Amine syntheses
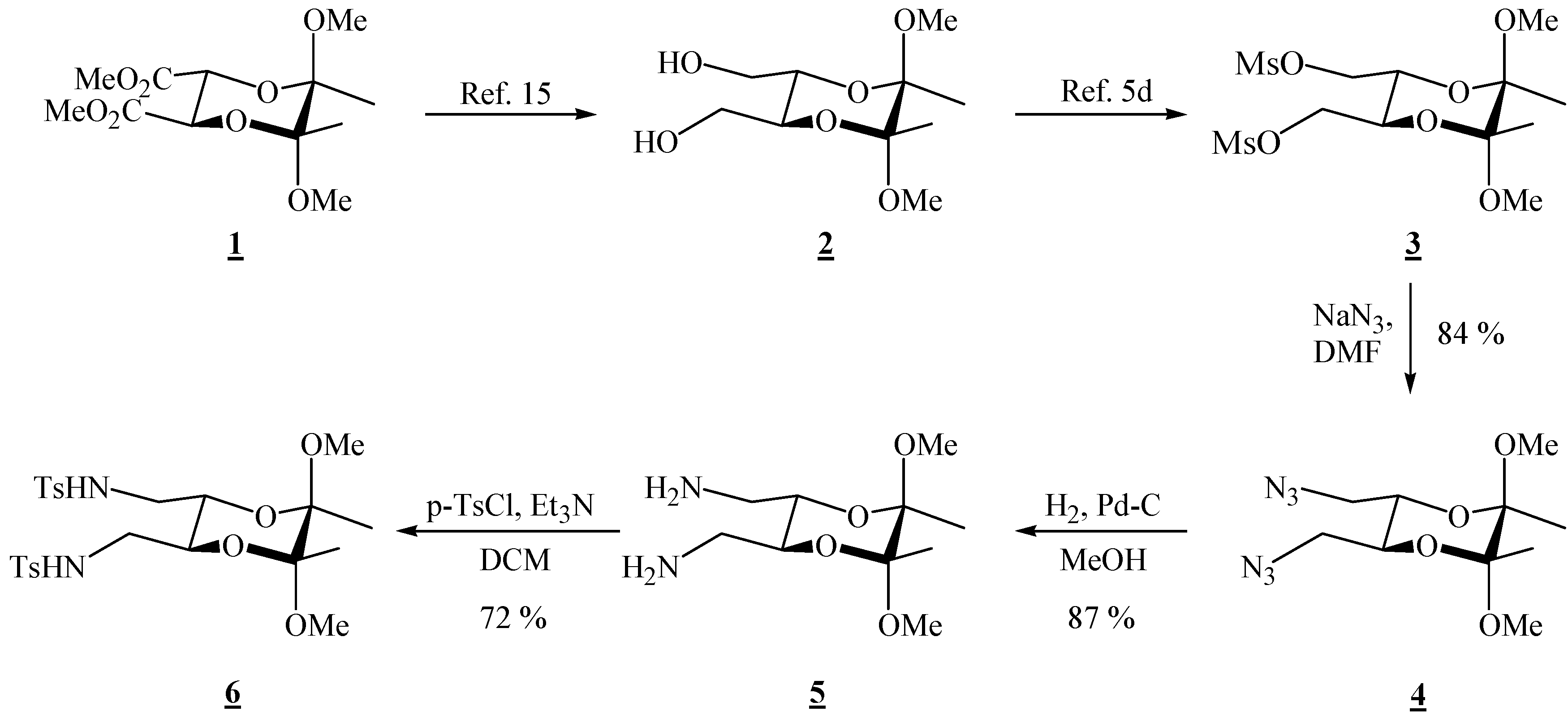
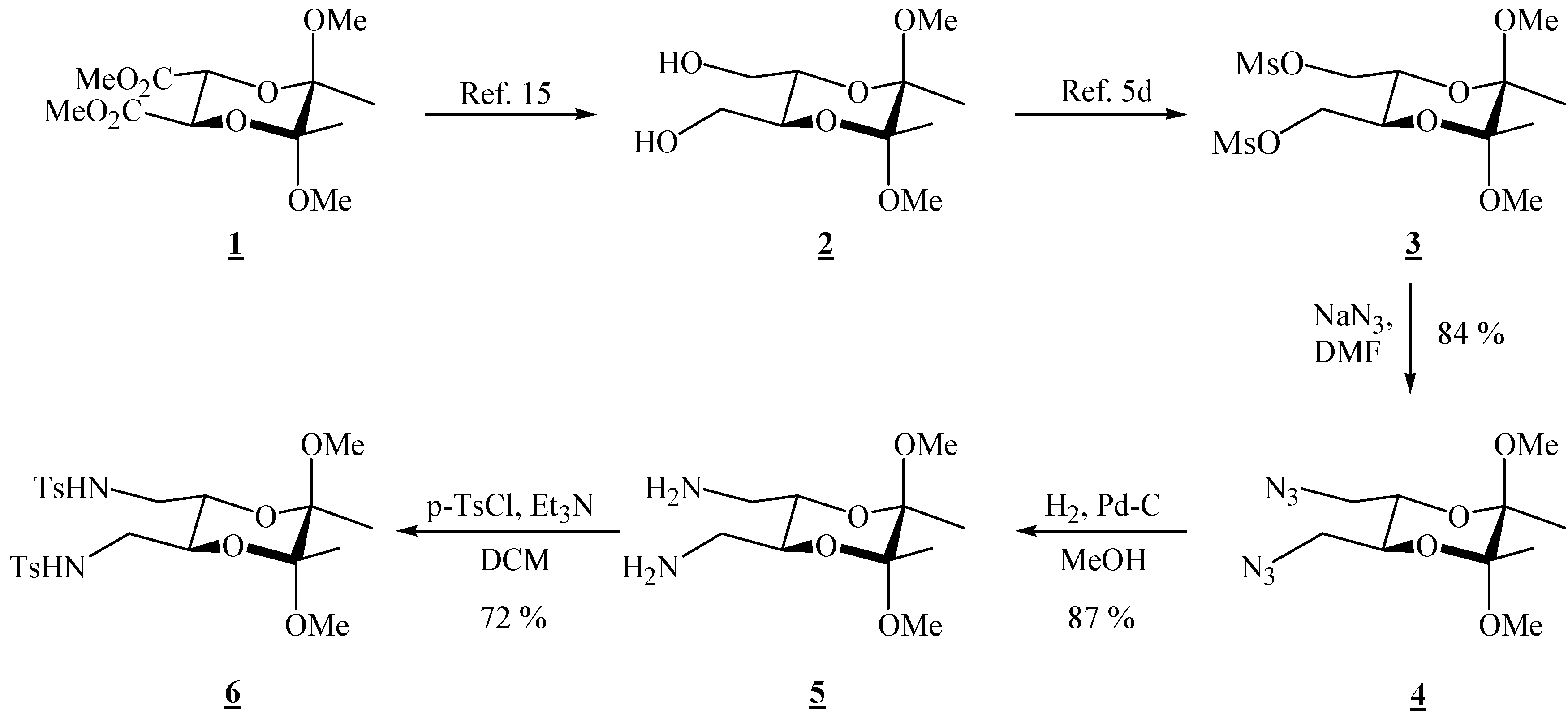
(2R,3R,5S,6S)-5,6-Bis(azidomethyl)-2,3-dimethoxy-2,3-dimethyl[1,4]dioxane (4).
Bismesylate 3 (0.984 g, 2.51 mmol), sodium azide (1.98 g, 30.5 mmol) and dry dimethylformamide (14.0 mL) were mixed. The resulting suspension was heated up to 80 °C and stirred for 2 days. The reaction mixture was cooled to room temperature, and ether and water were added. The layers were separated, and the aqueous layer was extracted twice more with ether. The combined ether extracts were then washed four times with water and dried through anhydrous sodium sulfate. Evaporation of the solvent gave a clear colourless liquid (0.604 g, 84 %), which was used in the next reaction without further purification; for elemental analysis, the product was purified by preparative chromatography on silica gel (1:4 EtOAc-hexane): [α]26D = –102.7 (c 2.00, CHCl3); 1H-NMR: δ 1.33 (s, 3 H, 2 × CH3), 3.21 (br d, 2 H, J = 12.8 Hz, CH2), 3.31 (s, 3 H, OCH3), 3.31–3.38 (m, 2 H, CH2), 3.87–3.88 (m, 2 H, 2 × CH) ppm; 13C-NMR: δ 17.3 (2 × CH3), 48.1 (2 × OCH3), 50.8 (2 × CH2), 69.0 (2 × CH), 99.1 (2 × acetal-C) ppm; IR: ṽ (CHCl3) 3009, 2948, 2932, 2837, 2103, 1444, 1379, 1294, 1253, 1229, 1143, 1133, 1037, 962, 909, 862, 651, 559 cm-1; MS (EI) m/z (%): 271 (M+-15, 0.12), 255 (0.33), 197 (46), 116 (55), 101 (99), 95 (29), 89 (23), 84 (11), 82 (30), 81 (89), 76 (99.8), 75 (96), 73 (100), 70 (11), 69 (22), 68 (44), 67 (88), 59 (36), 57 (19), 56 (20), 55 (62), 54 (95), 53 (11); Anal. Calcd for C10H18N6O4 (286.29): C 41.95, H 6.34, N 29.36; found C 41.78, H 6.32, N 29.48.
(2R,3R,5S,6S)-(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)dimethanamine (5).
Diazide 4 (0.270 g, 0.943 mmol) dissolved in dry MeOH (13.5 mL) was transferred to a hydrogenation flask and Pd on charcoal (0.034 g, 10 %, 0.032 mmol) was added. The flask was connected to a hydrogenation apparatus and the hydrogenation was performed at room temperature and at 5 psi for 4 h. The hydrogen was then released, and the reaction mixture was filtered through Celite®. The solvent was removed under reduced pressure to give the diamine as the only product (0.191 g, 87 %). It was used as it is in the next reaction. This product deteriorated with time at room temperature, and it was kept in the refrigerator at ca. 7 °C; 1H-NMR: δ 1.32 (s, 6 H, CH3), 2.03 (s, 4 H, exchange with D2O, 2 × NH2), 2.79 (s, 4 H, 2 × CH2), 3.28 (s, 6 H, 2 × OCH3), 3.61 (s, 2 H, 2 × CH) ppm; 13C-NMR: δ 17.6 (2 × CH3), 42.5 (2 × CH2), 47.9 (2 × OCH3), 71.0 (2 × CH), 98.6 (2 × acetal-C) ppm.
