOAc), yielding 15 (26 mg, 20%) and 16 (74 mg, 58%).
Compound 15: a colourless oil; +38.9 (c 1.2, CHCl3); UV: 238 nm (EtOH); IR (film) ν ( cm-1) 3046, 1738, 1454, 1371, 1254, 1109, 1034; 1H-NMR (400 MHz) 5.58-5.42 (2H, m, H-1, H-6), 4.14 (1H, dt, J = 10.4, 6.1 Hz, H-12), 3.91 (1H, dt, J = 10.4, 6.1 Hz, H-12), 3.60 (3H, s, -COOMe), 2.60-2.43 (1H, m, H-5), 2.35-1.40 (8H, m), 2.02 (3H, s, MeCOO-), 1.33 (3H, s, Me-19), 1.00 (3H, s, Me-20), 0.80 (3H, d, J = 6.8 Hz, Me-17); 13C-NMR (100 MHz) 122.0 (C-1), 23.4 (C-2), 33.5 (C-3), 46.3 (C-4), 135.1 (C-5), 119.6 (C-6), 31.1 (C-7), 36.0 (C-8), 39.4 (C-9), 137.6 (C-10), 36.2 (C-11), 62.0 (C-12), 15.9 (C-17), 176.8 (C-18), 20.9 (C-19), 23.8 (C-20), 51.6 (-COOMe), 21.8 (MeCOO-), 171.0 (MeCOO-); EIMS m/z (%) 320 (M+, 1), 305 (1), 290 (1), 260 (1), 245 (1), 233 (5), 217 (1), 201(18), 185 (5), 173 (69), 159 (16), 145 (24), 131 (38), 119 (22), 105 (54), 91 (18), 84 (15), 59 (7), 43 (100); HRMS (EI) m/z calcd for C19H28O4 (M+ + Na): 343.1879, found 343.1862.
Compound 16: a colourless oil ; +0.01 (c 0.8, CHCl3); IR (film) ν ( cm-1) 2953, 1726, 1468, 1370, 1244, 1030; 1H-NMR (400 MHz) 5.5 (1H, s, H-20), 5.12 (1H, s, H-20), 5.05-4.92 (1H, dd, J = 12.4, 4.8 Hz, H-1), 4.21-4.05 (2H, m, H-12), 3.59 (3H, s, -COOMe), 2.82-2.70 (1H, t, J = 8.9 Hz, H-5), 2.25 (2H, t, J = 6.8 Hz, H-11), 2.25-1.15 (1H, m, H-2), 2.10-2.02 (1H, m, H-3), 2.08-2.01 (1H, m, H-8), 2.05 (3H, s, MeCOO-), 1.99 (3H, s, MeCOO-), 1.92-1.40 (2H, m, H-6), 1.70-1.60 (1H, m, H-2), 1.62-1.50 (1H, m, H-3), 1.28-1.22 (2H, m, H-7), 1.05 (3H, s, Me-19), 0.35-0.20 (3H, d, J = 6.5 Hz, Me-17); 13C-NMR (100 MHz) 71.0 (C-1), 26.2 (C-2), 28.2 (C-3), 43.3 (C-4), 49.9 (C-5), 23.3 (C-6), 28.7 (C-7), 42.0 (C-8), 142.4 (C-9), 55.1 (C-10), 30.9 (C-11), 65.3 (C-12), 15.2 (C-17), 177.1 (C-18), 26.3 (C-19), 118.1 (C-20), 171.0 (MeCOO-), 170.6 (MeCOO-), 51.3 (-COOMe), 21.7 (MeCOO-), 21.0 (MeCOO-); EIMS m/z (%) 403 (M+ + Na, 473), 321 (130), 261 (105), 201 (70); HRMS (EI) m/z calcd for C21H32O6 (M+ + Na): 403.2091, found 403.2116.
Compounds 17, 18, 19, 20 and 21.
A solution of 3 (403 mg, 1.19 mmol) in dry C6H6 (13 mL) was treated under argon with BF3·Et2O (0.33 mL), and the reaction mixture was stirred at room temperature for 2 h. Following the same procedure described above, the residue was obtained and purified by chromatography on silica gel (95:5→9:1→85:15 n-hexane/EtOAc), affording 15 (90 mg, 22%), 17 (82 mg, 20%), 18 (8 mg, 3%), 19 (36 mg, 9%), 20 (138 mg, 34%) and 21 (9 mg, 2%).
