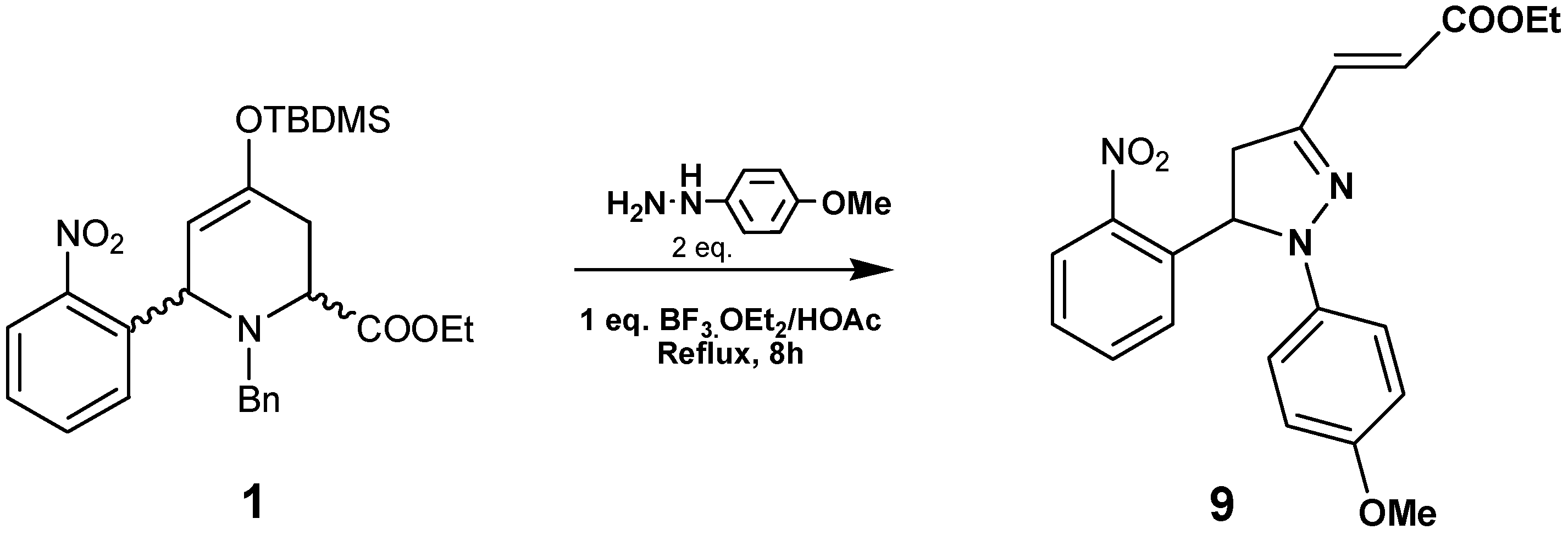Results and Discussion
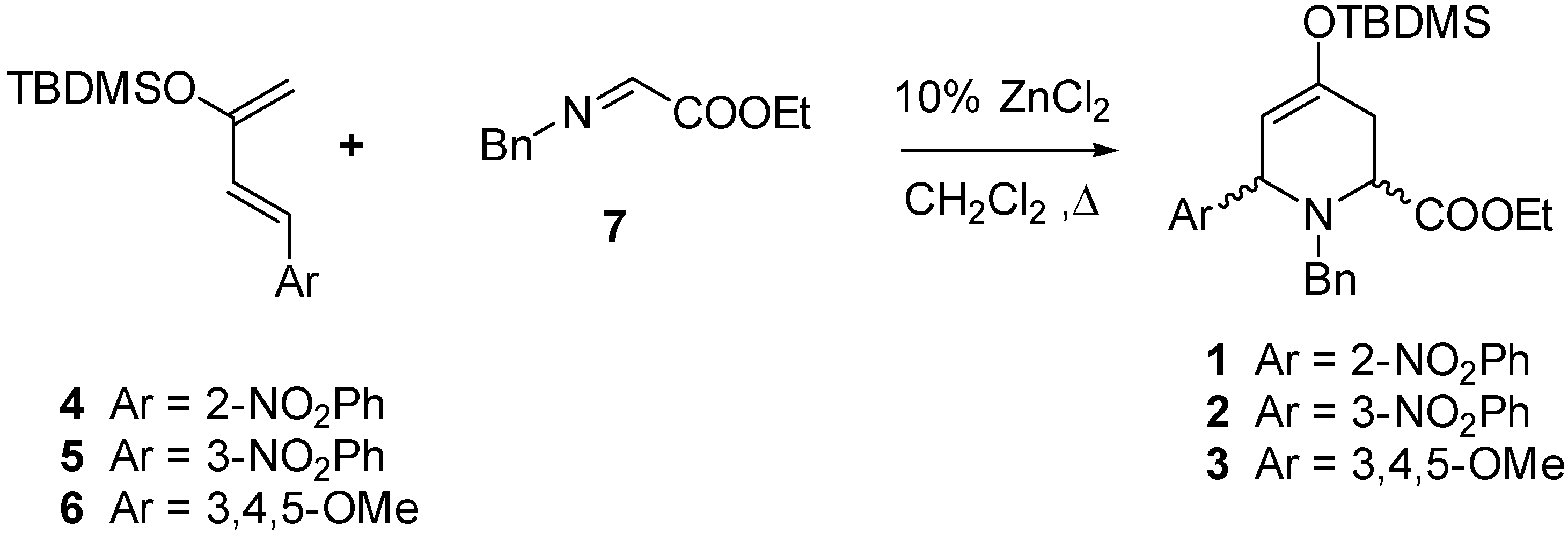
As starting materials we used ethyl 1-benzyl-4-(
tert-butyldimethylsiloxy)-6-(nitro or methoxyphenyl)-1,2,3,6-tetrahydropyridine-2-carboxylates
1-3, obtained by a hetero Diels-Alder reaction between the siloxydienes
4-6 and the imine
7 (
Scheme 1).
As previously mentioned, carbolines can be obtained by the reaction of 4-piperidones with phenylhydrazines under acid conditions. We have used the tetrahydropyridine 4-sylilenolether directly instead of the corresponding ketone, because the reaction conditions should lead to the hydrolysis of the silylenolether to the corresponding ketone. In fact, in previous work we had carried out the Fischer indolization of other related silylenolethers with good results [
12]. Encouraged by those findings, compound
1 was reacted at reflux with
p-methoxyphenylhydrazine, 1:1 glacial acetic acid/EtOH and a few drops of 37% HCl, producing the hydrazone
8 (
Scheme 2). The structure of
8 was established by
1H- and
13C-NMR spectroscopy, which revealed the presence of the two isomeric hydrazones (
Z/
E).
Different conditions to facilitate tautomerization to the enehydrazine and diaza-Cope breakdown of the N-N bond have been examined [
13]. We used H
2SO
4/HOAc at 75ºC, but only degradation of the starting material was detected.