(2R,3R,5S,6S)-5,6-Dimethoxy-5,6-dimethyl[1,4]dioxane-2,3-bis(methyl p-toluene sulfonamide) (6).
Diamine 5 (0.167 g, 0.714 mmol) was dissolved in dry dichloromethane (1.50 mL) and triethylamine (0.13 mL, 9.33 mmol) was added. The solution was cooled to 0 °C, and p-tosyl chloride (0.297g, 1.56 mmol) was added. After stirring for 10 min. at 0 °C, the cooling bath was removed, and the solution was stirred at room temperature for 30 min. Hydrochloric acid (1 M) was added, and the product was extracted three times with dichloromethane. The combined organic extracts were washed with water, and the solvent was removed under reduced pressure. The product was purified by chromatography on silica gel (1:1 EtOAc-hexane, adsorption from CHCl3) to give a colourless crystalline solid (0.288 g, 72 %), m.p. 194-195 °C (EtOAc-hexane); [α]24D = –97.7 (c 1.54, CHCl3); 1H-NMR: δ 1.14 (s, 6 H, 2 ° CH3), 2.38 (s, 6 H, 2 × CH3 of Ts), 2.88 (d, 2 H, CH2), 2.98 (d, 2 H, CH2), 3.07 (s, 6 H, 2 × OCH3), 3.63–3.64 (m, 2 H, 2 × CH), 4.80 (s, br, 2 H, 2 × NH), 7.27 (d, J = 8.0 Hz, 4 H, Ts), 7.68 (d, J = 8.0 Hz, 4 H, Ts) ppm; 13C-NMR: δ 17.3 (2 × CH3), 21.5 (2 × CH3 of Ts), 43.3 (2 × CH2), 48.1 (2 × OCH3), 67.4 (2 × CH), 98.9 (2 × acetal-C), 127.1 (4 × CH, Ts), 129.9 (4 × CH, Ts), 136.5 (2 × Cq, Ts), 143.7 (2 × Cq, Ts) ppm; IR (KBr): ṽ 3294, 3272, 2985, 2946, 2900, 2892, 2834, 1599, 1455, 1438, 1386, 1327, 1169, 1154, 1129, 1092, 1050, 1030, 970, 882, 856, 816, 723, 661, 585, 551, 529 cm-1; MS (EI) m/z (%): 527 (M+ - 15), 479 (70), 239 (88), 224 (73), 223 (100), 222 (92), 210 (60), 184 (63), 155 (51), 139 (28), 101 (37), 92 (20), 91 (18), 84 (22), 83 (35), 73 (21), 69 (10) 68 (88), 65 (38), 56 (25); Anal. Calcd for C24H34N2O8S2 (542.66): C 53.12, H 6.32, N 5.16, S 11.82; found C 53.34, H 6.38, N 4.99, S 11.62.
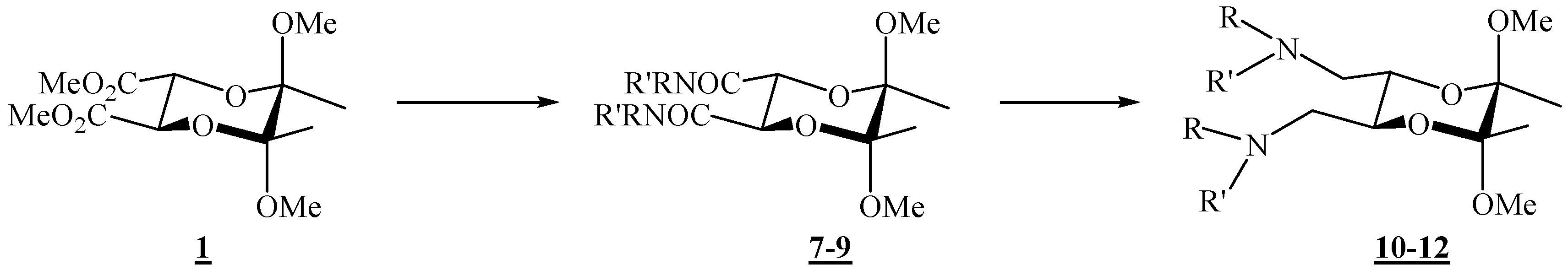
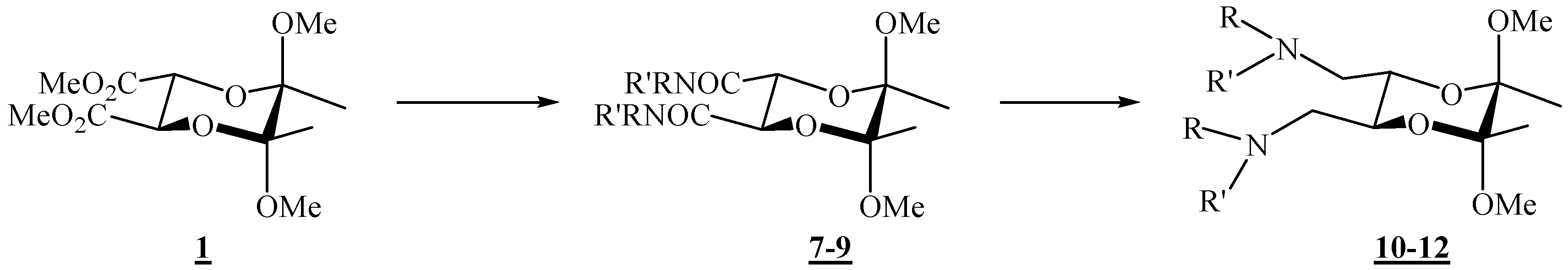
(2R,3R,5S,6S)-5,6-Dimethoxy-5,6-dimethyl-N,N-diphenyl[1,4]dioxane-2,3-dicarboxamide (7).
Diester 1 (1.20 g, 4.10 mmol) and aniline (0.75 mL, 8.23 mmol) were mixed in a round bottom flask topped up with a reflux condenser, heated up to 140 °C, and stirred for 4 days. After cooling to room temperature, the reaction mixture was chromatographed on silica gel (3:2 ether-hexane, adsorption from CHCl3) to give the product as colourless crystals (0.142 g, 84 %), m. p. 209 °C (ether-hexane); [α]26D = –100.5 (c 2.00, CHCl3); 1H-NMR: δ 1.45 (s, 6 H, 2 × CH3), 3.27 (s, 6 H, 2 × OCH3), 4.69 (s, 2 H, 2 × CH), 7.13 (t, 1 H, J = 7.6 Hz, Ph), 7.34 (t, 2 H, J = 7.2 Hz, Ph), 7.60 (d, 2 H, J = 7.6 Hz, Ph), 8.27 (s, br, 2 × NH) ppm; 13C-NMR: δ 18.0 (2 × CH3), 48.6 (2 × OCH3), 70.9 (2 × CH), 99.8 ( 2 × acetal-C), 119.9 (4 × CH, Ph), 124.4 (2 × CH, Ph), 129.0 (4 × CH, Ph), 137.3 (2 × i-C, Ph), 166.4 (2 × CONH) ppm; IR (CHCl3): ṽ 3469, 3389, 3366, 3329, 3198, 3136, 3081, 3000, 2947, 2836, 1687, 1600, 1531, 1499, 1445, 1377, 1312, 1142, 1115, 1045, 1034, 929, 906, 755, 693, 633, 457 cm-1; MS (EI) m/z (%): 416 (M+ + 2, 0.27), 415 (M+ + 1, 1.55), 414 (M+, 7.44), 174 (23), 146 (23), 145 (19), 116 (20), 115 (18), 101 (16), 93 (100), 92 (9), 77 (17), 73 (11); Anal. Calcd for C22H26N2O6 (414.46): C 63.76, H 6.32, N 6.76; found C 63.70, H 6.29, N 6.81.
(2R,3R,5S,6S)- N,N-Dibenzyl-5,6-dimethoxy-5,6-dimethyl- [1,4]dioxane-2,3-dicarboxamide (8).
Diester 1 (0.34 g, 1.16 mmol) and benzylamine (0.28 mL, 2.56 mmol) were mixed in a round bottom flask topped up with a reflux condenser, heated up to 120 °C, and stirred for 21 h. After cooling to room temperature, the reaction mixture was chromatographed on silica gel (96:4 CHCl3-acetone) to give the product as colourless crystals (0.32 g, 63 %), m. p. 120–121 °C (CHCl3 / acetone); [α]16D = –108.6 (c 0.28, CHCl3); 1H-NMR: δ 1.32 (s, 6 H, 2 × CH3), 3.23 (s, 6 H, 2 × OCH3), 4.41 (s, 2 H, 2 × CH), 4.54 (2 × dd, ABX system, 4 H, JAB 14.8 Hz, 2 × CH2), 6.70–6.82 (m, br, 2 × NH), 7.28–7.37 (m, 10 H, 2 × Ph) ppm; 13C-NMR: δ 17.58 (2 × CH3), 43.1 (2 × CH2), 48.4 (2 × OCH3), 71.3 (2 × CH), 99.4 (2 × acetal-C), 127.4 (2 × p-CH, Ph), 127.9 (4 × m-CH, Ph), 128.7 (4 × o-CH, Ph), 138.1 (2 × i-C, Ph), 167.5 (2 × CON) ppm; IR (CHCl3): ṽ 3426, 3067, 3010, 2950, 2838, 1677, 1604, 1527, 1500, 1455, 1379, 1240, 905, 700 cm-1; MS (EI) m/z (%): 444 (M+ + 2, 0.2), 443 (M+ + 1, 1), 442 (M+, 5), 202 (24), 117 (13), 116 (30), 115 (21), 106 (100), 101 (16), 98 (11), 92 (14), 91 (76); Anal. Calcd for C24H30N2O6 (442.508): C 65.14, H 6.83, N 6.33; found C 65.03, H 7.07, N 6.55.