Methyl (10R)-12-acetoxy-1-oxo-13,14,15,16-tetranor-ent-haliman-18-oate (17): a colourless oil; -4.5 (c 0.5, CHCl3); IR (film) ν ( cm-1) 1734, 1238; 1H-NMR (400 MHz) 4.16 (1H, dt, J = 10.4, 6.4 Hz, H-12A), 3.91 (1H, dt, J = 10.4, 6.1 Hz, H-12B), 3.70 (3H, s, -COOMe), 2.50-2.18 (6H, m, H-2, H-3A, H-5, H-10, H-11A), 2.15-1.90 (2H, m, H-3B, H-11B), 2.04 (3H, s, MeCOO-), 1.80-1.58 (2H, m, H-6), 1.55-1.41 (2H, m, H-7A, H-8), 1.40 (3H, s, Me-19), 1.33-1.15 (1H, m, H-7B), 1.15 (3H, s, Me-20), 0.90 (3H, d, J = 6.8 Hz, Me-17); 13C-NMR (100 MHz) 209.9 (C-1), 39.3 (C-2), 31.1 (C-3), 46.5 (C-4), 43.1 (C-5), 22.3 (C-6), 27.2 (C-7), 35.3 (C-8), 37.3 (C-9), 52.5 (C-10), 36.6 (C-11), 61.5 (C-12), 14.2 (C-17), 177.2 (C-18), 18.1 (C-19), 24.7 (C-20), 52.1 (-COOMe), 21.0 (MeCOO-), 171.1 (MeCOO-); EIMS m/z (%) 278 (M+ - 60, 20), 263 (16), 219 (22), 201 (12), 178 (82), 163 (34), 123 (100), 107 (14), 55 (90); HRMS (EI) m/z calcd for C19H30O5 (M+ + Na): 361.1985, found 361.1970.
12-acetoxy-13,14,15,16-tetranor-ent-halim-5-en-18(1)-olide (18): a colourless oil; IR (film) ν (cm‑1) 3403, 2926, 1737, 1463, 1370, 1249, 1107, 1036; 1H-NMR (400 MHz) 5.49 (1H, m, H-6), 4.82 (1H, s, H-1), 4.15 (2H, m, H-12), 2.68 (1H, br s, H-12), 2.06 (3H, s, MeCOO-), 2.05-0.95 (9H, m,), 1.31 (3H, s, Me-19), 0.86 (3H, d, J = 6.8 Hz, Me-17), 0.80 (3H, s, Me-20); 13C-NMR (100 MHz) 76.9 (C-1), 23.8 (C-2), 33.9 (C-3), 44.4 (C-4), 136.3 (C-5), 117.1 (C-6), 32.4 (C-7), 34.6 (C-8), 35.7 (C-9), 49.5 (C-10), 34.4 (C-11), 60.1 (C-12), 14.3 (C-17), 176.3 (C-18), 17.0 (C-19), 15.2 (C-20), 21.0 (MeCOO-), 171.1 (MeCOO-); HRMS (EI) m/z calcd for C18H26O4 (M+ + Na): 329.1831, found 329.1720.
Methyl 12-acetoxy-1β-hydroxy-13,14,15,16-tetranor-ent-labd-7-en-18-oate (19): a colourless oil; -19.0 (c 0.7, CHCl3); IR (film) ν ( cm-1)3419, 2921, 1742, 1468, 1364, 1238, 1041; 1H-NMR (400 MHz) 5.34 (1H, s, H-7), 4.35-4.05 (2H, m, H-12), 3.80 (1H, s, H-1), 3.64 (3H, s, -COOMe), 2.55 (1H, br s, H-9), 2.45-2.30 (1H, m, H-5), 2.30-2.20 (1H, m, H-3), 2.10 (3H, s, MeCOO-), 2.00-1.15 (6H, m, H-2, H-6, H-11), 1.70 (3H, s, Me-17), 1.33-1.20 (1H, m, H-3), 1.24 (3H, s, Me-19), 0.75 (3H, s, Me-20); 13C-NMR (100 MHz) 69.8 (C-1), 24.7 (C-2), 29.4 (C-3), 46.2 (C-4), 38.7 (C-5), 24.8 (C-6), 121.5 (C-7), 134.1 (C-8), 42.3 (C-9), 40.1 (C-10), 25.5 (C-11), 66.0 (C-12), 21.8 (C-17), 17.0 (C-19), 13.8 (C-20), 170.8 (MeCOO-), 20.9 (MeCOO-), 178.7 (-COOMe), 51.9 (-COOMe); EIMS m/z (%) 361 (M+ + Na, 325), 339 (180), 329 (140), 324 (160), 307 (145), 261 (260), 247 (112), 201 (208), 159 (38); HRMS (EI) m/z calcd for C19H30O5 (M+ + Na): 361.1985, found 361.1975.
Methyl 12-acetoxy-1β-hydroxy-13,14,15,16-tetranor-ent-labd-8-en-18-oate (20): a colourless oil; -51.2 (c 0.8, CHCl3); IR (film) ν ( cm-1) 3438, 2932, 1732, 1468, 1375, 1238, 1118, 1041; 1H‑NMR (400 MHz) 4.29 (1H, ddd, J = 10.8, 8.7, 7.6 Hz, HA-12), 4.09 (1H, ddd, J = 10.8, 8.6, 7.4 Hz, HB-12), 4.01 (1H, br s, H-1), 3.67 (3H, s, -COOMe), 2.40-1.05 (11H, m), 2.05 (3H, s, MeCOO-), 1.66 (3H, s, Me-17), 1.21 (3H, s, Me-19), 1.01 (3H, s, Me-20); 13C-NMR (100 MHz) 70.5 (C-1), 26.5 (C-2), 29.5 (C-3), 43.6 (C-4), 39.2 (C-5), 21.1 (C-6), 33.2 (C-7), 132.3 (C-8), 133.1 (C-9), 47.4 (C-10), 24.9 (C-11), 63.9 (C-12), 20.9 (C-17), 178.9 (C-18), 20.3 (C-19), 16.4 (C-20), 51.9 (-COOMe), 21.0 (MeCOO-), 171.1 (MeCOO-); EIMS m/z (%) 278 (M+ - 60, 8), 207 (15), 149 (100), 121 (9), 91 (14), 73 (44); HRMS (EI) m/z calcd for C19H30O5 (M+ + Na): 361.1985, found 361.1994.
