Solid Phase Synthesis of a Metronidazole Oligonucleotide Conjugate

{kind=link}
{kind=link}
Abstract
:Introduction
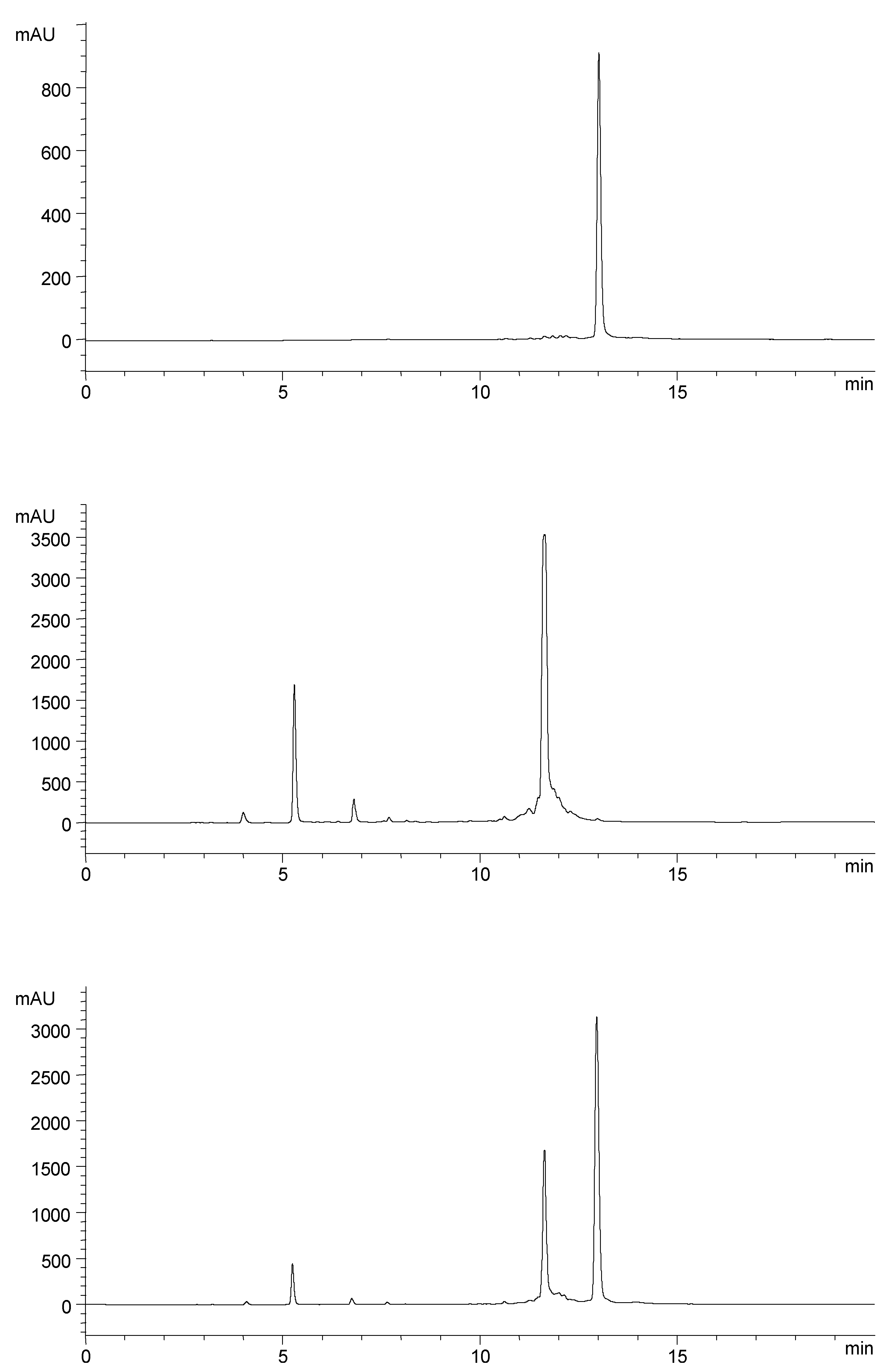
Results and Discussion







Experimental
General
Preparation of 2-(2-methyl-5-nitroimidazoyl)-ethyl-(2-cyanoethyl)-N,N-diisopropyl)-phosphoramidite (2)
Preparation of protected, solid supported metronidazole oligonucleotide conjugate 3
Preparation of aminoethyl oligonucliotide conjugate 4
Preparation of metronidazole oligonucleotide conjugate 5
Acknowledgements
References
- Edwards, D. I. Nitroimidazole drugs - action and resistance mechanisms I. Mechanisms of action. J. Antimicrob. Chemother. 1993, 31, 9–20. [Google Scholar] [CrossRef]
- Tocher, J. H. Reductive activation of nitroheterocyclic compounds. Gen. Pharmacol. 1997, 28, 485–487. [Google Scholar]
- Pendland, S. L; Piscitelli, S. C.; Schreckenberger, P. C.; Danziger, L. H. In vitro activities of metronidazole and its hydroxyl metabolite against Bacteroides spp. Antimicrob. Agents Chemother. 1994, 38, 2106–2110. [Google Scholar] [CrossRef]
- Oliveira, R. B.; Passos, A. P. F.; Alves, R. O.; Romanha, A. J.; Prado, M. A. F.; de Souza Filho, J. D.; Alves, R. J. In vitro evaluation of the activity of aromatic nitrocompounds against Trypanosoma cruzi. Memorias-Inst. Oswaldo Cruz, Rio de Janeiro 2003, 98, 141–144. [Google Scholar]
- Sóki, J.; Gal, M.; Brazier, J. S.; Rotimi, V. O.; Urbán, E.; Nagy, E.; Duerden, B. I. Molecular investigation of genetic elements contributing to metronidazole resistance in Bacteroides strains. J. Antimicrob. Chemother. 2006, 57, 212–220. [Google Scholar] [CrossRef]
- Sisson, G.; Jeong, J.-Y.; Goodwin, A.; Bryen, L.; Rossler, N.; Lim-Morrison, S.; Raudonikiene, A.; Berg, D. E.; Hoffman, P. S. Metronidazole activation is mutagenic and causes DNA fragmentation in Helicobacter pylori and in Escherichia coli containing a cloned H. pylori rdxA+ (nitroreductase) gene. J. Bacteriol. 2000, 182, 5091–5096. [Google Scholar] [CrossRef]
- Osato, M. S; Reddy, R.; Graham, D. Y. Metronidazole and clarithromycin resistance amongst Helicobacter pylori isolates from a large metropolitan hospital in the Unites States. Int. J. Antimicrob. Agents 1999, 12, 341–347. [Google Scholar] [CrossRef]
- Reysset, G. Genetics of 5-nitroimidazole resistance in Bacteroides species. Anearobe 1996, 2, 59–69. [Google Scholar] [CrossRef]
