Syntheses and Structural Studies of Schiff Bases Involving Hydrogen Bonds



1
Departamento de Química Orgánica y Bio-Orgánica, Facultad de Ciencias, UNED, Senda del Rey 9, E-28040 Madrid, Spain
2
Instituto de Química Médica (CSIC), Centro de Química Orgánica 'Manuel Lora Tamayo', Juan de la Cierva 3, E-28006 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2006, 11(6), 453-463; https://doi.org/10.3390/11060453
Submission received: 7 June 2006
/
Revised: 20 June 2006
/
Accepted: 20 June 2006
/
Published: 21 June 2006
Abstract
:New Schiff bases have been prepared by reacting 3-hydroxy-4-pyridine-carboxaldehyde with various amines. NMR spectroscopic methods provided clear evidence that the Schiff bases exist in the solid state and in solution as hydroxyimino tautomers with the E-configuration. A study of the stabilities of the tautomeric forms and the different conformers has been carried out using density functional calculations at the B3LYP/6-31G** level.
Introduction
It is well established that hydrogen bonds (HB) and proton transfer (PT) reactions play a crucial role in many areas of physical, chemical and biological phenomena. It appears that the low-barrier hydrogen bond (LBHB) may act by stabilizing intermediates in enzymatic reactions and lowering the energy of transition states [1]. Schiff bases derived from o-hydroxyarylaldehydes have attracted much attention because of the tautomeric equilibria between their hydroxyimino and oxoenamino forms, as shown in Scheme 1 [2,3,4,5,6,7], their ability to act as ligands with many different metals in various oxidation states, to the point of being considered “privileged ligands” [8], and also due to their biological properties [9,10].
Scheme 1.

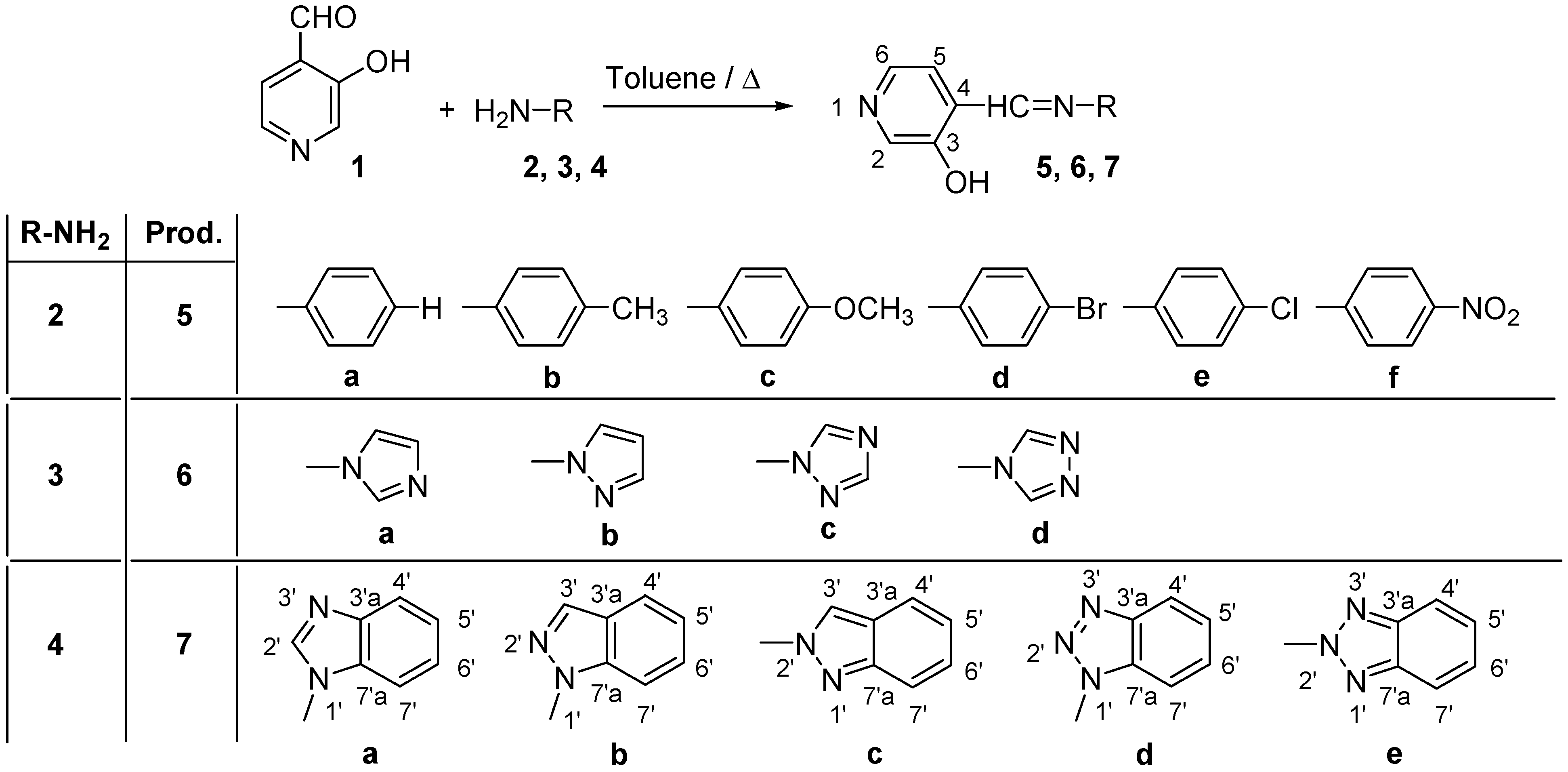
Within the framework of a general project investigating mechanisms of enzymatic reactions involving hydroxypyridines [11], we have already studied the new Schiff bases 5a-5f and 6a-6d shown in Scheme 2, obtained from 3-hydroxy-4-pyridinecarboxaldehyde (1), and 4-R-substituted anilines 2a-2f or N-aminoazoles 3a-3d, including their preparation and structural studies in solution (1H-, 13C- and 15N-NMR spectroscopy) and in solid state (13C- and 15N-CPMAS NMR) [6,12]. As an extension of that work, we now present our results concerning derivatives 7a-7e, obtained from 1 and the corresponding N-aminobenzazoles 4a-4e.
Results and Discussion
Like the previously reported reactions of 3-hydroxy-4-pyridinecarboxaldehyde (1) with the amines 2a-2f and 3a-3d to give the series of compounds 5 and 6, reaction of 1 with 4a-4e afforded the imines 7a-e in nearly quantitative yields (Scheme 2).
Scheme 2.