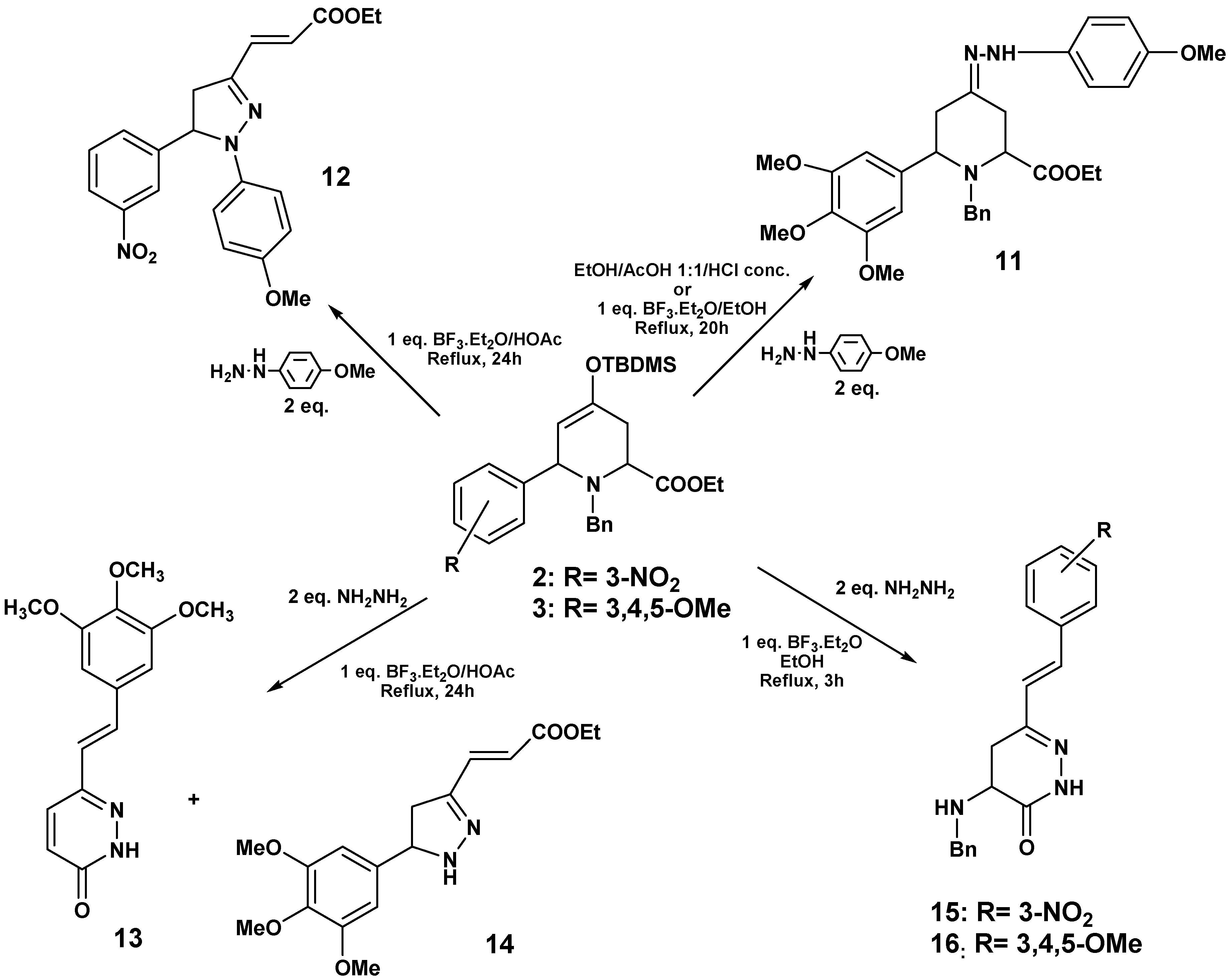
By reacting compound
1 with
p-methoxyphenylhydrazine in the presence of BF
3·Et
2O/HOAc at reflux pyrazoline
9 was obtained in 65% yield (
Scheme 3). This compound was characterized by HRMS,
1H- and
13C-NMR data. In the
1H-NMR spectra we observed three double doublets at 2.91, 3.85 and 5.88 ppm, corresponding to the pyrazoline ring. The olefinic protons resonate at 7.66 and 5.82 ppm as doublets with
J=15.8Hz, indicating an
E stereochemistry. The regiochemistry of compound
9 was established by HMBC connectivities: the olefinic proton at 5.82 ppm with the quaternary carbon (C=N) at 145 ppm; the olefinic proton at 7.66 ppm with the CH
2 carbon at 41.5 ppm; and the aromatic CH at 7.29 ppm (dd
J=8.0, 1.2Hz) with the CH at 61.4 ppm.
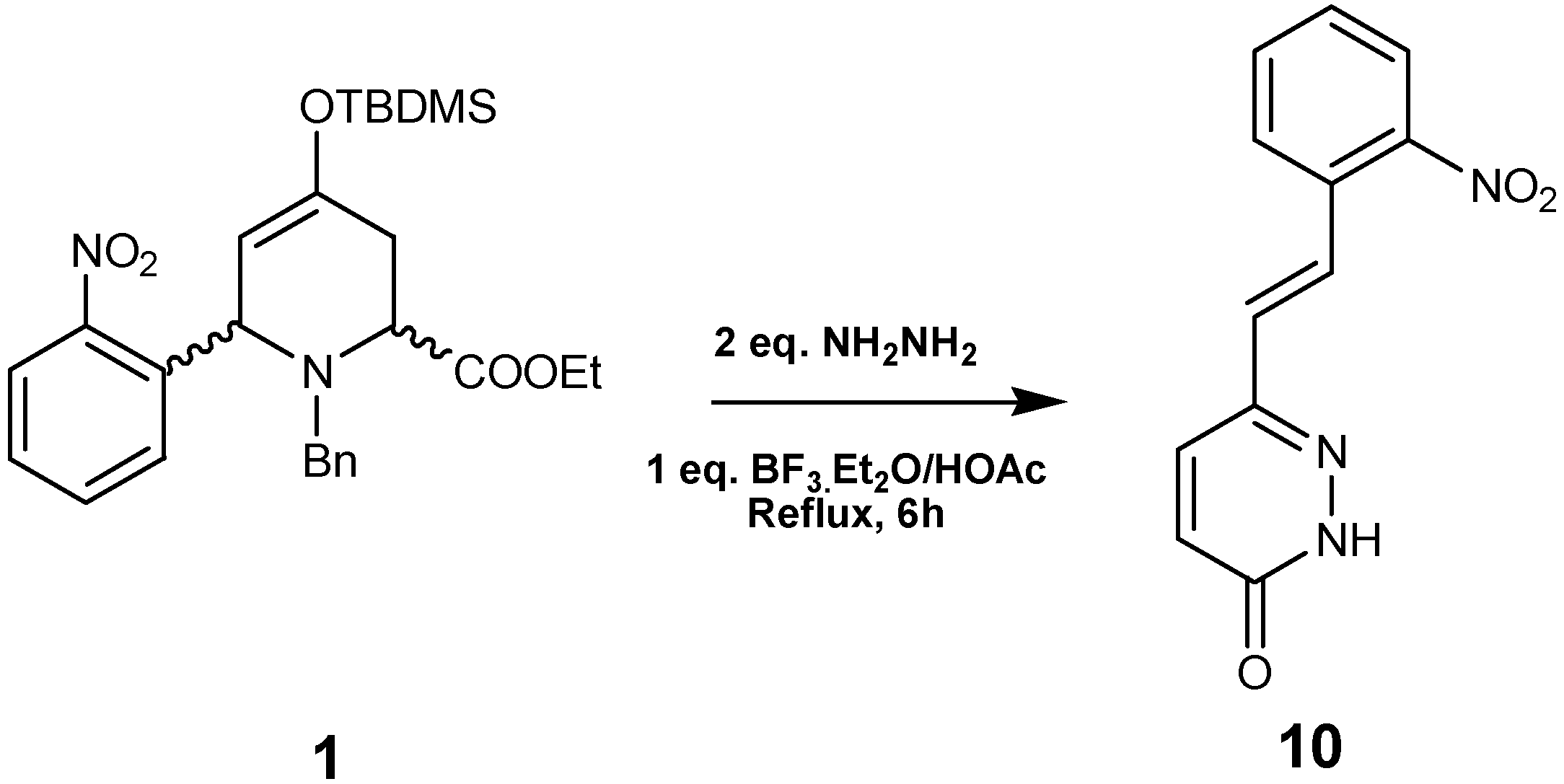
Based on these results, we carried out the reaction between
1 and hydrazine monohydrate under the same reaction conditions. The resulting reaction mixture afforded
10 in 63% yield after purification by crystallization from ether/hexane (
Scheme 4). The
1H and
13C-NMR data of
10 showed signals representative of a monosubstituted pyridazinone.