- Dachs, G. U.; Abratt, V. R.; Woods, D. R. Mode of action of metronidazole and a Bacteroides fragilis metA resistance gene in Escherichia coli. J. Antimicrob. Chemother. 1995, 35, 483–496. [Google Scholar] [CrossRef]
- Bowden, K.; Izadi, J. Multifunctional derivatives of metronidazole. Farmaco 1998, 53, 58–61. [Google Scholar] [CrossRef]
- Parrick, J.; Porssa, M. Synthesis of polyamine derivatives of 2-nitroimidazole as DNA-directed radiosensitisers. J. Chem. Res. (S) 1995, 186–187. [Google Scholar]
- Ali, H.; Ouellet, R.; DaSilva, J. N.; van Lier, J. E. Synthesis of steroidal nitroimidazoles as site-selective radiosensitisers. J. Chem. Res. (S) 1993, 92–93. [Google Scholar]
- Menéndez, D.; Rojas, E.; Herrera, L. A.; López, M. C.; Sordo, M.; Elizondo, G.; Ostrosky-Wegman, P. DNA breakage due to metronidazole treatment. Mutat. Res. 2001, 478, 153–158. [Google Scholar] [CrossRef]
- Connor, T. H.; Stoeckel, M.; Evrard, J.; Legator, M. S. The contribution of metronidazole and two metabolites to the mutagenic activity detected in urine of treated humans and mice. Cancer Res. 1977, 37, 629–633. [Google Scholar]
- Parrick, J.; Porssa, M. Synthesis of a nitro oligo-N-methylimidazole carboxamide derivative: A radiosensitiser targeted to DNA. Tetrahedron Lett. 1993, 34, 5011–5014. [Google Scholar] [CrossRef]
- Parrick, J.; Porssa, M.; Jenkins, T. M. The synthesis of radiosensitizers designed to bind to the minor groove of duplex DNA. J. Chem. Soc., Perkin Trans. 1 1993, 2681–2685. [Google Scholar] [CrossRef]
- Parrick, J.; Porssa, M.; Davies, L. K.; Dennis, M. F.; Patel, K. B.; Stratford, M. R. L.; Wardman, P. Targeting radiosensitizers to DNA by minor groove binding: Nitroarenes based on netropsin and distamycin. Bioorg. Med. Chem. Lett. 1993, 3, 1697–1702. [Google Scholar] [CrossRef]
- Da Ros, T.; Spalluto, G.; Prato, M.; Saison-Behmoaras, T.; Boutorine, A.; Cacciari, B. Oligonucleotides and oligonucleotide conjugates: A new approach for cancer treatment. Curr. Med. Chem. 2005, 12, 71–88. [Google Scholar] [CrossRef]
- Neeley, W. L.; Henderson, P. T.; Essigmann, J. M. Efficient synthesis of DNA containing the guanine oxidation-nitration product 5-guanidino-4-nitroimidazole: Generation by a postsynthetic substitution reaction. Org. Lett. 2004, 6, 245–248. [Google Scholar] [CrossRef]
- Atkinson, T.; Smith, M. Oligonucleotide Synthesis. A Practical Approach; Gait, M. J., Ed.; IRL Press: Oxford, 1984; Chapter 3; pp. 35–81. [Google Scholar]
- Grimmett, G. M. Comprehensive Heterocyclic Chemistry; Katrizky, A. R., Rees, C. W., Scriven, E. F. V., Eds.; Elsevier: Oxford, 1984; Vol. 5, Chapter 4.07; pp. 373–456. [Google Scholar]