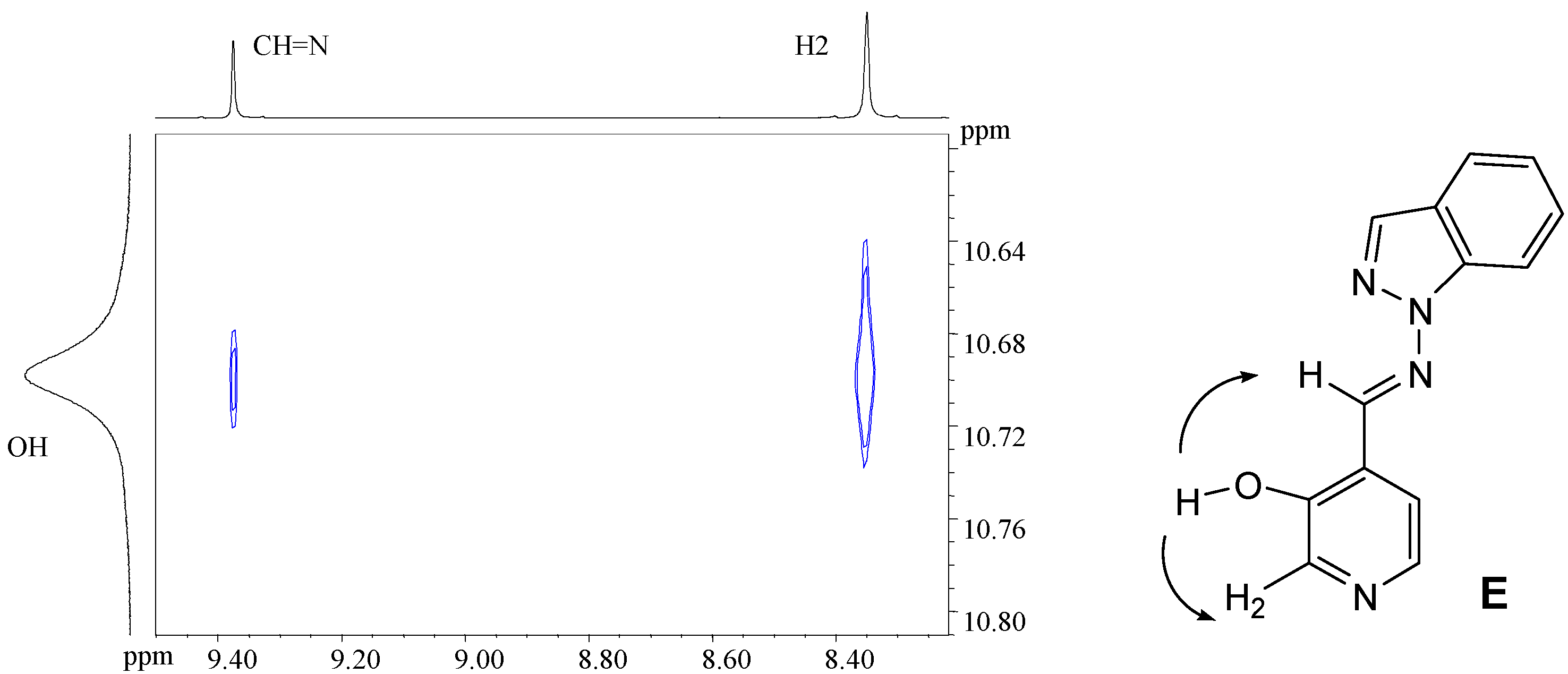
The relative stabilities of the hydroxyimino/oxoenamino tautomers have also been approached using B3LYP/6-31G** calculations; in the case of compound 7a the hydroxyimino tautomer with E configuration is more stable than the oxoenamino one by about 48.3 kJ mol–1. The presence of a N atom in the α position, as in compounds 7b-7e, further increases the stability difference to the point that only the hydroxyimino tautomer is at an energy minimum. In all cases only the E isomer has been observed, as proven by 2D-NOESY NMR experiments showing the correlation between the imino proton and the pyridine H5 proton (Scheme 3). These results are in full agreement with the B3LYP/6-31G** calculations, which favor the E isomer over the Z isomer by 52.4 kJ mol–1 in compound 7a, 49.0 kJ mol–1 in 7b, 45.3 kJ mol–1 in 7c, 50.0 kJ mol–1 in 7d and 56.3 kJ mol–1 in 7e, respectively.
Scheme 3.

These results are in agreement with the experimental NMR observations. The signals providing more information on the type of tautomer are those corresponding to the CH=N atom, with 15N-NMR chemical shifts between –63.8 ppm and –90.2 ppm in DMSO-d6. These values are typical of the non-protonated nitrogen atom of a Schiff base and indicate that compounds 7a-7e exist in the hydroxyimino tautomeric form.
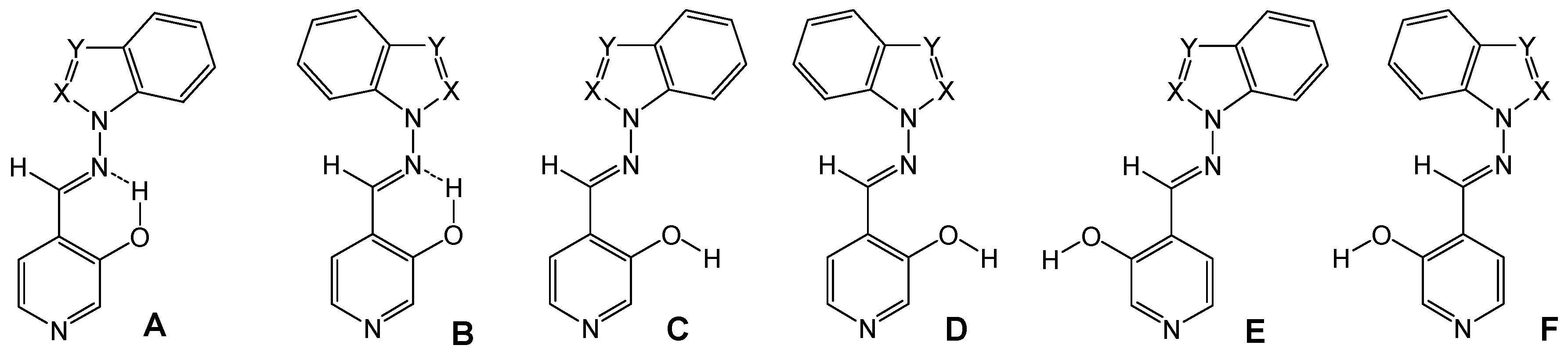
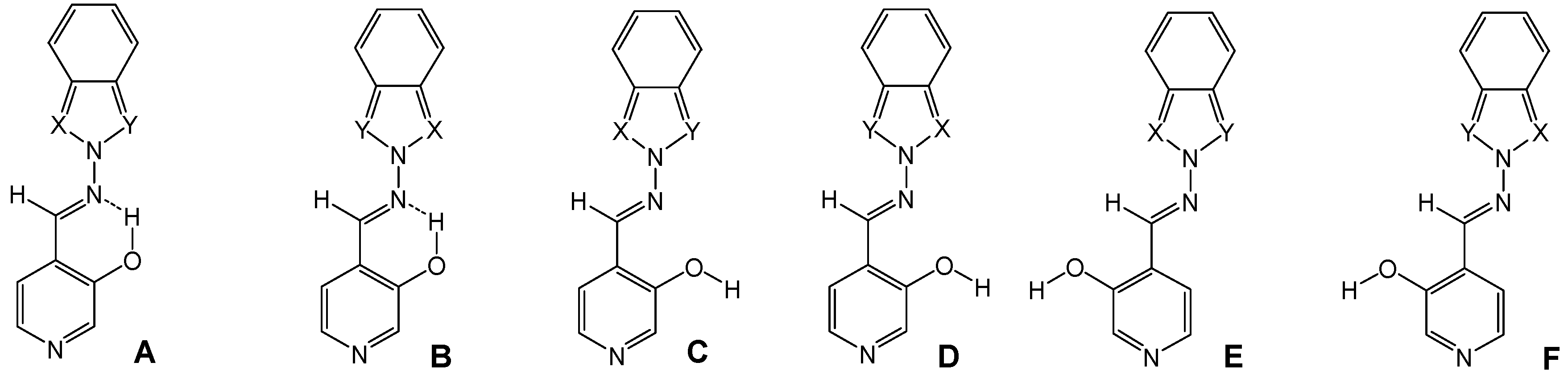
The OH group can form either an intramolecular hydrogen bond with the imino nitrogen, as in conformations A and B, or intermolecular hydrogen bonds. The latter structures can in turn present several different conformations (C-F) by rotation around the N-N and C-C bonds (Figure 1 and Figure 2).