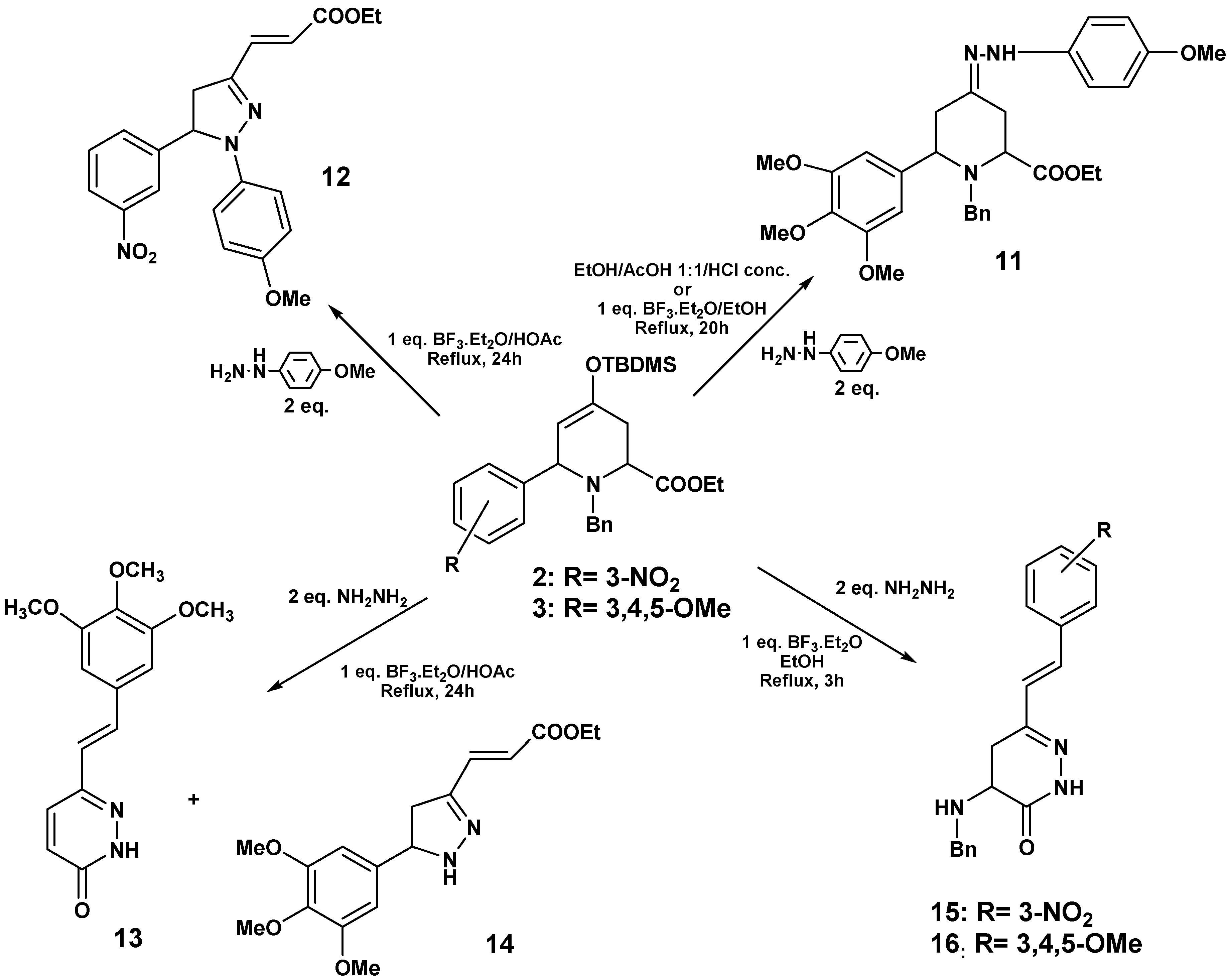
To investigate whether the substituents on the aryl group of the cycloadduct might have some influence in the observed reactivity, we studied the reactions of
2 and
3 with hydrazines. When
p‑methoxyphenylhydrazine was added to
3, under the same conditions described above for
8, the formation of hydrazone
11 was observed. The same results were detected upon replacing the acetic acid by the Lewis acid BF
3·Et
2O. However, when the acid medium was generated by one equivalent of BF
3·Et
2O in acetic acid, the major reaction product was the pyrazoline
12 (77%) (
Scheme 5).
When hydrazine monohydrate and 3 were used under these conditions, compound 13 was isolated (41%) by crystallization (from ether/hexane), and hence the reaction outcome was similar to the case of 1. 1H-NMR of the mother liquor revealed signals corresponding to a minor pyrazoline 14, which could not be purified, and this structure was proposed by comparison with related compounds.
When
2 and
3 were treated with hydrazine and BF
3·OEt
2/EtOH in the absence of other inorganic acids at reflux,
15 (54%) and
16 (60%) were isolated. The structure of these pyridazinones was confirmed through NMR experiments (
Scheme 6).