(2R,3R,5S,6S)-5,6-Dimethoxy-N,N,5,6-tetramethyl[1,4]dioxane-2,3-dicarboxamide (9).
To diester 1 (1.05 g, 3.44 mmol) was added methylamine (1.3 mL ca. 30 % solution in ethanol, ca. 32.6 mmol), and the mixture was stirred at 120 °C in an autoclave for 46 h. After cooling to room temperature, the mixture was then transferred to a round-bottom flask, and the volatiles were removed under reduced pressure. After column chromatography on silica gel (7:3 acetone-CHCl3) the product was obtained as colourless crystals (0.998 g, 96 %), m.p. 189 – 190 °C (CHCl3 / acetone); [α]18D = –79.8 (c 1.00, CHCl3); 1H-NMR: δ 1.34 (s, 6 H, 2 × CH3), 2.87 (d, J = 4.8 Hz, 6 H, 2 × NCH3), 3.26 (s, 6 H, 2 × OCH3), 4.28 (s, 2 H, 2 × CH), 6.52 (s, br, 2 × NHCO) ppm; 13C-NMR: δ 17.5 (2 × CH3), 25.9 (2 × NHCH3), 48.7 (2 × OCH3), 71.1 (2 × CH), 99.3 (2 × acetal-C), 168.0 (2 × CONH) ppm; IR (CHCl3): ṽ 3442, 3008, 2949, 2838, 1679, 1537, 1458, 1416, 1378, 1144, 1115, 1074, 1037, 903, 889 cm-1; MS (EI) m/z (%): 290 (M+, 0.07), 259 (11), 227 (30), 142 (55), 116 (100), 115 (95), 113 (98), 112 (100), 102 (74), 101 (96), 100 (14), 85 (49), 84.9 (36), 84 (91), 82.9 (47), 73 (55), 58 (90); Anal. Calcd for C12H22N2O6 (290.32): C 49.65, H 7.64, N 9.65; found C 49.38, H 7.86, N 9.63.
(2R,3R,5S,6S)-N,N’-[(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)bismethylene)]dianiline (10).
To a stirred suspension of LiAlH4 (0.119 g, 3.16 mmol) in dry tetrahydrofuran (0.78 mL) was added dropwise amide 10 (0.260 g, 0.627 mmol) in dry tetrahydrofuran (2.4 mL). The mixture was heated up and refluxed for 4.5 h, and then cooled on ice. Water (0.13 mL), KOH (10 %, 0.38 mL), and again water (0.13 mL) were added successively while the flask was swirled vigorously by hand. The resulting granular precipitate was filtered off through a sintered glass funnel. The filtrate was kept, the solid was returned to the reaction vessel, more tetrahydrofuran was added, and the mixture was refluxed for 30 min. The solids were filtered off once more, the filtrates were combined, and the product was obtained after removal of the solvent on a rotary evaporator. Purification by chromatography on silica gel (1:3 EtOAc-hexane) gave the product as colourless crystals (0.171 g, 71 %), m.p. 104–105 °C (EtOAc- hexane); [α]26D = –119.8 (c 0.93, CHCl3); 1H-NMR: δ 1.25 (s, 6 H, 2 × CH3), 3.12 (s, 6 H, 2 × OCH3), 3.05–3.15 (m, partially overlapped, 2 H, CH2N), 3.28 (d, 2 H, J = 13.1 Hz, CH2N), 3.93–3.94 (m, 2 H, 2 × CH), 6.58 (d, 2 H, J = 7.9 Hz, 2 × o-CH, Ph), 6.67 (t, 1 H, J = 7.3 Hz, p-CH, Ph), 7.10 (t, 2 H, J = 7.9 Hz, 2 × m-CH, Ph) ppm; 13C-NMR: δ 17.6 (2 × CH3), 44.7 (2 × CH2), 48.1 (2 × OCH3), 68.1 (2 × CH), 99.0 (2 × acetal-C), 113.7 (4 × CH, Ph), 118.2 (2 × CH, Ph), 129.3 (4 × CH, Ph), 147.7 (2 × i-C, Ph) ppm; IR (CHCl3): ṽ 3399, 3056, 3009, 2949, 2909, 2836, 1604, 1504, 1463, 1434, 1379, 1316, 1281, 1254, 1228, 1181, 1135, 1080, 1069, 1038, 959, 866, 732, 694, 664, 509 cm-1. MS (EI) m/z (%): 388 (M+ + 2, 0.4), 387 (M+ + 1, 6.0), 386 (M+, 80), 323 (34), 248 (36), 188 (63), 148 (71), 146 (15), 145 (98), 144 (70), 132 (18), 130 (15), 118 (17), 116 (71), 106 (100), 104 (28), 101 (43), 93 (29), 77 (58), 73 (12); Anal. Calcd for C22H30N2O4 (386.49): C 68.37, H 7.82, N 7.25; found C 68.20, H 7.92, N 7.28.
(2R,3R,5S,6S)-(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)bis(N-benzylmethanamine) (11).
Following the reduction procedure used for the synthesis of compound 10, diamine 11 was obtained as the only product from dicarboxamide 8 (0.205 g, 0.464 mmol) and LiAlH4 (0.183 g, 5.22 mmol) in THF (2.03 ml), after 17 h reflux. Yield (0.187 g, 97 %). For analytical purposes it was purified by column chromatography on silica gel (2:1 acetone-CHCl3 + 1 % Et3N). A viscous liquid was obtained: [α]18D = –140.7 (c = 0.85, CHCl3); 1H-NMR: δ 1.29 (s, 6 H, 2 × CH3), 2.64–2.72 (m, 4 H, 2 × CH2), 3.27 (s, 6 H, 2 × OCH3), 3.74, 3.80 (AB system, 4 H, JAB = 13.1 Hz, 2 × N–CH2Ph), 3.85–3.87 (m, 2 H, 2 × CH), 7.22–7.34 (m, 12 H, Ph) ppm; 13C-NMR: δ 17.6 (2 × CH3), 48.0 (2 × OCH3), 49.7 (2 × CH2), 54.0 (2 × CH2Ph), 69.4 (2 × CH), 98.6 (2 × dioxane-CH), 126.9 (2 × p-CH, Ph), 128.1 (4 × m-CH, Ph), 128.3 (4 × o-CH, Ph), 140.1 (2 × i-C, Ph) ppm; IR (neat, salt plates): ṽ 3328, 3085, 3062, 3025, 2991, 2946, 2908, 2830, 1604, 1495, 1454, 1374, 1198, 1125, 1041, 961, 864, 739, 699 cm –1. MS (EI) m/z (%): 414 (M+, 0.30), 351 (3), 188 (3), 174 (3), 162 (22), 159 (9), 158 (3), 144 (8), 120 (12), 116 (13), 106 (4), 101 (7), 92 (5), 91 (100), 73 (3), 65 (5); Anal. Calcd for C24H34N2O4 (414.55): C 69.54, H 8.27, N 6.76; found C 69.31, H 8.46, N 6.73.
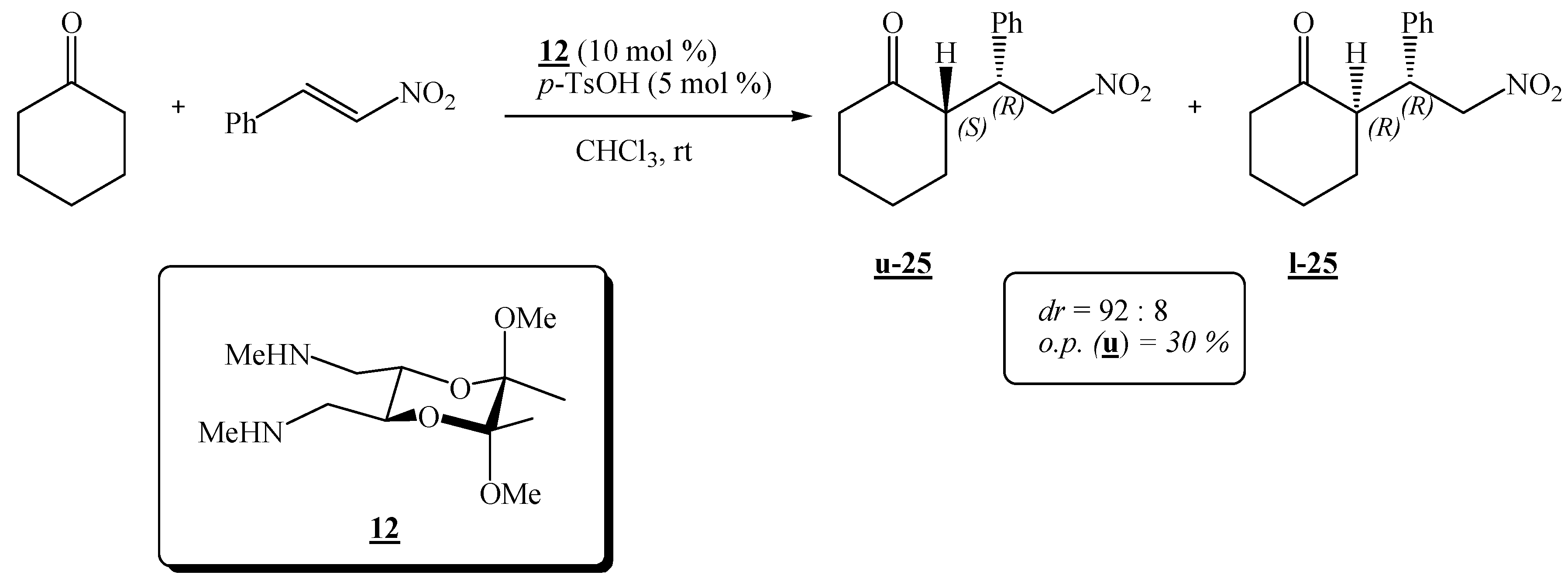
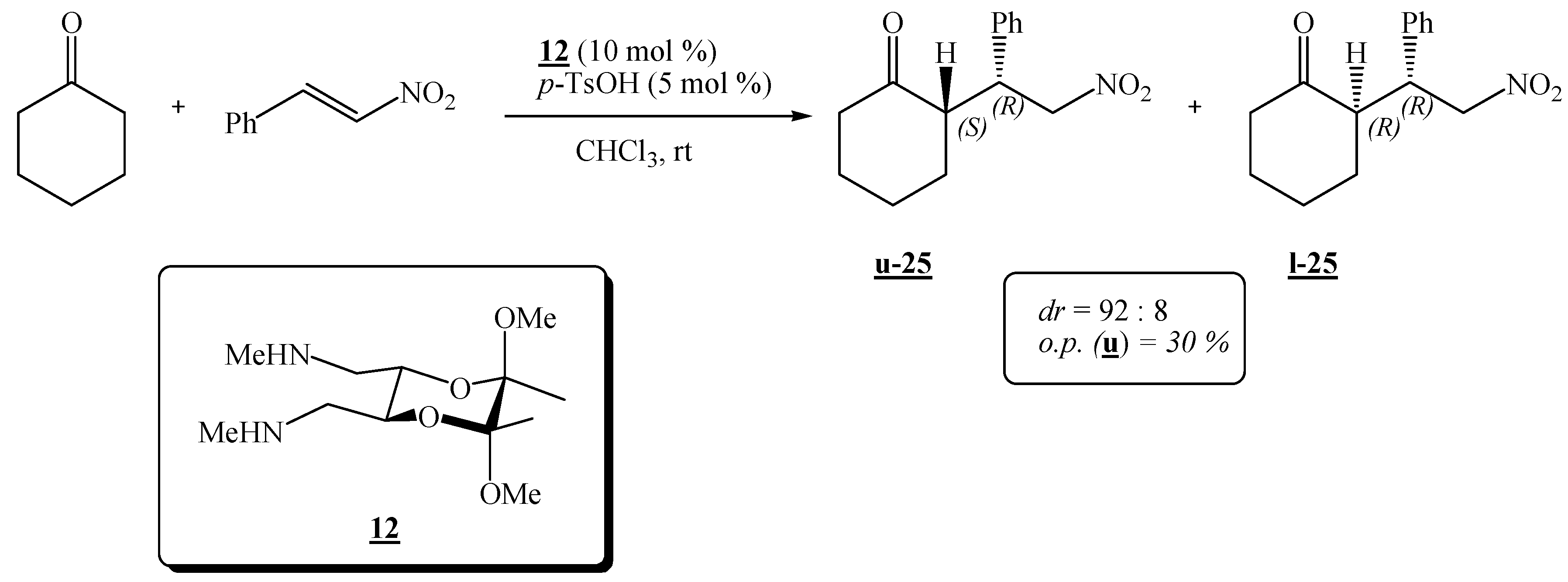
(2R,3R,5S,6S)-(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)bis(N-methylmethanamine) (12).