Figure 1.
Calculated conformations A-F for (E)-4-[1H-benzazol-1-ylimino)methyl]-pyridin-3-ols (7a, X=CH, Y=N; 7b, X=N, Y=CH; 7d, X=Y=N).
Figure 1.
Calculated conformations A-F for (E)-4-[1H-benzazol-1-ylimino)methyl]-pyridin-3-ols (7a, X=CH, Y=N; 7b, X=N, Y=CH; 7d, X=Y=N).

Figure 2.
Calculated conformations A-F for (E)-4-[(2H-benzazol-2-ylimino)methyl]-pyridine-3-ols (7c, X=N, Y=CH; 7e, X=Y=N).
Figure 2.
Calculated conformations A-F for (E)-4-[(2H-benzazol-2-ylimino)methyl]-pyridine-3-ols (7c, X=N, Y=CH; 7e, X=Y=N).

The B3LYP/6-31G** calculated energies of the different conformers are summarized in Table 1, where it is seen that the most stable form is the A one, with an intramolecular hydrogen bond and the azole part of the benzazole moiety directed towards the same side of the imino proton.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
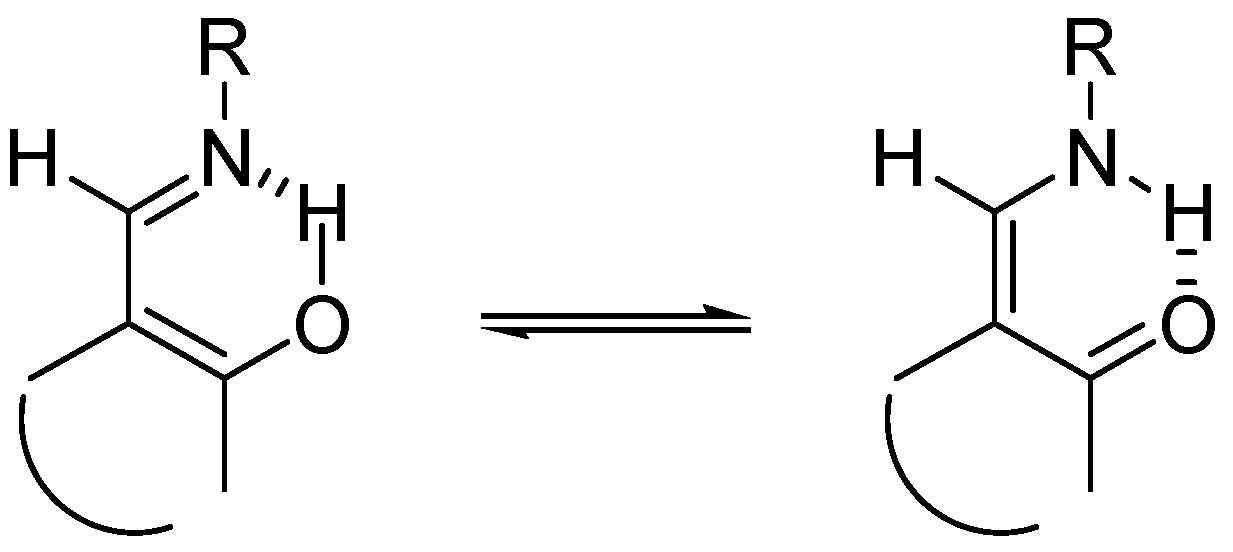{kind=link}
{kind=link}
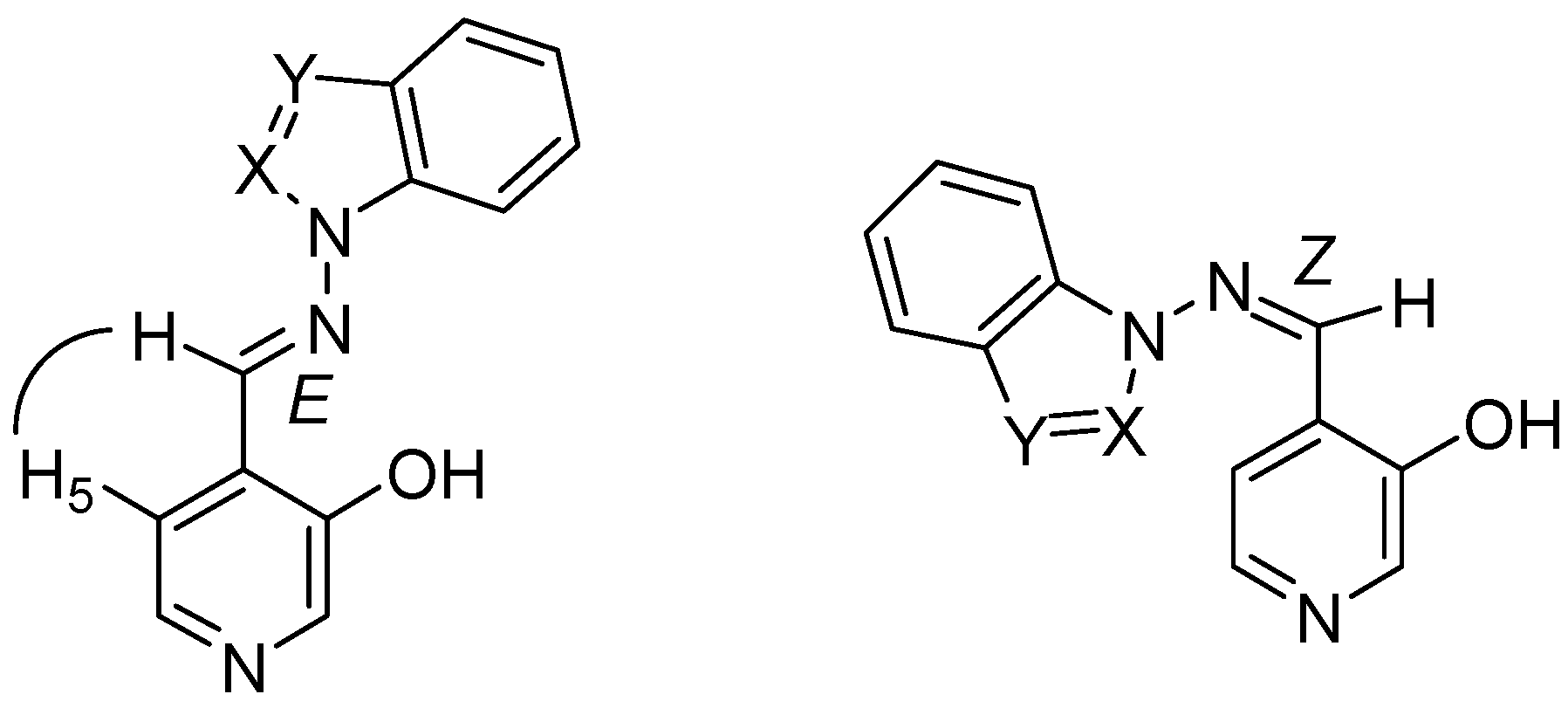{kind=link}
{kind=link}
| Compound | A | B | C | D | E | F | |
|---|---|---|---|---|---|---|---|
| 7a | -795.6184 (0.0) | 1.5 | 36.1 | 38.6 | 22.3 | 23.8 | |
| +ZPE | -795.4048 (0.0) | 2.6 | 34.6 | 37.4 | 21.2 | 23.1 | |
| 7b | -795.6071 (0.0) | 29.1 | 36.6 | 74.4 | 25.5 | 57.4 | |
| +ZPE | -795.3931 (0.0) | 28.8 | 35.0 | 72.1 | 23.9 | 55.4 | |
| 7c | -795.5988 (0.0) | 26.0 | 38.3 | 70.5 | 26.8 | 54.7 | |
| +ZPE | -795.3850 (0.0) | 24.6 | 36.3 | 67.5 | 25.2 | 51.4 | |
| 7d | -811.6246 (0.0) | 22.0 | 35.1 | 63.7 | 24.1 | 47.1 | |
| +ZPE | -811.4232 (0.0) | 21.7 | 33.6 | 61.6 | 22.4 | 45.6 | |
| 7e | -811.6255 (0.0) | 0.0 | 42.8 | 42.8 | 29.2 | 29.2 | |
| +ZPE | -811.4236 (0.0) | 0.0 | 40.4 | 40.4 | 27.5 | 27.5 |