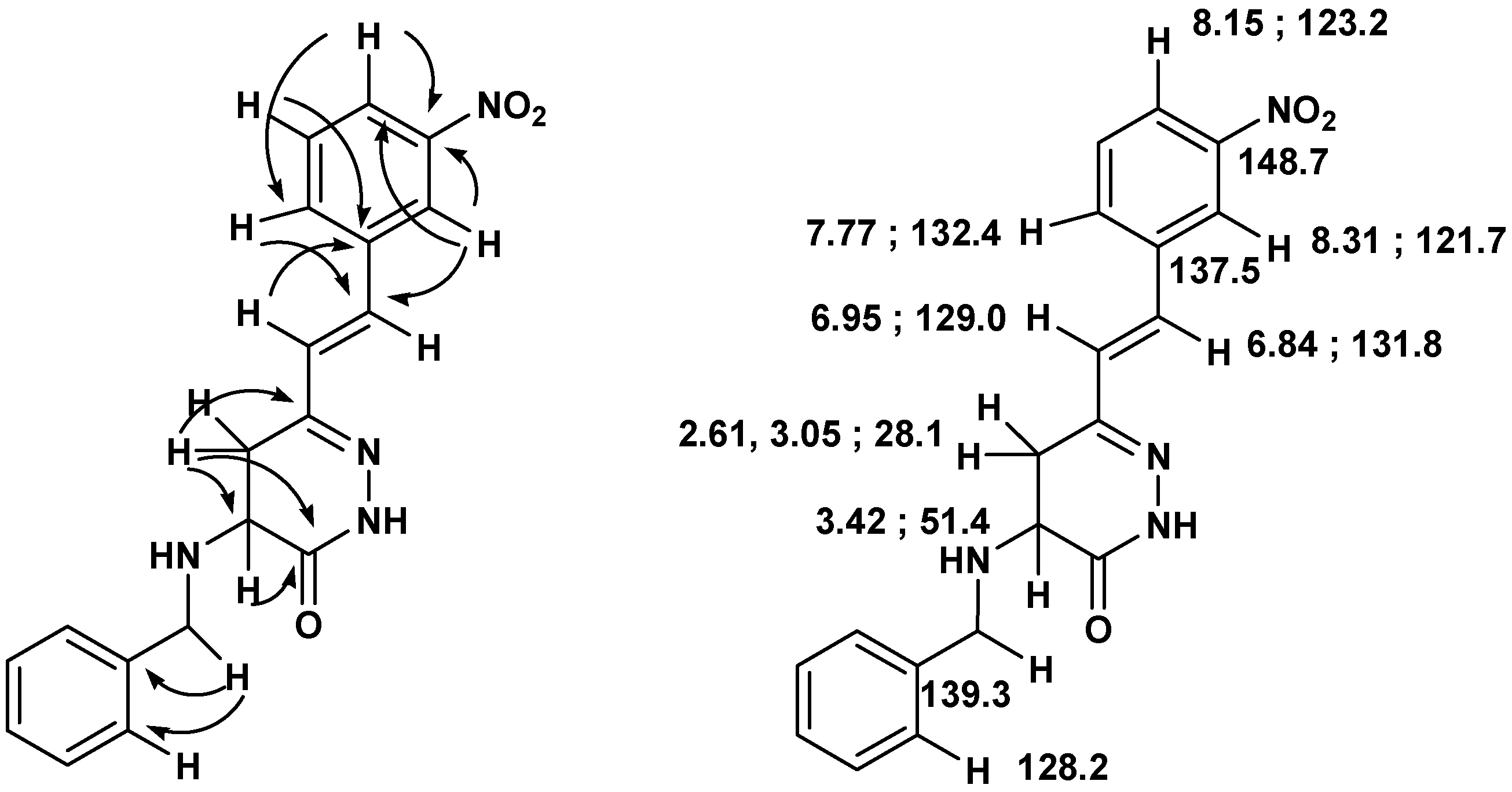
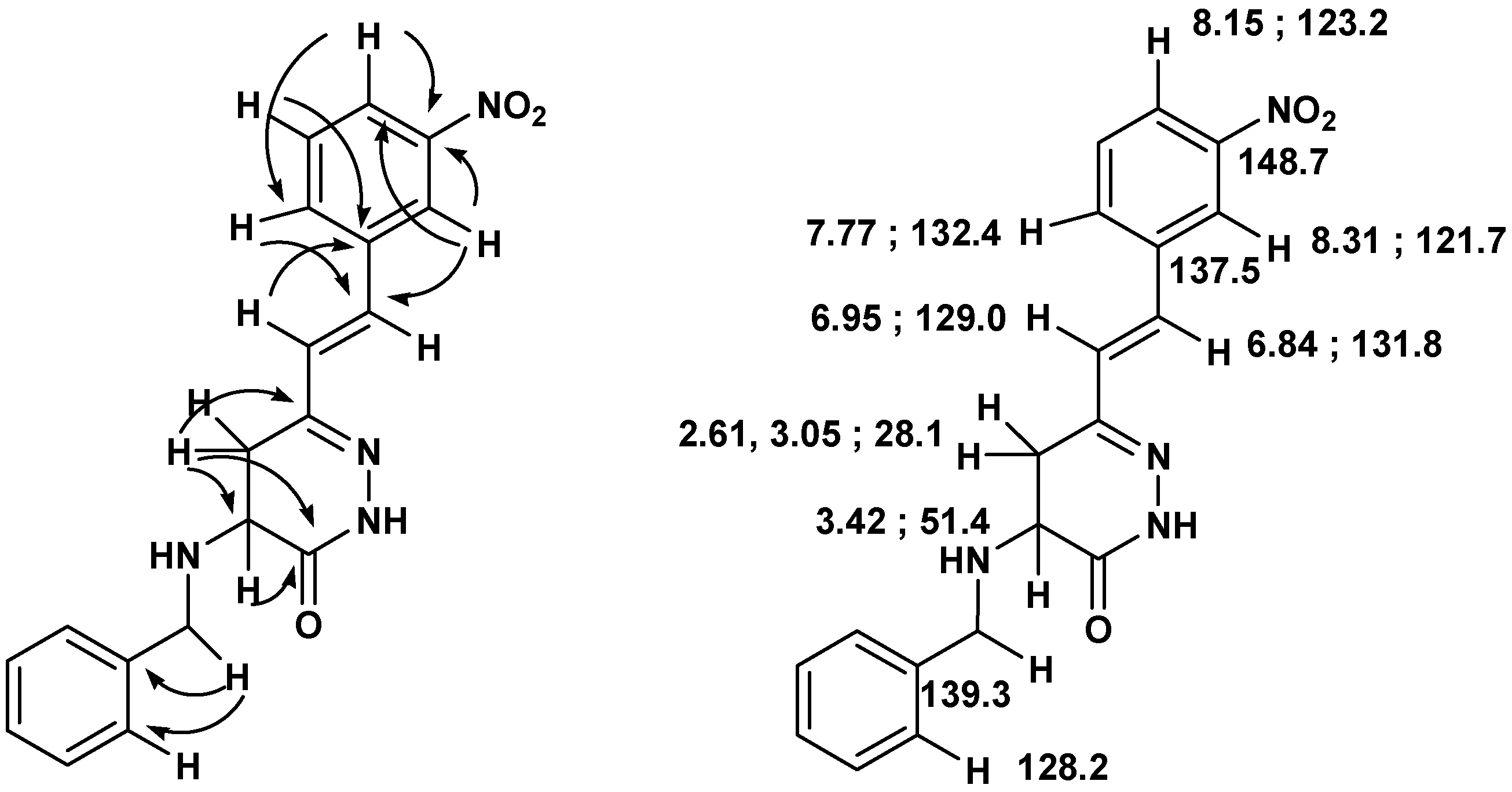
Scheme 6.
Important connectivities found in the HMBC spectra of 15, and 1H- and 13C- NMR assignments.
Scheme 6.
Important connectivities found in the HMBC spectra of 15, and 1H- and 13C- NMR assignments.