Methyl 12-acetoxy-1β-hydroxy-13,14,15,16-tetranor-ent-labd-8(17)-en-18-oate (21): a colourlees oil; IR (film) ν ( cm-1) 3438, 2932, 1732, 1468, 1375, 1238, 1118, 1041; 1H-NMR (400 MHz) 4.90 and 4.59 (1H, s each, Me-17), 4.36-4.05 (2H, m, H-12), 4.01 (1H, s br, H-1), 3.68 (3H, s, -COOMe), 2.40-1.05 (12H, m), 2.06 (3H, s, MeCOO-), 1.16 (3H, s, Me-19), 0.70 (3H, s, Me-20); HRMS (EI) m/z calcd for C19H30O5 (M+ + Na): 361.1985, found 361.1991.
Acknowledgements
The authors gratefully acknowledge Dr. A. Lithgow and Dr. C. Raposo, from the NMR and Mass Spectrometry Services, respectively, of the Universidad de Salamanca and the CICYT (CTQ2005-04406) for financial support. F.A.H. is grateful to the Universidad de Salamanca for a fellowship.
References
- Marcos, I. S.; Pedrero, A. B.; Sexmero, M. J.; Díez, D.; Basabe, P.; Hernández, F. A.; Urones, J. G. Synthesis and absolute configuration of three natural ent-halimanolides with biological activity. Tetrahedron Lett. 2003, 44, 369–372. [Google Scholar] [CrossRef]
- Marcos, I. S.; Hernández, F. A.; Sexmero, M. J.; Díez, D.; Basabe, P.; Pedrero, A. B.; García, N.; Urones, J. G. Synthesis and absolute configuration of (-)-chettaphanin I and (-)-chettaphanin II. Tetrahedron 2003, 59, 685–694. [Google Scholar]
- Marcos, I. S.; García, N.; Sexmero, M. J.; Basabe, P.; Díez, D.; Urones, J. G. Synthesis of (+)-agelasine C. A structural revisión. Tetrahedron 2005, 61, 11672–11678. [Google Scholar] [CrossRef]
- Marcos, I. S.; Pedrero, A. B.; Sexmero, M. J.; Díez, D.; Basabe, P.; Hernández, F. A.; Broughton, H. B.; Urones, J. G. Synthesis and Absolute Configuration of the Supposed Structure of Cladocoran A and B. Synlett 2002, 105–109. [Google Scholar] Marcos, I. S.; Pedrero, A. B.; Sexmero, M. J.; Díez, D.; Basabe, P.; García, N.; Moro, R. F.; Broughton, H. B.; Mollinedo, F.; Urones, J. G. Synthesis of Bioactive Sesterterpenolides from ent-Halimic Acid. 15-Epi-ent-cladocoran A and B. J. Org. Chem. 2003, 68, 7496–7504. [Google Scholar]
- Polonsky, J. Quassinoid Bitter Principles. Fortschr. Chem. Org. Naturst. 1973, 30, 101–150. [Google Scholar] [CrossRef]
- Hanson, J. R. Diterpenoids. Nat. Prod. Rep. 2005, 22, 594–602. [Google Scholar] [CrossRef]
- Demnitz, F. W. J.; Philippini, C.; Raphael, R. A. Unexpected Rearrangement in the Peroxytrifluoroacetic Acid-Mediated Baeyer-Villiger Oxidation of trans-3.beta.-Hydroxy-4,4,10.beta.-trimethyl-9-decalone Forming a 7-Oxabicyclo[2.2.1]heptane. Structure Proof and Total Synthesis of (±)-Farnesiferol-C. J. Org. Chem. 1995, 60, 5114–5120. [Google Scholar] Cooper, M. S.; Heaney, H.; Newbold, A.; Sanderson, W. R. Oxidation Reactions Using Urea-Hydrogen Peroxide; A Safe Alternative to Anhydrous Hydrogen Peroxide. Synlett 1990, 533–535. [Google Scholar]
- Corey, E. J.; Houpis, I. N. Total synthesis of glycinoeclepin A. J. Am. Chem. Soc. 1990, 112, 8997–8998. [Google Scholar] [CrossRef]
- Ley, S. V.; Norman, J.; Griffith, W. P.; Marsden, S. P. Tetrapropylammonium Perruthenate, Pr4N+RuO4-, TPAP: A Catalytic Oxidant for Organic Synthesis. Synthesis 1994, 639–666. [Google Scholar]
- Sample Availability: Samples of compounds 1, 2, 7, 9, 10, 11, 15, 16, 17, 19 and 20 are available from the authors
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.