In 7a the two conformers with intramolecular hydrogen bond (A and B) are of similar energy and the existence of both in CDCl3 has been experimentally observed by means of a NOESY spectrum showing the correlations existing between the imino proton and H2’ and H7’ of the benzimidazole ring (Figure 3).
Figure 3.
NOESY spectrum of compound 7a in CDCl3.

The NOESY spectrum of derivative 7b in CDCl3 also demonstrates that it exists in the most stable form A, with the OH forming an intramolecular hydrogen bond with the imino N atom (Figure 4).
Figure 4.
NOESY spectrum of compound 7b in CDCl3.

In DMSO-d6 solution, where the intramolecular hydrogen bonds are disrupted, the observed conformation is the E form, as theoretical calculations predict and NOESY experiments have confirmed (Figure 5).
Figure 5.
NOESY spectrum of compound 7b in DMSO-d6

For all compounds 7a-7e, the complete assignment of the 1H-, 13C- and 15 N-NMR signals in CDCl3 and DMSO-d6 has been achieved taking into account the chemical shift values, their multiplicity, as well as homonuclear (COSY and NOESY) and heteronuclear (HMQC and HMBC) correlations [13]. The values are given in the Experimental section.
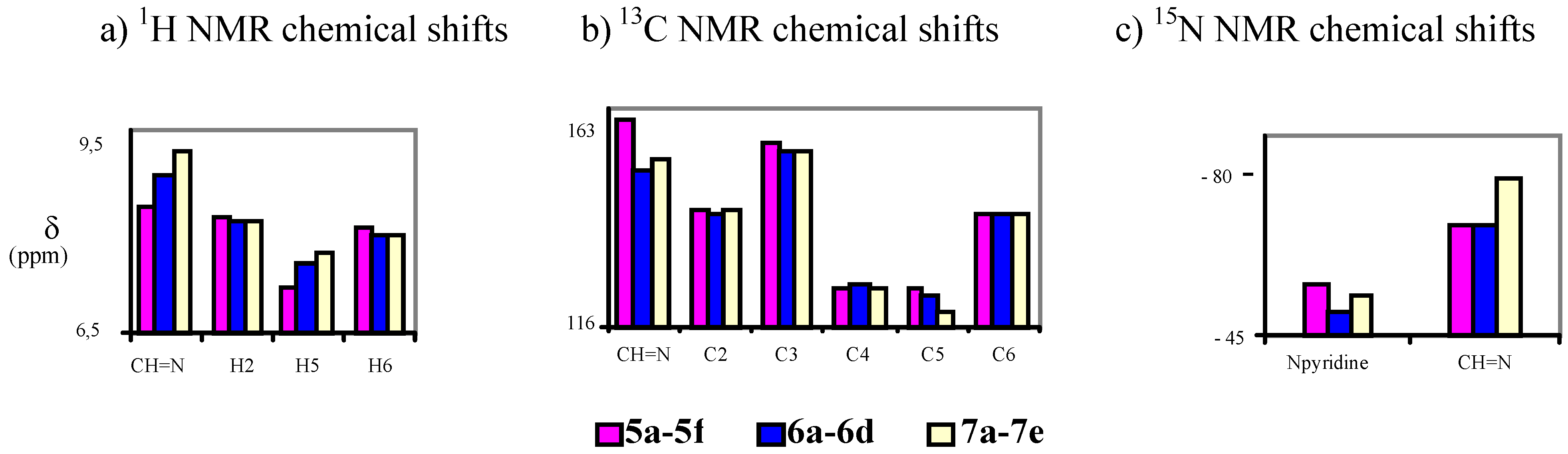
In addition to what was stated previously, the most relevant NMR data for our study have been: i) in 1H-NMR, the chemical shifts of the imino proton CH=N with mean values of 9.40 ppm in CDCl3 and 9.60 ppm in DMSO-d6, while the OH proton is around 10.2 ppm in CDCl3 and 10.93 ppm in DMSO-d6; ii) in 13C-NMR, the imino carbon CH=N at around 152.5 ppm in CDCl3 and at around 147.2 ppm in DMSO-d6; iii) in 15N-NMR, the imino nitrogen CH=N in the range of –63.8 to –90.2 ppm and the pyridine N atom at about -51 ppm in DMSO-d6.