Amide 9 (0.200 g, 0.689 mmol) dissolved in dry tetrahydrofuran (10.0 mL) was added dropwise to a suspension of LiAlH4 (0.290 g, 7.64 mmol) in dry tetrahydrofuran (1.0 mL). The mixture was refluxed for ca. 24 h, cooled, and more LiAlH4 (0.43 g, 11.3 mmol) was added. The mixture was refluxed for another 24 h. The product was then hydrolysed as described for amine 10 and it was purified by preparative chromatography on neutral alumina (8:1:1 CHCl3-CH2Cl2-MeOH). Colourless crystals were obtained (0.086 g, 39 %). It was recrystallized from diethyl ether, m. p. 53-54 °C; 1H-NMR: δ 1.26 (s, 6 H, 2 × CH3), 2.03 (s, 2 H, 2 × NH), 2.40 (s, 6 H, 2 × NCH3), 2.54–2.64 (m, 4 H, 2 × CH2), 3.24 (s, 6 H, 2 × OCH3), 3.77–3.78 (m, 2 H, 2 × CH) ppm; 13C-NMR: δ 17.6 (2 × CH3), 36.4 (2 × NCH3), 48.0 (2 × OCH3), 52.3 (2 × CH2), 69.2 (2 × CH), 98.6 (2 × acetal-C) ppm; IR (CHCl3): ṽ 3338, 3005, 2949, 2855, 2837, 2804, 1670, 1462, 1451, 1378, 1236, 1219, 1137, 1122, 1039, 957, 885, 869, 817, 777, 663, 562, 423 cm –1; MS (EI) m/z (%): 232 (1.3), 231 (1.5), 218.1 (38), 199.1 (50), 188 (38), 187 (41), 126 (13), 116 (93), 115 (26), 101 (88), 87 (30), 86 (100), 84 (18), 83 (87), 82 (67), 73 (42), 70 (47), 68 (72); Anal. Calcd for C12H26N2O4 (262.35): C 54.94, H 9.99, N 10.68; found C 54.73, H 10.00, N 10.60.
(2R,3R,5S,6S)-1,1’-[(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)bis(methylene)]dipyrrolidine (13).
Bismesylate 3 (0.300 g, 0.765 mmol), freshly distilled pyrrolidine (0.51 mL, 6.11 mmol) and xylene (1.20 mL) were mixed and heated to 120 oC for 3 h. After cooling to room temperature, the volatiles were removed on high vacuum. The residue was dissolved in CH2Cl2, and the product was washed with a saturated solution of sodium chloride and three times with water. The solution was filtered through anhydrous sodium sulfate, and the solvent was removed on a rotary evaporator, to give colourless crystals (0.104 g, 40 %). For analytical purposes the product was purified by column chromatography on silica gel (2:1 acetone-CHCl3 + 1 % Et3N), m.p. 52–53 °C; [α]16D = –133.2 (c 0.82, CHCl3); 1H-NMR: δ 1.28 (s, 6 H, 2 × CH3), 1.77 (s, br, 8 H, 4 × CH2), 2.54–2.63 (d + s, br, 10 H, 2 × CHHN + 2 × CH2N of pyrrolidine), 2.82 (s, br, 2 H, 2 × CHHN), 3.29 (s, 6 H, 2 × OCH3), 3.77 (s, br, 2 H, 2 × CH) ppm; 13C-NMR: δ 17.8 (2 × CH3), 23.6 (4 × CH2), 48.3 (2 × OCH3), 54.9 (4 × CH2N), 57.2 (2 × CH2N), 69.8 (2 × CH), 98.4 (2 × acetal-C) ppm; IR (CHCl3): ṽ 3000, 2963, 2912, 2881, 2832, 2808, 1461, 1375, 1353, 1221, 1209, 1178, 1162, 1141, 1122, 1104, 1084, 1073 cm‑1. MS (EI) m/z (%): 311 (M+- OCH3, 25), 126 (94), 123 (66), 108 (20), 101 (10), 84 (100); Anal. Calcd for C18H34N2O4 (342.48): C 63.13, H 10.01, N 8.18; found C 63.01, H 10.15, N 7.97.
(2R,3R,5S,6S)-1,1’-[(5,6-Dimethoxy-5,6-dimethyl-1,4-dioxane-2,3-diyl)bis(methylene)]dipiperidine (14).
Bismesylate 3 (0.656 g, 1.67 mmol), piperidine (1.31 mL, 13.2 mmol) and xylene (2.6 mL) were mixed, stirred 1h at room temperature, then refluxed for 2.5 h. After cooling to room temperature, the volatiles were evaporated off. The residue was dissolved in CH2Cl2, and the product was washed 4 ° with water. The solution was filtered through anhydrous sodium sulphate, and the solvent was removed on a rotary evaporator, to give colourless crystals (0.619 g, 81 %); the product was recrystallized from hexane, m. p. 84.5–85.0 °C; [α]17D = –187.0 (c 0.37, CHCl3); 1H-NMR: δ 1.27 (s, 6 H, 2 × CH3), 1.34–1.46 (m, 4 H, 2 × CH2), 1.48–1.63 (m, 8 H, 4 × CH2), 2.38–2.48 (m, 10 H, 2 × CHHN, 4 × CH2N), 2.64 (d, 2 H, J = 9.64 Hz, 2 × CHHN), 3.28 (s, 6 H, 2 × OCH3), 3.73 (s, br, 2 H, 2 × CH) ppm; 13C-NMR: δ 17.8 (2 × CH3), 24.3 (2 × CH2), 26.1 (4 × CH2), 48.1 (2 × OCH3), 55.1 (4 × CH2), 60.2 (2 × CH2), 69.2 (2 × CH), 98.4 (2 × acetal-C) ppm; IR (CHCl3): ṽ 3003, 2939, 2855, 2833, 2785, 1468, 1454, 1375, 1303, 1261, 1199, 1174, 1155, 1147, 1137, 1115, 1097, 1088, 1078, 1065, 1039, 991, 864, 748, 737, 663 cm-1; MS (EI) m/z (%): 339 (51), 272 (35), 140 (98), 138 (16), 137 (81), 122 (41), 116 (13), 99 (8), 98.8 (16), 98 (100); Anal. Calcd for C20H38N2O4 (370.53): C 64.83, H 10.34, N 7.56; found C 64.66, H 10.32, N 7.63.