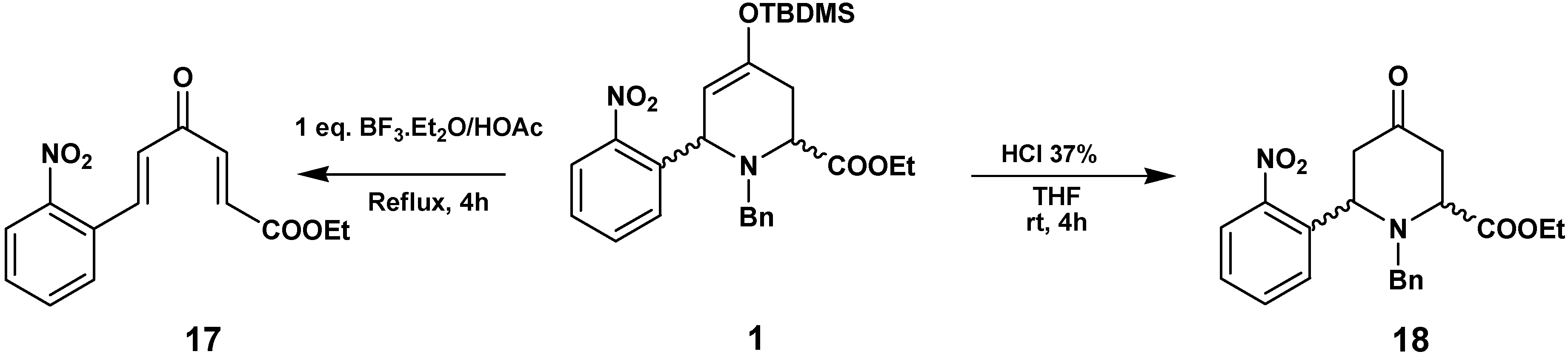
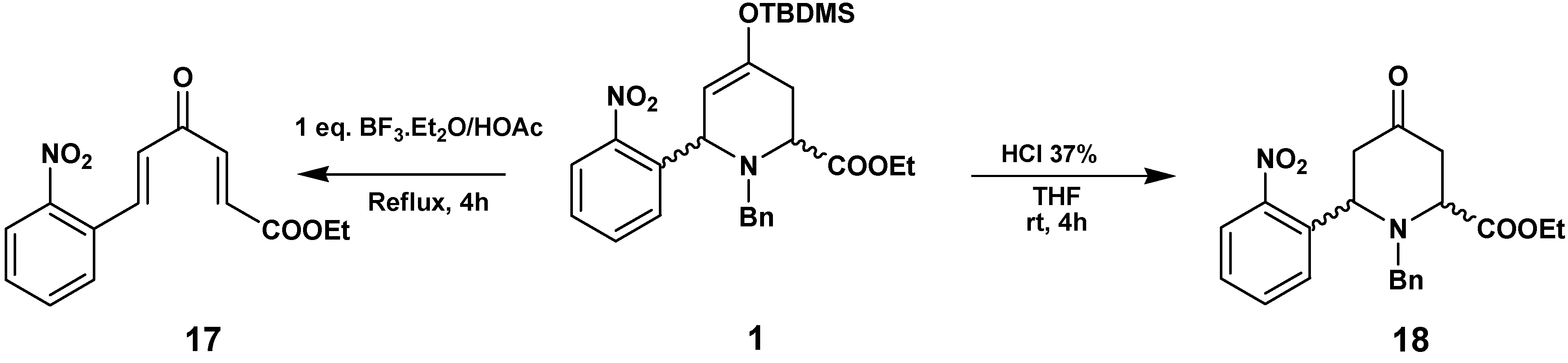
In order to understand these transformations we checked the reactivity of
1 in the different acid media used in the previous transformations, without added hydrazines (
Scheme 7). The reaction of
1 with BF
3·Et
2O/HOAc yielded the enone
17, produced by hydrolysis of the enol-ether, ring opening and the elimination of BnNH
2. Treatment of
1 with HCl led to the hydrolysis of the enol to the ketone
18, with conservation of the piperidine structure.
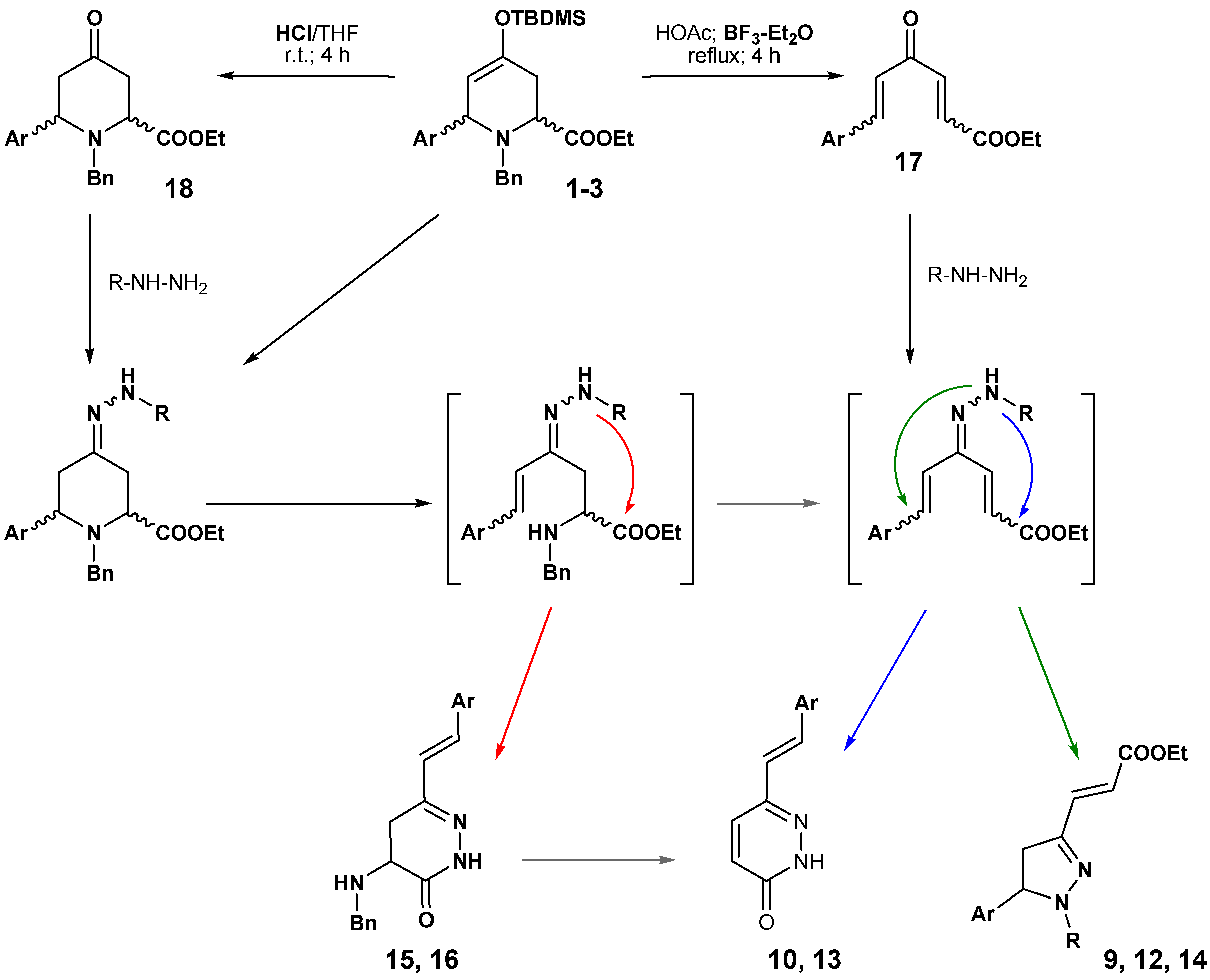
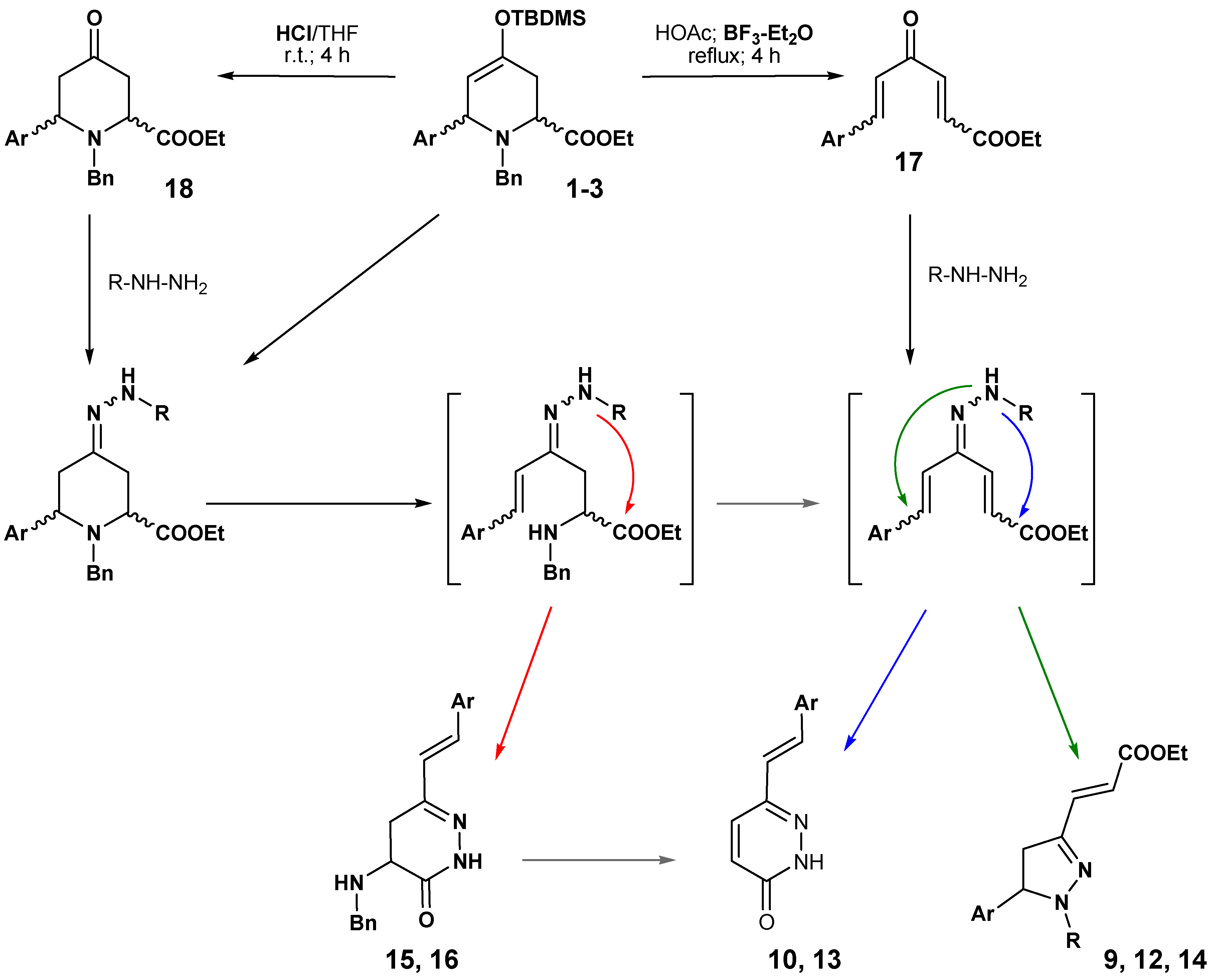
The results reported here are summarized in
Scheme 8. The carbonyl derivatives can be postulated as the precursors of most of the isolated derivatives, for example, through the route from
18 to
8 and
11, and then to other products, or a similar pathway through compound
17 or intermediate hydrazones. This mechanism is also supported by the described syntheses of these types of compound from conjugated ketones [
14,
15,
16,
17].
The major differences are due to the presence of Lewis acid (BF3·Et2O), which facilitates the evolution to pyrazolines and pyridazinones, and to the use of hydrazine or phenylhydrazine as the nucleophilic reagent. The stronger nucleophilic character of the unsubstituted nitrogen of the hydrazones from the hydrazine must be responsible for the formation of the final products 10, 13, 15 or 16, whereas the lower reactive nitrogen of the hydrazones derived from the phenylhydrazine only reacts, with the double bond conjugated with the aromatic ring, after the double elimination of benzylamine. In this case, the absence of reaction with the carboxylate favours the formation of pyrazolines as the major reaction product.