Finally, 13C- and 15N-CPMAS NMR studies have also been performed for all five derivatives 7a-7e, demonstrating that in solid state there is no intramolecular hydrogen bond and that they exist predominantly as E rotamers. Two beautiful 13C-NMR spectra of 7b and 7e are shown in Figure 6, where the upper part presents only the quaternary carbon atoms (NQS spectra) and the lower part presents the full spectrum.
Figure 6.
13C-CPMAS NMR spectra of compounds 7b and 7e.

The splitting observed in some signals could be explained as due to the presence of more than one rotamer or to the different disposition of a same isomer in the solid state.
Conclusions
All Schiff bases studied show a similar behavior, existing as the corresponding E-isomers in the hydroxyimino tautomeric form in solution and in the solid state. An overview of the 1H-NMR, 13C- NMR and 15N-NMR chemical shifts (δ in ppm) is shown in Scheme 3.
Scheme 3.

Experimental
General
Melting points were determined both under an Axiolab "Zeiss" microscope with a TMS 92 LINKAN heating stage and by DSC on a SEIKO DSC 220C connected to a Model SSC5200H Disk Station. Thermograms (sample size 0.003-0.010 g) were recorded at the scanning rate of 2.0 ºC min-1. Unless otherwise stated, column chromatography was performed on silica gel (Merck 60, 70-230 mesh). The Rf values were measured on aluminium backed TLC plates of silica gel 60 F254 (Merck, 0.2 mm) with the indicated eluent. Elemental analyses were performed using Perkin-Elmer 240 by “Centro de Microanálisis Elemental-UCM, Madrid”.
NMR spectroscopy [13]
Solution NMR spectra were recorded on a Bruker DRX 400 spectrometer (9.4 Tesla, 400.13 MHz for 1H-, 100.62 MHz for 13C- and 40.56 MHz for 15N-) with a 5-mm inverse-detection H-X probe equipped with a z-gradient coil, at 300 K. Chemical shifts (δ in ppm) are given from internal solvent, CDCl3 7.26 for 1H- and 77.0 for 13C-, DMSO-d6 2.49 for 1H- and 39.5 for 13C- and for 15N-NMR nitromethane (0.00) was used as external standard. Typical parameters for 1H-NMR spectra were spectral width 4000 Hz and pulse width 7.5 µs at an attenuation level of 0 dB and resolution 0.15-0.25 Hz per point. Typical parameters for 13C-NMR spectra were as follows: spectral width 21 kHz, pulse width 10.6 µs at an attenuation level of -6 dB, resolution 0.6 Hz per point and relaxation delay 2 s; WALTZ-16 was used for broadband proton decoupling; the FIDS were multiplied by an exponential weighting (lb = 2 Hz) before Fourier transformation. 2D (1H-1H) gs-COSY and inverse proton detected heteronuclear shift correlation spectra, (1H-13C) gs-HMQC, (1H-13C) gs-HMBC and (1H-15N) gs-HMBC, were acquired and processed using standard Bruker NMR software and in non-phase-sensitive mode. Gradient selection was achieved through a 5% sine truncated shaped pulse gradient of 1 ms. Selected parameters for (1H-1H) gs-COSY were spectral width 2500-3500 Hz, the acquisition data size was 1024 points and one transient was accumulated per increment, with a 1 s relaxation delay, for a total of 256 experiments, data processing using zero filling in the F1 domain and shifted sine-bell apodization of factor 0 in both dimensions. Selected parameters for (1H-1H) gs-NOESY were spectral width 2000-3000 Hz, the acquisition data size was 1024 points and 32-64 transient was accumulated per increment, with a 1 s relaxation delay, 1000-2000 ms for the mixing time, for a total of 512 experiments, data processing using zero filling in the F1 domain and shifted sine-bell apodization of factor 0 in both dimensions. Selected parameters for (1H-13C) gs-HMQC and gs-HMBC spectra were spectral width 2500-3500 Hz for 1H and 12.0-20.5 kHz for 13C, 1024 x 256 data set, number of scans 2 (gs-HMQC) or 4 (gs-HMBC) and relaxation delay 1s. The FIDs were processed using zero filling in the F1 domain and a sine-bell window function in both dimensions was applied prior to Fourier transformation. In the gs-HMQC experiments GARP modulation of 13C was used for decoupling. Selected parameters for (1H-15N) gs-HMBC spectra were spectral width 2500-3500 Hz for 1H and 12.5 kHz for 15N, 1024 x 256 data set, number of scans 4, relaxation delay 1s, 50-75 ms delay for the evolution of the 15N-1H long-range coupling. The FIDs were processed using zero filling in the F1 domain and a sine-bell window function in both dimensions was applied prior to Fourier transformation. Solid state 13C- (100.73 MHz) and 15N- (40.60 MHz) CPMAS NMR spectra have been obtained on a Bruker WB 400 spectrometer at 300 K using a 4 mm DVT probehead. Samples were carefully packed in a 4-mm diameter cylindrical zirconia rotor with Kel-F end-caps. Operating conditions involved 3.2 µs 90° 1H pulses and decoupling field strength of 78.1 kHz by TPPM sequence. 13C-NMR spectra were originally referenced to a glycine sample and then the chemical shifts were recalculated to the Me4Si [for the carbonyl atom δ(glycine) = 176.1 ppm] and 15N- spectra to 15NH4Cl and then converted to nitromethane scale using the relationship: δ 15 (MεNO2) = δ 15N(NH4Cl) – 338.1 ppm. Typical acquisition parameters for 13C-CPMAS were: spectral width, 40 kHz; recycle delay, 5-120 s; acquisition time, 30 ms; contact time, 2-6 ms; and spin rate, 12 kHz. In order to distinguish protonated and unprotonated carbon atoms, the NQS (Non-Quaternary Suppression) experiment by conventional cross-polarization was recorded; before the acquisition the decoupler is switched off for a very short time of 25 μs [14,15,16]. Typical acquisition parameters for 15N CPMAS were: spectral width, 40 kHz; recycle delay, 5-120 s; acquisition time, 35 ms; contact time, 5-8 ms; and spin rate, 6 kHz.
DFT calculations
Synthesis of compounds 7a-7e
The compounds were prepared in nearly quantitative yields (85-90%) by refluxing equimolar amounts of 1 [6] and the corresponding amines 4a-4e [23] in toluene during 7 h and then stirring overnight.