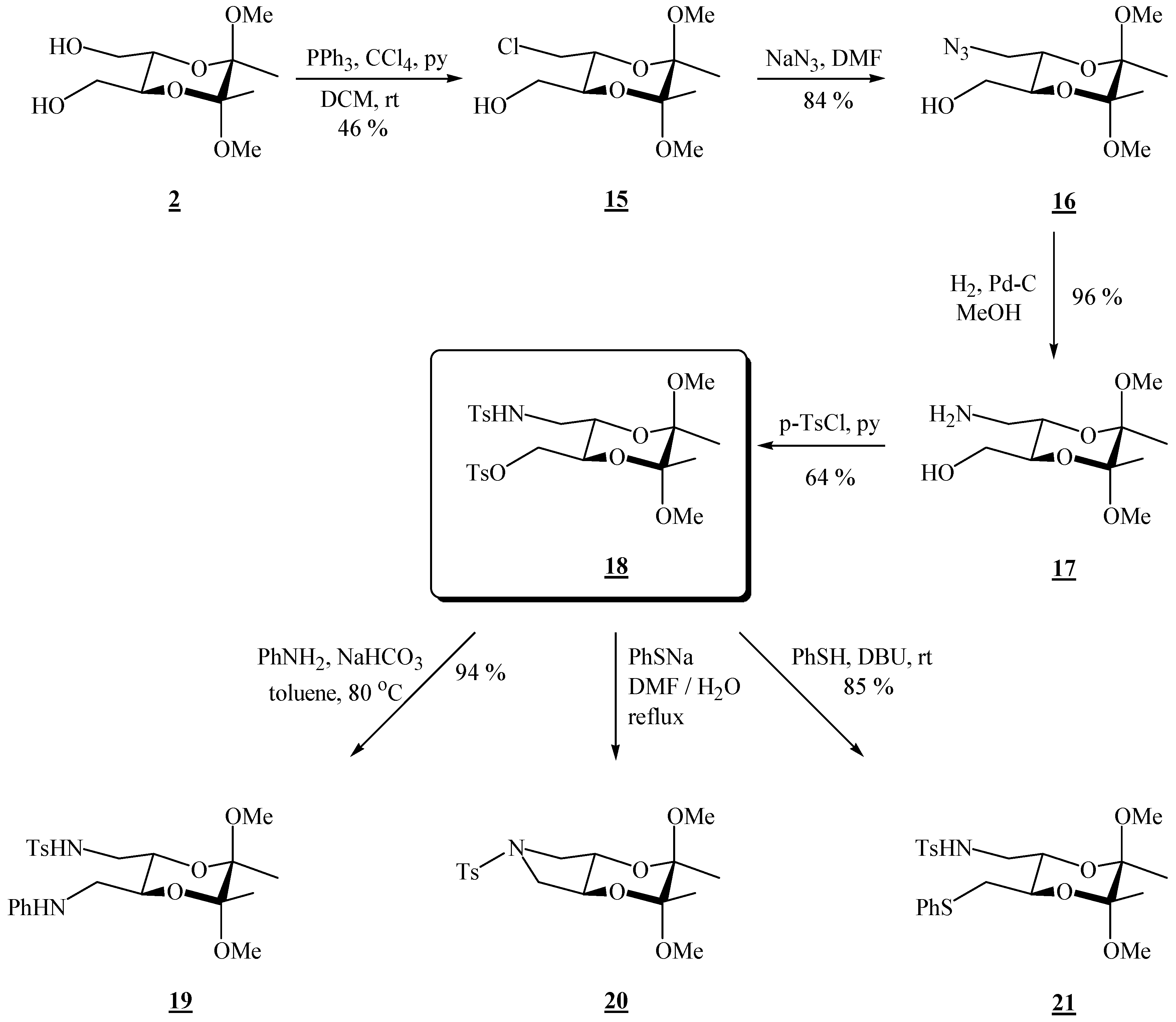
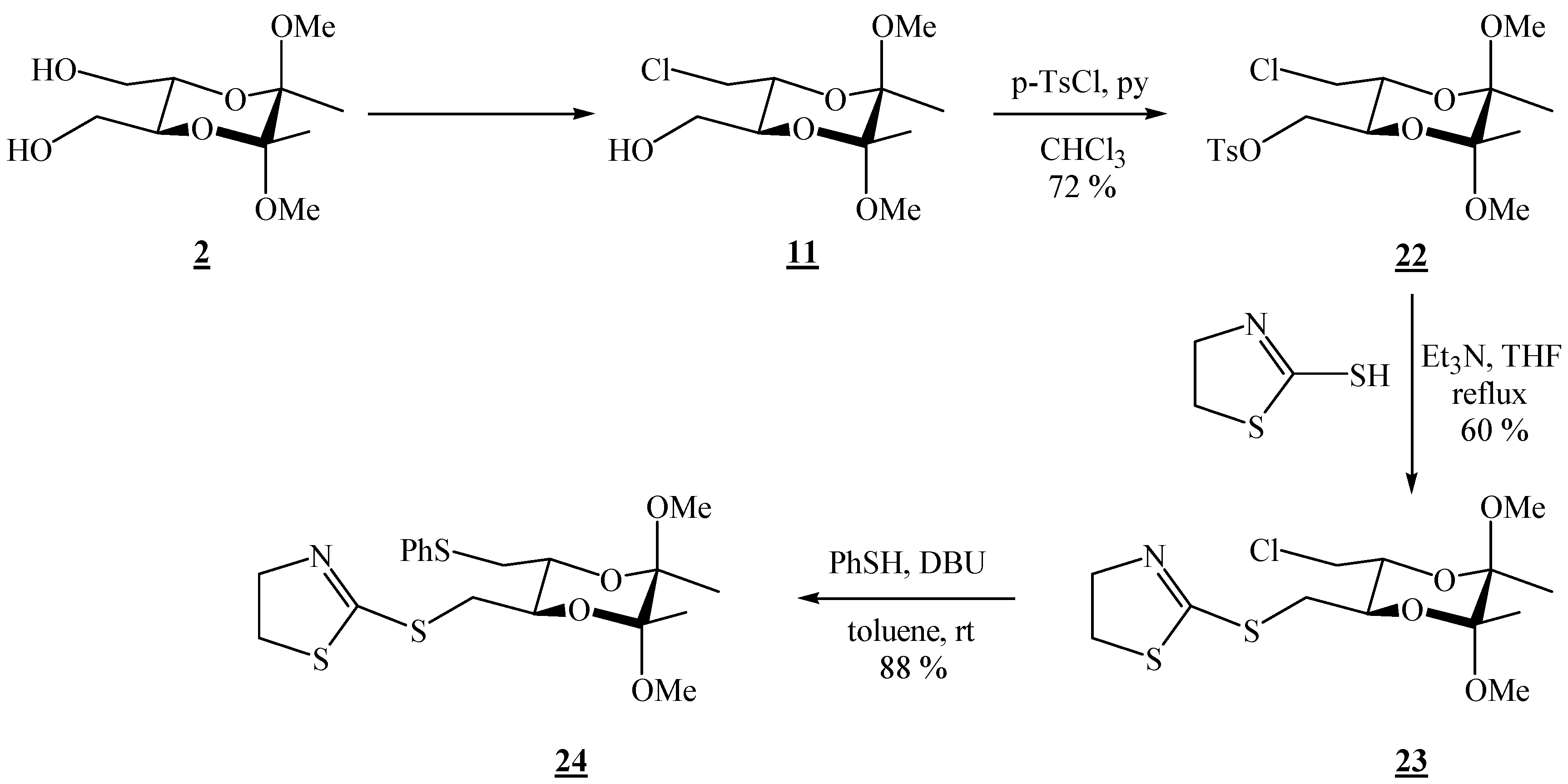
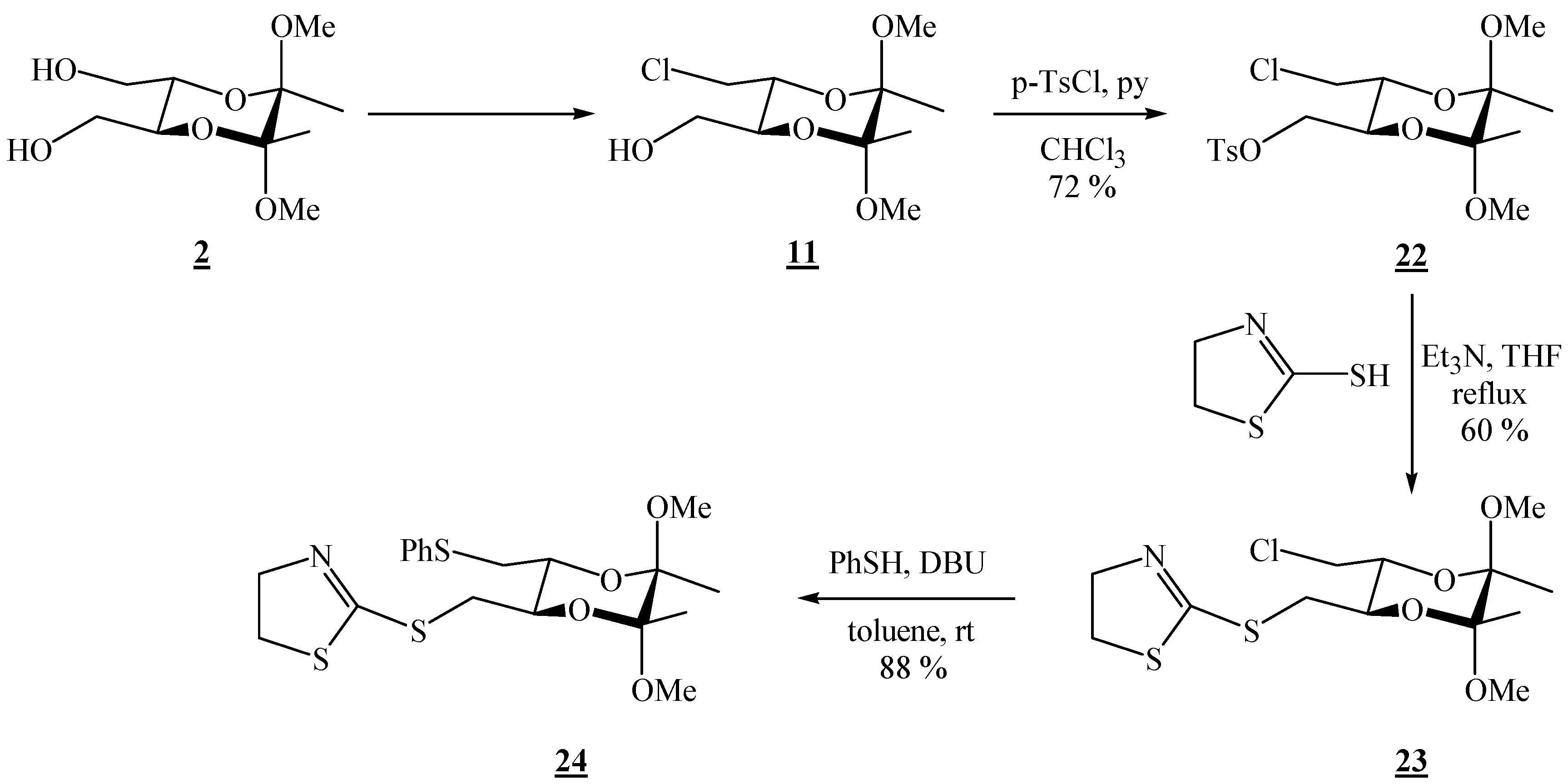
(2R,3R,5S,6S)-3-Chloromethyl-5,6-dimethoxy-5,6-dimethyl[1,4]dioxane-2-methanol (15).