Scheme 8.
Proposal for the formation of pyrazolines and pyridazinones.
Scheme 8.
Proposal for the formation of pyrazolines and pyridazinones.
Experimental
General
Melting points were determined on a Büchi 510 instrument and are uncorrected. NMR spectra were recorded on a Bruker ARX 400 and on a Bruker AC 200 spectrometer with TMS as internal standard. Mass spectra were obtained on a VGTS-250 mass spectrometer by using the electrospray ionization technique (ESI). Flash chromatography was performed with Merck 60 silica gel (0.063-0.2 or 0.040-0.063 mm).
Synthesis of Ethyl 1-benzyl-4-(2-(4-methoxyphenyl)hydrazono)-6-(2-nitrophenyl)piperidine-2-carboxylate (8)
Compound 1 (100 mg, 0.20 mmol) was dissolved in EtOH/glacial AcOH (1:1 v/v, 16 mL) containing HCl (37%, 180 µL). Then, p-methoxyphenylhydrazine (2 equivalents) was added at reflux for 6 h. Ethanol was evaporated in vacuo and the residue was washed with a saturated solution of Na2CO3. The mixture was extracted with EtOAc and the organic layer was washed with a NaCl solution to pH 7, dried with Na2SO4, and evaporated in vacuo. The product was purified by flash chromatography (hexane/ether 1:1), affording 104 mg (54%) of 8 as a brown oil. 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.10 (1H, m), 7.30-7.70 (8H, m), 6.72 (4H, s), 5.48 (1H, m), 4.20 (2H, c, J=7.0 Hz), 3.85 (1H, m), 3.70 (3H, s), 3.68 (2H, m), 3.52 (1H, s), 2.90-2.60 (2H, m), 2.60 (1H, m), 1.29 (3H, t, J=7.0 Hz); 13C-NMR (100 MHz) δ (ppm): 174.1, 148.4, 147.5 x 2, 139.9, 139.5, 138.2, 134.4, 128.7 x 5, 127.3, 126.8, 125.2, 114.5 x 2, 114.3 x 2, 61.9, 61.1, 58.4, 55.7, 52.0, 46.1, 33.8, 14.4; HRMS (ESI) m/z calcd for C28H30N4O5 [M+1] 503.2288, found 503.2279.