4-[(E)-(1H-benzimidazol-1-ylimino)methyl]pyridin-3-ol (7a). TLC [Rf 0.44 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from C2H5OH), mp 239 ºC (microscope) and 239.1 ºC with decomposition at 255.5 (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 64.48; H, 4.33; N, 23.42; 1H-NMR (DMSO-d6) δ: 10.80 (s vbr, 1H, OH), 9.29 (s, 1H, CH=N), 9.07 (s, 1H, C2’-H), 8.38 (s br, 1H, C2-H), 8.18 (d br, 1H, 3J5-6= 4.8, C6-H), 7.84 (d, 1H, C5-H), 7.79 (d, 1H, 3J6’-7’= 8.0, C7’-H), 7.72 (d, 1H, 3J5’-4’= 7.9, C4’-H4), 7.40 (t, 1H, C6’-H), 7.31 (t, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 152.6 (C3), 146.9 (CH=N, 1J=169.6), 141.8 (C3’a), 140.5 (C6, 1J=181.2, 3J=11.9), 139.9 (C2, 1J=179.2, 3J=11.3), 137.0 (C2’, 1J=214.0), 132.1 (C7’a), 125.6 (C4), 124.0 (C6’, 1J=161.0, 3J=7.8), 123.0 (C5’, 1J=160.4, 3J=7.8), 120.1 (C4’, 1J=161.8, 3J=8.0), 119.4 (C5, 1J=161.9), 110.7 (C7’, 1J=165.0, 3J=8.3); 15N-NMR (DMSO-d6) δ: -181.2 (N1’), -134.4 (N3’), -70.3 (CH=N), -53.2 (N1); 1H-NMR (CDCl3) δ: 10.20 (s br, 1H, OH), 8.90 (s, 1H, CH=N), 8.58 (s br, 1H, C2-H), 8.41 (s, 1H, C2’-H), 8.34 (d , 1H, 3J5-6= 5.0, C6-H), 7.86 (ddd, 1H, 3J5’-4’= 8.0, 4J6’-4’= 1.3, 5J7’-4’= 0.7, C4’-H), 7.69 (ddd, 1H, 3J6’-7’= 7.9, 3J5’-7’= 1.3, C7’-H), 7.46 (ddd, 1H, C6’-H), 7.41 (ddd, 1H, C5’-H), 7.31 (d, 1H, C5-H); 13C-NMR (CDCl3) δ: 152.9 (C3), 152.1 (CH=N), 142.6 (C3’a), 141.4 (C6), 141.3 (C2), 135.9 (C2’), 131.0 (C7’a), 125.1 (C6’), 124.2 (C5’), 123.5 (C5), 121.9 (C4), 121.4 (C4’,), 110.2 (C7’); 15N-NMR (CDCl3) δ:-186.5 (N1’); 13C-CP/MAS NMR δ: 154.0 (C3), 140.7 (CH=N), 140.7 (C3’a), 140.1 (C6), 138.2 (C2), 137.3 (C2’), 133.7 (C7’a), 128.1 (C4), 122.8 (C6’), 122.0 (C5’), 119.1 (C4’), 118.6 (C5), 113.0 (C7’); 15N- CP/MAS NMR δ: -177.7 (N1’), -135.1 (N3’), -87.1 (CH=N), -67.6 (N1).
4-[(E)-(1H-indazol-1-ylimino)methyl]pyridin-3-ol (7b). TLC [Rf 0.82 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from CHCl3-C2H5OH), mp 190 ºC (microscope) and 186.0 ºC (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 63.25; H, 4.24; N, 22.76; 1H-NMR (DMSO-d6) δ: 10.69 (s br, 1H, OH), 9.37 (s, 1H, CH=N), 8.35 (s, 1H, C2-H), 8.34 (t, 1H, 4J4’-3’=5J7’-3’=0.8, C3’-H), 8.16 (d, 1H, 3J5-6= 5.0, C6-H), 7.89 (d, 1H, C5-H), 7.87 (ddd, 1H, 3J6’-7’= 8.1, 4J5’-7’= 0.9, C7’-H), 7.84 (ddd, 1H, 3J5’-4’=8.0, 4J6’-4’= 1.0, C4’-H), 7.56 (ddd, 1H, 3J5’-6’= 6.9, C6´-H), 7.29 (ddd, H5’); 13C-NMR (DMSO-d6) δ: 152.4 (C3), 140.5 (C6, 1J=181.6, 3J=11.1), 139.8 (CH=N, 1J=172.0), 139.6 (C2, 1J=178.5, 3J=11.3), 137.6 (C7’a, 3J=3J=9.1), 134.2 (C3’, 1J=192.8), 128.3 (C6’, 1J=159.6, 3J=7.7, 2J=2.2), 125.8 (C4), 123.5 (C3’a), 122.6 (C5’, 1J=161.3, 3J=7.0), 121.5 (C4’, 1J=163.8, 3J=8.2), 118.9 (C5, 1J=163.9, 3J=9.8, 2J=4.1), 110.0 (C7’, 1J=167.5, 3J=8.4); 15N-NMR (DMSO-d6) δ: -157.5 (N1’), -81.8 (N2’), -70.1 (CH=N), -54.4 (N1); 1H-NMR (CDCl3) δ: 10.37 (s, 1H, OH), 9.18 (s, 1H, CH=N), 8.51 (s, 1H, C2-H), 8.28 (d, 1H, 3J5-6= 4.9, C6-H), 8.11 (t, 1H, 4J4’-3’=5J7’-3’=0.9, C3’-H), 7.75 (td, 1H, 3J5’-4’=8.1, 4J6’-4’= 0.9, C4’-H), 7.70 (qd, 1H, 3J6’-7’= 8.4, 4J5’-7’= 0.9, C7’-H), 7.55 (ddd, 1H, 3J5’-6’= 7.0, C6´-H), 7.29 (d, 1H, C5-H), 7.29 (ddd, H5’); 13C-NMR (CDCl3) δ: 152.6 (C3), 141.2 (C6, 1J=181.2, 3J=11.5), 146.2 (CH=N, 1J=172.4, 3J=6.3), 140.6 (C2, 1J=181.1), 137.5 (C7’a), 134.6 (C3’, 1J=191.4, 3J=2.5), 128.7 (C6’, 1J=161.0, 3J=7.7), 123.6 (C5, 1J=158.0), 123.1 (C5’, 1J=162.6), 122.9 (C4), 124.0 (C3’a), 121.5 (C4’, 1J=163.5, 3J=8.1), 109.5 (C7’, 1J=167.3, 3J=7.9); 15N-NMR (CDCl3) δ: -162.8 (N1’), -86.2 (N2’), -83.9 (CH=N), -59.7 (N1); 13C-CP/MAS NMR δ: 155.6/153.8 (C3), 137.9 (C7’a), 137.5 (C6 and CH=N), 135.9 (C2), 134.5 (C3’), 130.3 (C6’), 128.9 (C4), 123.9/122.4 (C3’a), 121 (C5’ and C4’), 119.1 (C5), 109.4 (C7’); 15N- CP/MAS NMR δ: -154.5 (N1’), -88.7 (N2’), -80.8 (CH=N), -68.9 (N1).