Bis-alcohol 2 (1.76 g, 7.45 mmol) and triphenylphosphine (3.92 g, 15.0 mmol) were dissolved in dichloromethane (13.1 mL) and pyridine (1.22 mL, 15.1 mmol) and tetrachloromethane (1.50 mL, 15.6 mmol) were added. The reaction vessel was wrapped in foil and the solution was stirred at room temperature for 23 h. The volatiles were then evaporated off under reduced pressure. The product was purified by column chromatography on silica gel (3:2 EtOAc-hexane, adsorption from DCM) to give the product as colourless needle-like crystals (0.798 g, 47 %), m.p. 100–101 °C; [α]19D = –188.7 (c 2.05, CHCl3); 1H-NMR: δ 1.30 (s, 3 H, CH3), 1.32 (s, 3 H, CH3), 2.21 (s, br, 1 H, OH), 3.25 (s, 3 H, OCH3), 3.28 (s, 3 H, OCH3), 3.54–3.77 (m, 5 H, 2 × CH2 + 1 × dioxane-CH), 3.95–4.00 (m, 1 H, dioxane-CH) ppm; 13C-NMR: ×δ 17.4 (2 × CH3), 43.6 (CH2Cl), 48.0 (2 × OCH3), 62.1 (CH2OH), 68.5 (dioxane-CH), 70.1 (dioxane-CH), 98.9 (acetal-C), 99.2 (acetal-C) ppm; IR (KBr): ṽ 3232, 3025, 3008, 2991, 2964, 2938, 2884, 2836, 1462, 1425, 1384, 1266, 1221, 1144, 1131, 1090, 1040, 966, 954, 914, 892, 872, 860, 825, 737, 651, 564, 511, 459 cm-1; MS (EI) m/z (%): 225 (6), 223 (51), 193 (59), 192 (14), 191 (53), 165 (38), 151 (38), 116 (15), 113 (52), 105 (67), 101 (62), 88 (63), 76 (100), 75 (41), 73 (55), 71 (10), 70 (58), 69 (41), 57 (44); Anal. Calcd for C10H19ClO5 (254.71): C 47.16, H 7.52; found C 46.93, H 7.49.
(2R,3R,5S,6S)-3-Azidomethyl-5,6-dimethoxy-5,6-dimethyl[1,4]dioxane-2-methanol (16).
Monochloride 15 (0.708 g, 2.78 mmol), sodium azide (0.704 g, 10.8 mmol) and dry dimethylformamide (14.0 mL) were mixed. The resulting suspension was stirred at 80 °C, for 88 h. After cooling to room temperature, the product was extracted as described for the bisazide. A single product was obtained, as colourless crystals (0.607 g, 84 %), used as it is in the next reaction. For analytical purposes it was purified by chromatography on silica gel (2:3 EtOAc-hexane), m.p. 71 – 72 °C (EtOAc / hexane); [α]27D = –134.2 (c 2.02, CHCl3); 1H-NMR: δ 1.31 (s, 3 H, CH3), 1.33 (s, 3 H, CH3), 1.91 (s, br, 1 H, OH), 3.26 (dd, 1 H, J = 3.2, 12.0 Hz, CH2N3), 3.39 (dd, 1 H, J = 6.8, 12.0 Hz, CH2N3), 3.60 (dd, 1 H, J = 5.2, 12.0 Hz, CHHOH), 3.69 (dd, 1 H, J = 3.2, 12.0 Hz, CHHOH), 3.75 (ddd, 1 H, J = 3.2, 5.2, 9.6 Hz, CH-CH2OH), 3.97 (ddd, 1 H, J = 2.8, 6.8, 9.6 Hz, CH-CH2N3) ppm; 13C-NMR: δ 17.4 (2 × CH3), 48.0 (2 × OCH3), 50.9 (CH2), 62.0 (CH2), 68.4 (CH), 69.5 (CH), 99.0 (2 × acetal-C) ppm; IR (CH2Cl2): ṽ 3590, 2996, 2950, 2929, 2836, 2102, 1460, 1446, 1377, 1266, 1224, 1202, 1138, 1132, 1037, 962, 861, 754, 746, 726 cm-1; MS (EI) m/z (%): 172 (20), 129 (11), 116 (25), 110 (22), 101 (99), 85 (11), 84 (22), 76 (86), 75 (89), 73 (100), 70 (86), 69 (49), 68 (60), 67 (85), 59 (20), 57 (70), 56 (90), 55.0 (13), 54 (11); Anal. Calcd for C10H19N3O5 (261.28): C 45.97, H 7.33, N 16.08; found C 46.14, H 7.14, N 15.74.
(2R,3R,5S,6S)-3-Aminomethyl-5,6-dimethoxy-5,6-dimethyl[1,4]dioxane-2-methanol (17).
Azido alcohol 16 (0.270 g, 0.943 mmol) dissolved in dry MeOH (13.5 mL) was transferred to an hydrogenation flask and Pd on charcoal (0.034 g, 10 %, 0.032 mmol) was added. The flask was connected to a hydrogenation apparatus, and the hydrogenation was performed at room temperature and at 5 psi for 4 h. The reaction mixture was then treated as described for the diazide. A single product was obtained (0.260 g, 96 %), used as it is in the next reaction. For analytical purposes it was crystallized from ether, m.p. 109 °C; 1H-NMR: δ 1.16 (s, 6 H, 2 × CH3), 2.72 (s, br, 2 H, CH2N), 2.83 (s, br, 2 H, NH2), 3.12 (s, 6 H, 2 × OCH3), 3.47 (s, br, CH2O), 3.50 (s, br, 2 × CH) ppm; 13C-NMR: δ 17.3 (2 × CH3), 42.5 (CH2N), 47.6 (2 × OCH3), 62.0 (CH2OH), 70.6 (CH), 71.0 (CH), 98.4 (2 × acetal-C) ppm; IR (KBr): ṽ 3382, 3301, 3113, 3012, 2995, 2949, 2926, 2894, 2882, 2834, 1606, 1456, 1440, 1395, 1379, 1215, 1126, 1083, 1037, 957, 889, 858, 655, 566 cm-1; MS (EI) m/z (%): 172 (78), 116 (12), 101 (50), 87 (16), 86 (15), 75 (31), 73 (41), 70 (38), 69 (100), 68 (14), 56 (94); Anal. Calcd for C10H21NO5 (235.28): C 51.05, H 9.00, N 5.95; found C 50.87, H 8.81, N 6.03.
(2R,3R,5S,6S)-5,6-Dimethoxy-5,6-dimethyl-3-[(p-toluenesulfonylamino)-methyl]-[1,4]-dioxan-2-yl-methyl p-toluenesulfonate (18).
Amino alcohol 17 (0.296 g, 1.26 mmol) was dissolved in dry chloroform (1.50 mL) and pyridine (0.49 mL, 6.06 mmol) was added. The solution was cooled to 0 °C, and p-tosyl chloride (0.604 g, 3.17 mmol) was added in portions. The resulting solution was stirred for 2.5 h at 0 °C, under argon. The reaction mixture was then partitioned between ether and water, and the ether layer washed successively with HCl (2 m) and a saturated solution of sodium bicarbonate. The solvent was evaporated off under reduced pressure, and the residue was chromatographed on silica gel (2:3 EtOAc-hexane, adsorption from CHCl3), to give the product as colourless crystals (0.438 g, 64 %), m.p. 110 °C (EtOAc / hexane); [α]25D = –87.2 (c 1.00, CHCl3); 1H-NMR: δ 1.18 (s, 3 H, CH3), 1.20 (s, 3 H, CH3), 2.44 (s, 3 H, CH3 of Ts), 2.46 (s, 3 H, CH3 of Ts), 2.88–2.94 (m, 1 H, CH2N), 2.88–2.94 (m, 1 H, CH2N), 3.10 (s, 3 H, OCH3), 3.12 (s, 3 H, OCH3), 3.71–3.78 (m, 2 H, 2 × CH), 3.99 (dd, 1 H, J = 3.2, 10.8 Hz, CH2O), 4.11 (dd, 1 H, J = 4.4, 11.2 Hz, CH2O), 4.88 (t, br, 1 H, NH), 7.33 (d, 2 H, J = 8 Hz, aniline or Ts), 7.37 (d, 2 H, J = 8 Hz, aniline or Ts), 7.72 (d, 2 H, J = 8 Hz, aniline or Ts), 7.82 (d, 2 H, J = 8 Hz, aniline or Ts) ppm; 13C-NMR: δ 17.2 (CH3), 17.3 (CH3), 21.5 (CH2NTs), 21.6 (CH3, Ts), 43.2 (CH2NTs), 48.0 (2 × OCH3), 67.1 (CH), 67.2 (CH), 68.7 (CH2O), 98.9 (acetal-C), 99.0 (acetal-C), 127.0 (CH, aniline or Ts), 128.0 (CH, aniline or Ts), 129.8 (CH, aniline or Ts), 129.9 (CH, aniline or Ts), 132.6 (Cq, aniline or Ts), 136.5 (Cq, aniline or Ts), 143.6 (Cq, aniline or Ts), 145.0 (Cq, aniline or Ts) ppm; IR (KBr): ṽ 3368, 3031, 3009, 3000, 2951, 2930, 2837, 1599, 1495, 1456, 1414, 1402, 1375, 1337, 1307, 1291, 1212, 1190, 1177, 1162, 1137, 1095, 1036, 980, 930, 866, 815, 774, 662, 585, 553 cm-1; MS (EI) m/z (%): 482 (0.16), 481 (0.45), 480 (2.66), 294 (9), 224 (22), 223 (84), 155 (97), 101 (12), 92 (17), 91 (100), 68 (74), 65 (23); Anal. Calcd for C24H33NO9S2 (543.65): C 53.02, H 6.12, N 2.58; found C 53.34, H 2.45, N 6.15.