Synthesis of Ethyl 1-benzyl-4-(2-(4-methoxyphenyl)hydrazono)-6-(3,4,5-trimethoxyphenyl)piperidine-2-carboxylate (11)
Following the procedure described for 8, 50 mg (40 % yield) of 11 were obtained as a brown oil. 1H-NMR (200 MHz, CDCl3) δ (ppm): 7.0-7.50 (5H, m), 6.88 (2H, d, J=8.8 Hz), 6.72 (2H, d, J=8.8 Hz), 6.55 (1H, s), 6.54 (1H, s), 4.70 (1H, m), 4.20 (2H, m), 4.10 (2H, m), 3.88 (3H, s), 3.82 (6H, s), 3.71 (3H, s), 3.60 (1H, m), 2.50-2.80 (4H, m), 1.23 (3H, m); 13C-NMR (50.3 MHz) δ (ppm): 174.2, 153.7, 153.4, 148.5, 141.3, 139.6x2, 138.8, 137.0, 128.4x2, 127.5, 127.2 x 2, 115.2 x 2, 114.3 x 2, 102.8 x 2, 67.0, 61.1, 61.0, 58.5, 56.2 x 2, 55.6, 52.0, 46.9, 34.8, 14.4; HRMS (ESI) m/z calcd. for C31H37N3O6 [M+] 547.2685, found 547.2678.
Synthesis of (E)-Ethyl 3-(1-(4-methoxyphenyl)-5-(2-nitrophenyl)-4,5-dihydro-1H-pyrazol-3-yl)acrylate (9) and (E)-Ethyl 3-(1-(4-methoxyphenyl)-5-(3-nitrophenyl)-4,5-dihydro-1H-pyrazol-3-yl)acrylate (12)
p-Methoxyphenylhydrazine hydrate (2 equivalents) and BF3·OEt2 (1 equivalent) were added to a solution of 1 or 2 (0.79 mmol) in glacial HOAc (60 mL) and the mixture was stirred at reflux for 8 h. The reaction was cooled and neutralized with a saturated Na2CO3 solution, extracted with EtOAc and the organic layer was washed with a NaCl solution to pH 7, dried with Na2SO4, evaporated in vacuo, and purified by flash chromatography (hexane/ether 1:1) to give 9 and 12 respectively.
Compound 9: Brown oil (65% yield); 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.12 (1H, d, J= 8.0 Hz), 7.66 (1H, d, J=15.8 Hz), 7.55 (1H, t, J=8.0 Hz), 7.46 (1H, t, J=8.0 Hz), 7.29 (1H, d, J=8.0 Hz), 6.86 (2H, d, J=9.2 Hz), 6.75 (2H, d, J=9.2 Hz), 5.88 (1H, dd, J=12.7, 6.5 Hz), 5.82 (1H, d, J=15.8 Hz), 4.25 (2H, q, J=6.7 Hz), 3.85 (1H, dd, J=17.3, 12.7 Hz), 3.72 (3H, s), 2.91 (1H, dd, J=17.3, 6.5 Hz), 1.30 (3H, t, J=6.7 Hz); 13C-NMR (100 MHz) δ (ppm): 166.5, 154.3, 147.2, 145.0, 136.5 x2, 136.3, 134.5, 128.8, 127.9, 125.5, 120.3, 114.7 x2, 114.6 x2, 61.4, 60.4, 55.5, 41.5, 14.2; HRMS (ESI) m/z calcd. for C21H21N3O5 [M+1] 396.1553, found 396.1538.
Compound 12: Brown oil (77% yield); 1H-NMR (200 MHz, CDCl3) δ (ppm): 8.10 (1H, s), 8.11 (1H, m), 7.70 (1H, d, J=15.8 Hz), 7.50-7.60 (2H, m), 6.93 (2H, d, J=8.7 Hz), 6.75 (2H, d, J=8.7 Hz), 5.79 (1H, d, J= 15.8 Hz), 5.41 (1H, dd, J=12.7, 7.0 Hz), 4.23 (2H, q, J=7.4 Hz), 3.72 (3H, s), 3.67 (1H, dd, J=16.6, 12.7 Hz), 2.88 (1H, dd, J=16.6, 7.0 Hz), 1.30 (3H, t, J=7.4 Hz); 13C-NMR (50.3 MHz) δ (ppm): 166.6, 154.6, 148.9, 144.6, 136.7x2, 136.3, 131.9, 130.6, 123.1, 121.2, 120.4, 115.4x2, 114.7x2, 64.7, 60.6, 55.5, 41.4, 14.3; HRMS (ESI) m/z calcd. for C21H21N3O5 [M+] 395.1485, found 395.1470.
Synthesis of (E)-6-(2-nitrostyryl)pyridazin-3(2H)-one (10) and (E)-6-(3,4,5-trimethoxystyryl)-pyridazin-3(2H)-one (13)
Hydrazine monohydrate (2 equivalents) and BF3.OEt2 (1 equivalent) were added to a solution of 1 or 3 (0.13 mmol) in glacial HOAc (25 mL) and the resulting mixture was stirred at reflux for 8 h. The reaction was cooled and neutralized with a saturated Na2CO3 solution, then extracted with EtOAc and the organic layer was washed with NaCl solution to pH 7, dried with Na2SO4, evaporated in vacuo, and the product was purified by crystallization in ether.
Compound 10: Brown solid (63% yield); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 8.01 (1H, d, J=8.0 Hz), 7.95 (1H, d, J=8.0 Hz), 7.87 (1H, d, J=8.0 Hz), 7.75 (1H, t, J=8.0 Hz), 7.59 (1H, t, J=8.0 Hz), 7.58 (1H, d, J=16.4 Hz), 7.10 (1H, d, J=16.4 Hz), 6.95 (1H, d, J=9.9 Hz); 13C-NMR (50.3 MHz) δ (ppm): 162.9, 150.6, 145.7, 136.2, 133.7, 133.5, 132.4, 131.2, 131.9, 131.1, 129.2, 127.2; HRMS (ESI) m/z calcd. for C12H9N3O3 [M+] 243.0650, found 243.0643.
Compound 13: Brown solid purified by crystallization from ether (41% yield; 1H-NMR (200 MHz, CDCl3) δ (ppm): 7.63 (1H, d, J=9.9 Hz), 7.08 (1H, d, J=16.4 Hz), 7.00 (1H, d, J=9.9 Hz), 6.89 (1H, d, J=16.4 Hz), 6.73 (2H, s), 3.90 (6H, s), 3.87 (3H, s); 13C-NMR (50.3 MHz) δ (ppm): 161.1, 153.6 x 2, 145.0, 139.1, 133.2, 131.4, 130.6, 130.0, 123.0, 104.2 x 2, 61.0, 56.2 x 2; HRMS (ESI) m/z calcd. for C15H16N2O4 [M+Na] 311.1002, found 311.1018.
Synthesis of (E)-6-(3-nitrostyryl)-4-(benzylamino)-4,5-dihydropyridazin-3(2H)-one (15) and (E)-6-(3,4,5-trimethoxystyryl)-4-(benzylamino)-4,5-dihydropyridazin-3(2H)-one (16)
Hydrazine monohydrate (2 equivalents) and BF3.OEt2 (1 equivalent) were added to a solution of 2 or 3 (0.13 mmol) in EtOH (25 mL) and the mixture was stirred at reflux for 3 h. The reaction was cooled and neutralized with a saturated Na2CO3 solution. The solvent was evaporated in vacuo and the crude product was dissolved in EtOAc and washed with a solution of NaHCO3.