4-[(E)-(2H-indazol-2-ylimino)methyl]pyridin-3-ol (7c). TLC [Rf 0.79 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (from C2H5OH), mp 270 ºC (microscope) and 267.7 ºC with decomposition at 284.7 (DSC); Anal. Calcd for C13H10N4O: C, 65.54; H, 4.23; N, 23.52. Found: C, 64.75; H, 4.39; N, 23.30; 1H-NMR (DMSO-d6) δ: 10.94 (s, 1H, OH), 9.77 (s, 1H, CH=N), 8.70 (d, 1H, 5J7’-3’=0.9, C3’-H), 8.40 (s, 1H, C2-H), 8.20 (d, 1H, 3J5-6=5.0, C6-H), 7.81 (d, 1H, C5-H), 7.75 (td, 1H, 3J5’-4’=8.5, 4J6’-4’=5J7’-4’=1.1, C4’-H), 7.66 (qd, 1H, 3J6’-7’=8.8, 4J5’-7’=1.0, C7’-H), 7.36 (ddd, 1H, 3J5’-6’=6.6, C6’-H), 7.11 (ddd, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 153.8 (C3), 148.9 (CH=N, 1J=176.1), 146.0 (C7’a, 3J=3J=3J=7.3), 140.5 (C6, 1J=180.7, 3J=11.7), 140.2 (C2, 1J=179.8, 3J=11.1), 127.9 (C6’, 1J=158.5, 3J=7.3), 124.4 (C4), 123.7 (C3’, 1J=197.1), 122.2 (C5’, 1J=159.9, 3J=8.2), 121.3 (C3’a), 121.2 (C4’, 1J=163.9, 3J=7.4), 119.5 (C5, 1J=164.5, 3J=8.5), 117.2 (C7’, 1J=163.1, 3J=7.0); 15N-NMR (DMSO-d6) δ: -125.5 (N2’), -63.8 (CH=N), -50.5 (N1); 1H-NMR (CDCl3) δ: 10.14 (s, 1H, OH), 9.61 (s, 1H, CH=N), 8.56 (s, 1H, C2-H), 8.23 (d, 1H, 5J7’-3’=0.9, C3’-H), 8.33 (d, 1H, 3J5-6=4.9, C6-H), 7.69 (m, 2H, C4’-H and C7’-H), 7.39 (ddd, 1H, 3J5’-6’=6.6 3J7’-6’=8.9, 5J4’-6’=1.0, C6’-H), 7.37 (d, 1H, C5-H), 7.15 (ddd, 1H, 3J4’-5’=8.4, 4J7’-5’=0.8, C5’-H); 13C-NMR (CDCl3) δ: 154.5 (CH=N), 153.0 (C3), 147.0 (C7’a), 141.6 (C6), 141.2 (C2), 128.6 (C6’), 124.5 (C5), 123.1 (C5’), 122.5 (C3’), 121.8 (C3’a), 121.7 (C4), 120.7 (C4’), 117.5 (C7’); 15N-NMR (CDCl3) δ: -133 (N2’); 13C-CP/MAS NMR δ: 156.1 (C3), 146.9 (CH=N), 145.0 (C7’a), 141.3 (C6), 139.4 (C2), 129.3 (C4), 127.4 (C6’), 123.5 (C3’), 121.7 (C5’ and C3’a), 121.1 (C4’), 119.6 (C5), 118.4 (C7’); 15N-CP/MAS NMR δ: -122.9 (N2’), -110.6 (N1’), -84.0 (CH=N), ‑69.9/-66.7 (N1).
4-[(E)-(1H-1,2,3-benzotriazol-1-ylimino)methyl]pyridin-3-ol (7d). TLC [Rf 0.70 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (CHCl3), mp 209 ºC (microscope) and 199.9 ºC and 204.7 ºC with decomposition at 237.1 ºC (DSC); Anal. Calcd for C12H9N5O: C, 60.25; H, 3.79; N, 29.27. Found: C, 60.01; H, 4.28; N, 28.12; 1H-NMR (DMSO-d6) δ: 11.00 (s br, 1H, OH), 9.75 (s, 1H, CH=N), 8.41 (s, 1H, C2-H), 8.22 (d, 1H, 3J5-6=5.0, H6), 8.13 (td, 1H, 3J5’-4’=8.4, 4J6’-4’=5J7’-4’=1.0, C4’-H), 7.96 (td,1H, 3J6’-7’=8.3, 4J5’-7’=1.0, C7’-H) 7.93 (d, 1H, C5-H), 7.70 (ddd, 1H, 3J5’-6’=6.9, C6’-H), 7.51 (ddd, 1H, C5’-H); 13C-NMR (DMSO-d6) δ: 153.1 (C3), 147.2 (CH=N, 1J=173.4), 144.8 (C3’a, 3J=9.8, 3J=4.5), 140.5 (C6, 1J=181.3, 3J=11.5), 140.2 (C2, 1J=179.4, 3J=11.4), 130.4 (C7’a, 3J=10.6, 3J=6.5), 129.2 (C6’, 1J=163.5, 3J=7.8), 125.3 (C5’, 1J=164.0, 3J=7.7), 124.6 (C4), 119.8 (C4’, 1J=166.5, 3J=7.8), 119.2 (C5, 1J=163.2), 110.5 (C7’, 1J=168.9, 3J=8.2); 15N-NMR (DMSO-d6) δ: -121.6 (N1’), -77.1 (CH=N), -49.7 (N1); 1H-NMR (CDCl3) δ: 10.20 (s br, 1H, OH), 9.64 (s, 1H, CH=N), 8.59 (s, 1H, C2-H), 8.35 (d, 1H, 3J5-6=4.6, C6-H), 8.11 (d, 1H, 3J5’-4’=8.3, C4’-H), 7.76 (d, 1H, 3J6’-7’=8.2, C7’-H), 7.65 (t, 1H, 3J5’-6’=7.5, C6’-H), 7.49 (t, 1H, C5’-H), 7.43 (d, 1H, C5-H); 13C-NMR (CDCl3) δ: 153.2 (C3), 152.3 (CH=N, 1J=172.8, 3J=6.1), 145.5 (C3’a), 141.1 (C6, 1J=183.0, 3J=11.8), 141.0 (C2, 1J=181.5, 3J=11.3), 130.3 (C7’a), 129.4 (C6’, 1J=163.4, 3J=8.1), 125.4 (C5’, 1J=163.7, 3J=7.9), 123.8 (C5, 1J=164.1, 3J=8.6), 122.4 (C4), 120.5 (C4’, 1J=166.5, 3J=7.8), 109.6 (C7’, 1J=169.5, 3J=8.3); 15N-NMR (CDCl3) δ: -126.9 (N1’), -89.4 (CH=N), -55.8 (N1); 13C-CP/MAS NMR δ: 154.8 (C3), 145.5 (CH=N), 143.9 (C3’a), 141.1 (C6), 138.2 (C2), 130.8 (C7’a), 128.7 (C6’), 123.6 (C5’ and C4), 118.7 (C5), 118.0 (C4’), 109.7 (C7’); 15N-CP/MAS NMR δ: -159.0 (N1’), -80.5 (CH=N), -67.6 (N3’), -55.3 (N1), -17.6 (N2’).