(2R,3R,5S,6S)-5,6-Dimethoxy-5,6-dimethyl-3-(N-phenylamino)methyl[1,4]dioxan-2-ylmethyl p-toluene sulfonate (19).
Tosyl derivative 18 (0.207 g, 0.368 mmol) was dissolved in toluene (0.76 mL) and sodium bicarbonate (0.066 g, 0.786 mmol) was added. To the resulting suspension aniline (0.17 mL, 1.87 mmol) was added, and the mixture was left stirring at 80 °C for 48 h. More toluene (0.4 mL) was added and also aniline (0.17 mL, 1.87 mmol) and the mixture was stirred at 80 °C another 24 h. The reaction mixture was then cooled to room temperature, filtered, and dried. The product (0.161 g, 94 %) was obtained after chromatography on silica gel (2:3 EtOAc-hexane), m.p. 174-175 ºC (EtOAc / hexane); [α]29D = –113.7 (c 2.01, CHCl3); 1H-NMR: δ 1.17 (s, 3 H, CH3), 1.21 (s, 3 H, CH3), 2.35 (s, 3 H, CH3 of Ts), 2.88–3.02 (m, partially overlapped, 1 H, CHHNHSO2), 2.97 (s, 3 H, OCH3), 3.08 (s, 3 H, OCH3), 3.00–3.18 (m, partially overlapped, 3 H, CHHNHSO2 + 2 × H of CH2NHPh), 3.68–4.00 (m, 2 H, 2 × CH), 4.94 (t, 1 H, NHSO2), 6.64 (d, 2 H, J = 7.6 Hz, aniline), 6.71 (t, 1 H, J = 7.2 Hz, p-H, aniline), 7.13 (t, 2 H, J = 7.2 Hz, aniline), 7.22 (d, J = 7.6 Hz, 2 H, Ts), 7.65 (d, J = 7.6 Hz, 2 H, Ts); 13C-NMR: δ 17.3 (CH3), 17.4 (CH3), 21.4 (CH3 of Ts), 43.6 (CH2NHSO2), 44.5 (CH2NHPh), 47.9 (OCH3), 48.1 (OCH3), 67.2 (CH), 68.2 (CH), 98.85 (acetal-C), 98.91 (acetal-C), 113.7 (2 × CH, aniline), 118.3 (p-CH, aniline), 127.0 (2 × CH, Ts), 129.2 (2 × CH, aniline), 129.8 (2 × CH, Ts), 136.6 (Cq, Ts), 143.6 (CqSO2N), 147.4 (Cq, aniline) ppm; IR (KBr): ṽ 3369, 3060, 3016, 2990, 2947, 2904, 2876, 2852, 1605, 1505, 1463, 1393, 1378, 1335, 1260, 1253, 1237, 1208, 1184, 1164, 1135, 1076, 1050, 1034, 993, 951, 855, 816, 763, 697, 666, 655 cm-1; MS (EI) m/z (%): 466 (M+ + 2, 1), 465 (M+ + 1, 4), 464 (M+, 24), 401 (18), 223 (12), 188 (17), 187 (27), 155 (41), 145 (47), 132 (28), 116 (24), 106 (100), 101 (20), 93 (18), 91 (65), 73 (11); Anal. Calcd for C23H32N2O6S (414.46): C 59.46, H 6.94, N 6.03; found C 59.19, H 6.96, N 5.75.
(2R,3R,5S,6S)-2,3-Dimethoxy-2,3-dimethyl-6-(toluene-4-sulfonyl)-hexahydro-[1,4]dioxino[2,3-c]-pyrrole (20).
Tosylate 18 (0.061 g, 0.112 mmol) dissolved in dimethylformamide (0.15 mL) was added to sodium thiophenolate (0.045 g, 0.341 mmol) dissolved in H2O (0.15 mL), producing an exothermic reaction. The mixture was refluxed for 16 h. It was then cooled to room temperature and ether and water were added. The layers were separated, and the aqueous phase was extracted three times more with ether. Removal of the solvent gave a crude product which was chromatographed on silica gel (4:0.2 hexane- EtOAc). Colourless crystals were obtained
mp?; 1H-NMR: δ 1.21 (s, 6 H, 2 × CH3), 2.37 (s, 3 H, CH3 of Ts), 3.02 (t, 2 H, J = 9.2 Hz), 3.13 (s, 3 H, 2 × OCH3), 3.51 (t, 2 H, J = 6.8 Hz), 3.73 (t, 2 H, J = 6.0 Hz), 7.27 (d, 2 H, J = 8.0 Hz, Ts), 7.64 (d, 2 H, J = 8.0 Hz, Ts); MS (EI) m/z (%): 279 (0.74), 214 (39), 202 (11), 155 (70), 149 (11), 102 (32), 92 (9), 91 (100), 84 (11), 65 (20).
(2R,3R,5R,6R)-5,6-Dimethoxy-5,6-dimethyl-3-(phenylthio)methyl[1,4]dioxan-2-ylmethyl p-toluene sulfonamide (21).
Tosylate 18 (0.082 g, 0.151 mmol) was dissolved in toluene (0.17 mL) and thiophenol (0.020 mL, 0.195 mmol) was added. The components were mixed well. DBU (0.020 mL, 0.134 mmol) was added. The resulting solution was stirred under argon, at room temperature, overnight. Volatiles were then removed on a rotary evaporator and the remaining residue was chromatographed on silica gel plates (3:1 hexane-EtOAc) to give the product as colourless hygroscopic crystals (0.062 g, 85 %), m.p. 58-59 °C (hexane-EtOAc); [α]26D = –132.4 (c 0.79, CHCl3); 1H-NMR: δ 1.15 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 2.36 (s, 3 H, CH3 of Ts), 2.79–2.86 (m, 1 H, CH2N), 2.90–2.96 (m, 2 H, CH2S), 3.08 (s, 3 H, OCH3), 3.11 (s, 3 H, OCH3), 3.03–3.19 (m, partially overlapped, 1 H, CH2N), 3.63–3.71 (m, 2 H, 2 × CH), 4.84 (t, 1 H, NHSO2), 7.11 (t, 1 H, p-H, SPh), 7.10–7.29 (m, 6 H, 4 × CH, SPh + 2 × CH, Ts), 7.65 (d, 2 H, J = 8.4 Hz, 2 × CH, Ts) ppm; 13C-NMR: δ 17.3 (CH3), 17.4 (CH3), 21.5 (CH3 of Ts), 35.0 (CH2S), 43.7 (CH2N), 48.0 (OCH3), 48.1 (OCH3), 68.3 (CH), 69.9 (CH), 98.9 (acetal-C), 99.1 (acetal-C), 126.3 (CH, SPh), 127.1 (2 × CH, Ts), 128.9 (2 × CH, SPh), 129.5 (2 × CH, SPh), 129.8 (2 × CH, Ts), 136.1 (Cq, SPh or Ts), 136.6 (Cq, SPh or Ts), 143.6 (CqSO2N) ppm; IR (CHCl3): ṽ 3359, 3029, 3007, 2951, 2030, 2836, 1599, 1481, 1459, 1439, 1413, 1379, 1335, 1214, 1162, 1141, 1131, 1093, 1049, 1036, 1004, 961, 892, 856, 814, 778, 761, 748, 740, 691, 663, 552 cm-1; MS (EI) m/z (%): 481 (M+, 0.22), 333 (10), 294 (10), 224 (54), 223 (49), 184 (10), 162 (82), 155 (97), 123 (22), 110 (11), 109 (17), 101 (11), 91 (100), 68 (14), 65 (13); Anal. Calcd for C23H31NO6S2 (481.62): C 57.36, H 6.49, N 2.91; found C 57.05, H 6.50, N 2.62.
(2R,3R,5S,6S)-3-Chloromethyl-5,6-dimethoxy-5,6-dimethyl[1,4]-dioxan-2-ylmethyl-p-toluene sulfonate (22).
Monochloride 15 (0.541 g, 2.13 mmol) was dissolved in chloroform (2.18 mL) and pyridine was added (0.35 mL, 4.33 mmol). The solution was cooled to 0 °C, and p-tosyl chloride (0.623 g, 3.27 mmol) was added in portions. The mixture was then stirred for 2 h at 0 °C (ice bath), and then it was poured into a 1:1 ether / water mixture. Afterwards it was washed successively with 2 M HCl, a saturated solution of NaHCO3, and H2O. The solvent was removed under reduced pressure to give a white solid, which was purified by column chromatography on silica gel (1.5:8.5 EtOAc-hexane, adsorption from CHCl3). Colourless crystals were obtained (0.626 g, 72 %), m.p. 74-75 °C (EtOAc-hexane); [α]16D = –20.5 (c 2.01, CHCl3); 1H-NMR: δ 1.21 (s, 3 H, CH3), 1.28 (s, 3 H, CH3), 2.46 (s, 3 H, CH3 of Ts), 3.18 (s, 3 H, OCH3), 3.24 (s, 3 H, OCH3), 3.49–3.57 (m, 2 H, CH2Cl), 3.83–3.90 (m, 2 H, 2 × CH), 4.02–4.06 (m, 2 H, CH2OSO2), 7.36 (d, 2 H, J = 8.0 Hz, Ts), 7.81 (d, 2 H, J = 8.0 Hz, Ts) ppm. 13C-NMR: δ 17.2 (CH3), 17.3 (CH3), 21.6 (CH3 of Ts), 43.2 (CH2Cl), 48.0 (2 × OCH3), 67.8 (CH), 68.8 (CH), 68.9 (CH2OSO2), 99.1 (acetal-C), 99.2 (acetal-C), 128.0 (2 × CH, Ts), 129.9 (2 × CH, Ts), 132.7 (Cq – CH3), 145.0 (Cq – SO2–O) ppm. IR (KBr): ṽ 3019, 2993, 2951, 2909, 2838, 1597, 1455, 1370, 1353, 1190, 1170, 1127, 1097, 1037, 982, 931, 881, 859, 847, 819, 704, 680, 665, 619, 573, 556, 535, 504, 463 cm-1; MS (EI) m/z (%): 380 (0.13), 379 (M+ + 2 – OMe, 0.97), 225 (17), 155 (100), 91 (45), 88 (18), 73 (10); Anal. Calcd for C17H25ClO7S (408.89): C 49.94, H 6.16, S 7.84; found C 50.23, H 6.15, S 7.47.