Compound 15: White solid (54% yield); mp 139-140 ºC (ether/hexane); 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.70 (1H, br s), 8.31 (1H, s), 8.15 (1H, d, J=7.0 Hz), 7.77 (1H, d, J=7.0 Hz), 7.55 (1H, t, J=7.0 Hz), 7.35-7.37 (5H, m), 6.95 (1H, d, J=16.5 Hz), 6.84 (1H, d, J=16.5 Hz), 3.96 (2H, s), 3.42 (1H, dd, J=12.0, 6.7 Hz), 3.05 (1H, dd, J=16.6, 6.8 Hz), 2.61 (1H, dd, J=16.6, 11.6 Hz); 13C-NMR (100 MHz) δ (ppm): 168.1, 150.9, 148.7, 139.3, 137.5, 132.4, 131.8, 129.8, 129.0, 128.6 x 2, 128.2 x 2, 127.3, 123.2, 121.7, 51.6, 51.4, 28.1; HRMS (ESI) m/z calcd. for C19H18N4O3 [M+Na] 373.1271, found 373.1254.
Compound 16: Brown solid (60% yield); mp 126-127 ºC (ether/hexane); 1H-NMR (200 MHz, CDCl3) δ (ppm): 8.55 (1H, br s), 7.30-7.38 (5H, m), 6.77 (2H, s), 6.70 (2H, s), 3.86 (6H, s), 3.84 (3H, s), 3.80 (2H, s) 3.41 (1H, dd, J=12.4, 6.6 Hz), 3.08 (1H, dd, J=16.4, 6.9 Hz), 2.59 (1H, dd, J=16.4, 12.2 Hz); 13C-NMR (50.3 MHz) δ (ppm): 168.4, 153.5, 153.4, 152.1, 139.5, 139.1, 135.0, 131.3, 128.6 x 2, 128.2 x 2, 127.3, 125.4, 104.3 x 2, 61.0, 56.2 x 2, 51.6, 51.5, 28.1; HRMS (ESI) m/z calcd. for C22H25N3O4 [M+] 395.1853, found 395.1848.
Synthesis of (2E,5E)-ethyl 6-(2-nitrophenyl)-4-oxohexa-2,5-dienoate (17)
To a solution of 1 (120 mg, 0.24 mmol) in glacial HOAc (30 mL), BF3.OEt2 (1 equivalent) was added; the mixture was stirred at reflux for 3 h. The reaction was cooled and neutralized with a saturated Na2CO3 solution, extracted with EtOAc and the organic layer was washed with an NaCl solution to pH 7, dried with Na2SO4, evaporated in vacuo and purified by flash chromatography (hexane/ether 1:1) to give 45 mg (69 %) of 17 as a brown oil. 1H-NMR (200 MHz, CDCl3) δ (ppm): 8.13 (1H, d, J=15.8 Hz), 8.08 (1H, d, J=7.5 Hz), 7.5-7.7 (3H, m), 7.46 (1H, d, J=15.8 Hz), 6.83 (2H, d J=15.8 Hz), 4.28 (2H, q, J=7.0 Hz), 1.33 (3H, t, J=7.0 Hz); 13C-NMR (50.3 MHz) δ (ppm): 188.2, 165.4, 148.5, 141.0, 137.6, 133.8, 132.0, 130.9, 130.5, 129.5, 129.2, 125.2, 61.5, 14.2.
Synthesis of Ethyl 1-benzyl-6-(2-nitrophenyl)-4-oxopiperidine-2-carboxylate (18)
Compound 1 (105 mg, 0.21 mmol) dissolved in THF (5 mL) was treated with 37% HCl (20 μL) and then stirred for 4 h. The reaction mixture was washed with saturated NaHCO3, dried and evaporated. The residue was chromatographed (hexane/EtOAc 7:3) to afford 18 (60 mg, 75 %) as an orange oil. 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.03 (1H, d, J=8.0 Hz), 7.78 (1H, d, J=8.1 Hz), 7.67 (1H, t, J=8.0 Hz), 7.43 (1H, t, J=8.1 Hz), 7.30-7.24 (5H, m), 4.99 (1H, dd, J=8.0, 5.6 Hz), 4.26 (2H, q, J=7.2 Hz), 3.88 (1H, dd, J=6.4, 2.0 Hz), 3.61 (1H, d, J=13.6 Hz), 3.46 (1H, d, J=13.6 Hz), 2.99 (1H, dd, J=15.6, 5.6 Hz), 2.75 (1H, dd, J=14.8, 6.4 Hz), 2.60 (1H, dd, J=14.8, 2.0 Hz), 2.58 (1H, dd, J=15.6, 8.0 Hz), 1.33 (3H, t, J=7.2 Hz); 13C-NMR (100 MHz) δ (ppm): 204.7, 171.2, 149.9, 137.5, 137.4, 133.2, 128.8, 128.6 x5, 127.5, 124.1, 61.1, 58.2, 56.8, 55.0, 47.3, 42.7, 14.3; HRMS (ESI) m/z calcd. for C21H22N2O5 [M+] 382.1529, found 382.1536.

{kind=link}
{kind=link}
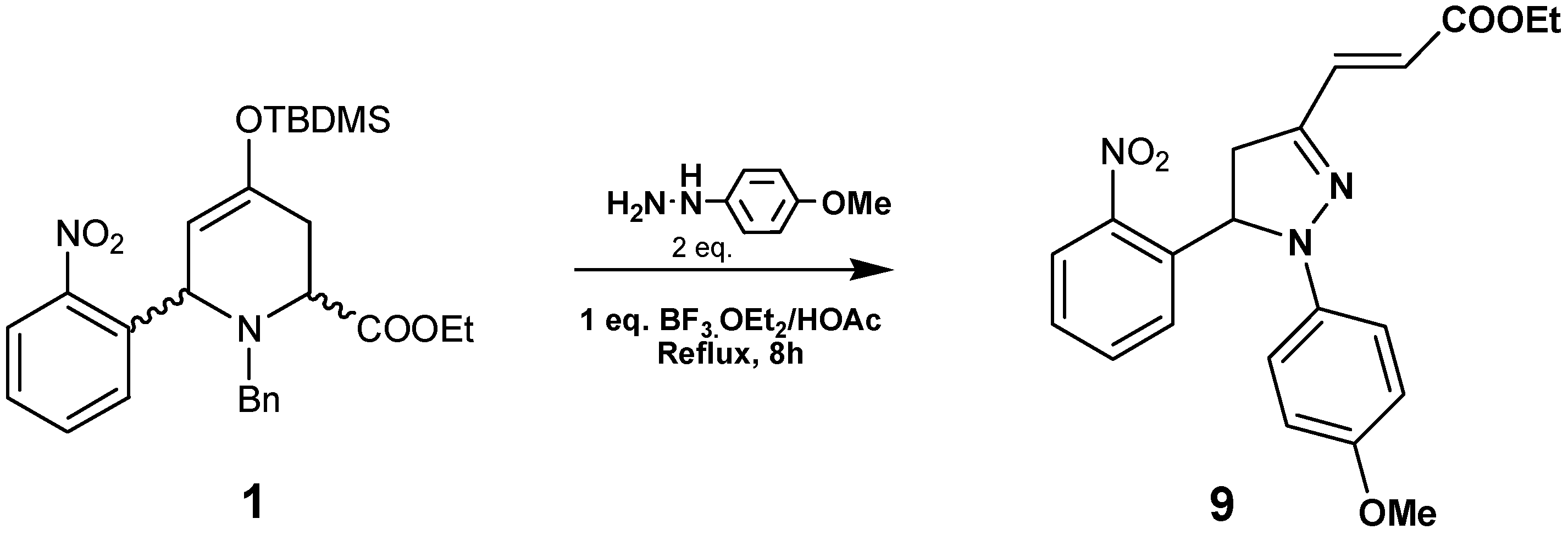{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}