4-[(E)-(2H-1,2,3-benzotriazol-2-ylimino)methyl]pyridin-3-ol (7e). TLC [Rf 0.80 (9:1 CHCl3-C2H5OH)]. The crystals were purified by crystallisation (CHCl3), this compound changes its appearance at 250°C and then at 280°C it decomposes, and 259.6 ºC (DSC); Anal. calcd for C12H9N5O: C, 60.25; H, 3.79; N, 29.27. Found: C, 59.90; H, 3.95; N, 28.80; 1H-NMR (DMSO-d6) δ: 11.20 (s br, 1H, OH), 9.86 (s, 1H, CH=N), 8.43 (s, 1H, C2-H), 8.20 (d, 1H, 3J5-6=5.0, C6-H), 7.97 (m, 2H, C4’-H and C7’-H), 7.84 (d, 1H, C5-H), 7.52 (m, 2H, C5’-H and C6’-H); 13C-NMR (DMSO- d6) δ: 153.9 (C3, 3J=2J=5.2), 153.2 (CH=N, 1J=173.0, 3J=3.1), 142.7 (C3’a and C7’a), 140.5 (C6 and C2, 1J=180.2, 3J=11.3), 128.0 (C5’ and C6’, 1J=162.0, 3J=8.3), 123.5 (C4, 3J=3J=2J=6.0), 119.3 (C5, 1J=164.3, 3J=9.7, 2J=3.8), 118.1 (C4’ and C7’, 1J=169.2, 3J=5.2); 15N-NMR (DMSO- d6) δ: -90.2 (CH=N), -47.2 (N1); 1H-NMR (CDCl3) δ: 10.02 (s vbr, 1H, OH), 9.68 (s, 1H, CH=N), 8.62 (s, 1H, C2-H), 8.36 (d, 1H, 3J5-6=4.8, C6-H), 7.90 (m, 2H, C4’-H and C7’-H), 7.49 (m, 2H, C5’-H and C6’-H), 7.42 (d, C5-H); 13C-NMR (CDCl3) δ: 157.5 (CH=N, 1J=169.7), 153.5 (C3), 143.6 (C3’a and C7’a), 141.8 (C2, 1J=182.1, 3J=11.0), 141.2 (C6, 1J=182.7, 3J=11.2), 128.4 (C5’ and C6’, 1J=161.4, 3J=8.2), 124.7 (C5, 1J=162.9), 121.0 (C4), 118.4 (C4’ and C7’, 1J=170.1, 3J=4.9); 13C-CP/MAS NMR δ: 155.2 (C3), 149.1 (CH=N), 142.8 (C3’a and C7’a), 139.8 (C6), 135.0 (C2), 129.7(C5’), 126.2, (C6’ and C4), 123.2 (C5), 120.3 (C4’), 113.3 (C7’); 15N-CP/MAS NMR δ: -90.6 (N1’), -88.0 (N3’), -84.5 (CH=N), -83.6 (N2’), -60.7 (N1).
Acknowledgements
This work was supported by DGES/MCyT (Project no. BQU2003-00976). One of us (A.P.) is indebted to the MCyT of Spain for an FPI grant.
References
- Cleland, D.; Kreevoy, M. W. Science 1994, 264, 1887–1890.
- Katrizky, A. R.; Ghiviriga, I.; Leeming, P.; Soti, F. Magn. Reson. Chem. 1996, 34, 518–526. [CrossRef]
- Dziembowska, T.; Rozwadowski, Z.; Filarowski, A.; Hansen, P. E. Magn. Reson. Chem. 2001, 39, S67–80. [CrossRef]
- Hansen, P. E.; Sitkowski, J.; Stefaniak, L.; Rozwadowski, Z.; Dziembowska, T. Ber. Bunsen. Phys. Chem. 1998, 102, 410–413. [CrossRef]
- Alarcón, S. H.; Olivieri, A. C.; Sanz, D.; Claramunt, R. M.; Elguero, J. J. Mol. Struct. 2004, 705, 1–9.
- Sanz, D.; Perona, A.; Claramunt, R. M.; Elguero, J. Tetrahedron 2005, 61, 145–154.
- That, Q. T.; Nguyen, K. P. P.; Hansen, P. E. Magn. Reson. Chem. 2005, 43, 302–308. [CrossRef]
- Cozzi, P. G. Chem. Soc. Rev. 2004, 33, 410–421. [CrossRef]
- Dao, V.-T.; Gaspard, C.; Mayer, M.; Werner, G. H.; Nguen, S. N.; Michelot, R. J. Eur. J. Med. Chem. 2000, 35, 805–813. [CrossRef]
- Zheng, B.; Brett, S.; Tite, J. P.; Brodie, T. A.; Rhodes, J. Science 1992, 256, 1560–1563.
- Sharif, S.; Denisov, G. S.; Toney, M. D.; Limbach, H.-H. J. Am. Chem. Soc. 2006, 128, 3375–3387.
- Sanz, D.; Perona, A.; Claramunt, R. M.; Pinilla, E.; Torres, M. R.; Elguero, J. Helv. Chim. Acta. accepted for publication.
- Berger, S.; Braun, S. “200 and more NMR Experiments”; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- Murphy, P. D. J. Magn. Reson. 1983, 52, 343–345.
- Murphy, P. D. J. Magn. Reson. 1985, 62, 303–308.
- Alemany, L. B.; Grant, D. M.; Alger, T. D.; Pugmire, R. J. J. Am. Chem. Soc. 1983, 105, 6697–6704.
- Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. [CrossRef]
- Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652.
- Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200–206.
- Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213–222. [CrossRef]
- Spartan 2002 for Windows, Wavefunction Inc.: 18401 Von Karman Ave., Suite 370, Irvine, CA 92612, USA.
- Foces-Foces, M. C.; Hernández-Cano, F.; Claramunt, R. M.; Sanz, D.; Catalán, J.; Fabero, F.; Fruchier, A.; Elguero, J. J. Chem. Soc. Perkin Trans. 2 1990, 237–244.
- Sample Availability: Samples of the compounds 5a-5f, 6a-6d and 7a-7e are available from the authors (or from MDPI).
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Perona, A.; Sanz, D.; Claramunt, R.M.; Elguero, J. Syntheses and Structural Studies of Schiff Bases Involving Hydrogen Bonds. Molecules 2006, 11, 453-463. https://doi.org/10.3390/11060453
AMA Style
Perona A, Sanz D, Claramunt RM, Elguero J. Syntheses and Structural Studies of Schiff Bases Involving Hydrogen Bonds. Molecules. 2006; 11(6):453-463. https://doi.org/10.3390/11060453
Chicago/Turabian StylePerona, Almudena, Dionisia Sanz, Rosa M. Claramunt, and José Elguero. 2006. "Syntheses and Structural Studies of Schiff Bases Involving Hydrogen Bonds" Molecules 11, no. 6: 453-463. https://doi.org/10.3390/11060453