(2R,3R,5R,6R)-2-(5,6-Dimethoxy-5,6-dimethyl-3-chloro[1,4]dioxan-2-ylmethylthio)-4,5-dihydro-thiazole (23).
Monotosylate 22 (0.330 g, 0.807 mmol) and 2-mercapto-2-thiazoline (0.100 g, 0.839 mmol) were dissolved in dry tetrahydrofuran. Triethylamine (0.220 mL, 1.58 mmol) was added, and the solution was refluxed for 20 h. After cooling to room temperature, the volatiles were removed on a rotary evaporator. The product was isolated by chromatography on silica gel (1:3 hexane-EtOAc). 0.172 g (60 % yield) of a clear colourless liquid were obtained, [α]26D = –123.3 (c 1.00, CHCl3); 1H-NMR: δ 1.23 (s, 3 H, CH3), 1.25 (s, 3 H, CH3), 3.01 (dd, 1 H, J = 8.6, 13.6 Hz, CH2S), 3.19 (s, 3 H, OCH3), 3.22 (s, 3 H, OCH3), 3.33 (t, J = 8.0 Hz, 2 H, SCH2 of thiazoline), 3.43 (dd, 1 H, J = 2.9, 13.6 Hz, CH2S), 3.45 (dd, 1 H, J = 7.2, 11.9 Hz, CH2Cl), 3.65 (dd, 1 H, J = 2.8, 12.0 Hz, CH2Cl), 3.74 ( td, 1 H, J = 2.8, 7.2 Hz, CH), 3.83 (td, 1 H, J = 2.9, 8.7 Hz, CH), 4.11 (td, 3 H, J = 3.0, 8.0 Hz, NCH2 of thiazoline) ppm; 13C-NMR: δ 17.3 (CH3), 33.5 (CH2S), 35.7 (SCH2), 43.7 (CH2Cl), 47.95 (OCH3), 47.98 (OCH3), 63.8 (CH2N), 68.8 (CH), 71.4 (CH), 99.0 (2 × acetal-C), 165.2 (C=N) ppm; IR (KBr): ṽ 3031, 3010, 2953, 2909, 2837, 1599, 1456, 1433, 1211, 1190, 1177, 1141, 1122, 1097, 1036, 985, 925, 859, 815, 780, 758, 744, 663, 555 cm-1; MS (EI) m/z (%): 324 (36), 292 (13), 224 (16), 206 (9.8), 208 (3.4), 174 (27), 173 (64), 172 (100), 170 (12), 119 (14), 116 (11), 101 (44), 91 (18), 89 (17), 85 (48), 83 (67), 75 (69), 73 (48), 72 (10), 60 (22), 59 (17), 53 (24); Anal. Calcd for C13H22ClNO4S2 (355.90): C 43.87, H 6.23, N 3.94, S 18.02; found C 44.14, H 6.02, N 3.75, S 17.84.
(2R,3R,5R,6R)-2-(5,6-Dimethoxy-5,6-dimethyl-3-phenylthiomethyl[1,4]dioxan-2-ylmethylthio)-4,5-dihydrothiazole (24).
Monochloride 23 (0.045 g, 0.126 mmol) was dissolved in dry toluene (0.26 mL); thiophenol (0.030 mL, 0.292 mmol) was added and the mixture was well stirred; finally 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (0.030 mL, 0.201 mmol) was added. The solution was then stirred at room temperature for 22 h. The solvent was evaporated off, and the product was isolated by chromatography on silica gel (1:9 EtOAc-CH2Cl2). Colourless hygroscopic crystals were obtained (0.048 g, 88 %), m.p. 58–59 °C (EtOAc-CH2Cl2); [α]27D = –179.7 (c 0.61, CHCl3); 1H-NMR: δ 1.21 (s, 6 H, 2 ° CH3), 2.94–3.01 (m, 2 H, CHHSPh + CHHS-thiazoline), 3.12 (s, 3 H, OCH3), 3.14 (dd, 1 H, J = 3.6, 13.6 Hz, CH2S), 3.20 (s, 3 H, OCH3), 3.32 (t, 2 H, J = 8.0 Hz, SCH2 of thiazoline), 3.55 (dd, 1 H, J = 2.0, 13.6 Hz), 3.71 (td, 1 H, J = 3.6, 9.6 Hz, CH), 3.82 (td, 1 H, J = 2.8, 9.2 Hz, CH), 4.10 (t, 2 H, J = 8.0 Hz, NCH2), 7.10 (t, 1 H, J = 7.2 Hz, Ph), 7.20 (t, 2 H, J = 7.6 Hz, Ph), 7.32 (d, 2 H, J = 7.6 Hz, Ph) ppm; 13C-NMR: δ 17.4 (2 × CH3), 34.1 (CH2), 35.3 (CH2), 35.4 (CH2 of thiazoline), 48.0 (OCH3), 48.1 (OCH3), 63.1 (NCH2 of thiazoline), 70.4 (CH), 70.5 (CH), 99.1 (2 × acetal-C), 126.1 (p-CH, Ph), 128.9 (2 × CH, Ph), 129.6 (2 × CH, Ph), 136.4 (i-C, Ph), 167.6 (SC=N) ppm; IR (CHCl3): ṽ 3077, 3062, 3007, 2951, 2854, 2836, 1569, 1481, 1460, 1439, 1378, 1306, 1234, 1212, 1161, 1138, 1122, 1091, 1036, 1018, 997, 968, 943, 922, 890, 857, 776, 764, 754, 738, 692, 665, 647, 565, 535 cm-1; MS(EI) m/z (%): 431 (M+ + 2, 0.21), 430 (M+ + 1, 0.32), 429 (M+, 2.8), 172 (100), 162 (20), 110 (9), 101 (13), 85 (14); Anal. Calcd for C19H27NO4S3.H2O (438.62): C 52.03, H 6.44, N 3.19; found C 51.99, H 6.34, N 3.47.



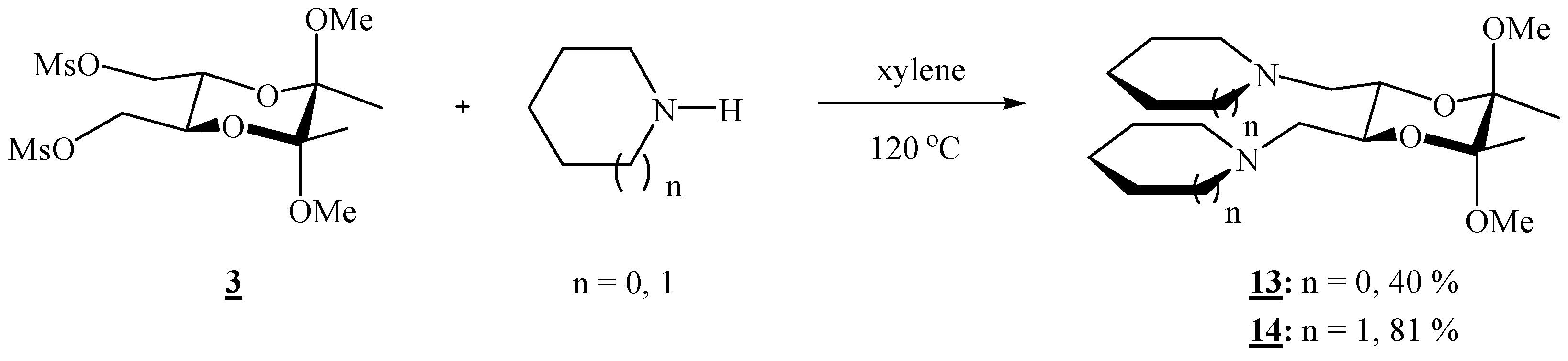

{kind=link}
{kind=link}
{kind=link}
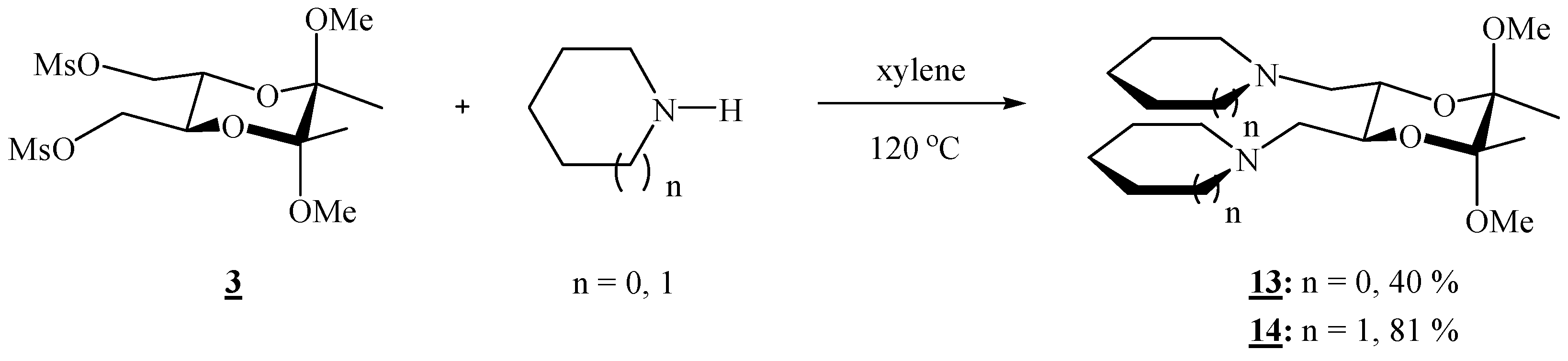{kind=link}
{kind=link}
{kind=link}
{kind=link}