- Apen, P. G.; Rasmussen, P. G. Nucleophilic aromatic-substitution in 4,5-dicyanoimidazoles. Heterocycles 1989, 29, 1325–1329. [Google Scholar] [CrossRef]
- Grimmett, G. M. Comprehensive Heterocyclic Chemistry; Katrizky, A. R., Rees, C. W., Scriven, E. F. V., Eds.; Elsevier: Oxford, 1996; Vol. 3, Chapter 3.02; pp. 373–456. [Google Scholar]
- Girard, M.; Clarimont, F.; Maneckjee, A.; Mousseau, N.; Dawson, B. A.; Whitehouse, L. W. 5-Nitroimidazoles II: Unexpected reactivity of ronidazole and dimetridazole with thiols. Can. J. Chem. 1993, 71, 1349–1352. [Google Scholar] [CrossRef]
- Beck, J. R. Nucleophilic displacement of aromatic nitro groups. Tetrahedron 1978, 34, 2057–2068. [Google Scholar] [CrossRef]
- Sunjić, V.; Fajdiga, T.; Japelj, M.; Rems, P. Nucleophilic substitutions in some derivatives of 4- and 5-nitroimidazoles. J. Heterocycl. Chem. 1969, 6, 53–60. [Google Scholar] [CrossRef]
- Goldman, P.; Ramos, S. M.; Wuest, J. D. Reactions of nitroimidazoles with hydrazine. J. Org. Chem. 1984, 49, 932–935. [Google Scholar]
- Chasseaud, L. F.; Henrick, K.; Matthews, R. W.; Scott, P. W.; Wood, S. G. Metabolic ring hydroxylation of tinidazole involving a novel nitro-group migration: X-ray structures of tinidazole and the NH4+ salt of its ring hydroxylated metabolite. Chem. Commun. 1984, 491–492. [Google Scholar]
- ‘2005 Product Guide’, Link Technologies, 2005. Available online: http://www.linktech.co.uk [Accessed April 2006].
- Surzhikov, S. A.; Timofeev, E. N.; Chernov, B. K.; Golova, J. B.; Mirzabekov, A. D. Advanced method for oligonucleotide deprotection. Nucleic Acids Res. 2000, 28, e29. [Google Scholar]
- Still, W. C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- ‘Oligo 1000 DNA Synthesiser Operating Instructions’, Beckman Instruments Incorporated, 1994.
- Sample availability: Please contact the authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Walsh, A.J.; Davis, M.L.; Fraser, W. Solid Phase Synthesis of a Metronidazole Oligonucleotide Conjugate. Molecules 2006, 11, 486-495. https://doi.org/10.3390/11060486
Walsh AJ, Davis ML, Fraser W. Solid Phase Synthesis of a Metronidazole Oligonucleotide Conjugate. Molecules. 2006; 11(6):486-495. https://doi.org/10.3390/11060486
Chicago/Turabian StyleWalsh, Andrew J., Michael L. Davis, and William Fraser. 2006. "Solid Phase Synthesis of a Metronidazole Oligonucleotide Conjugate" Molecules 11, no. 6: 486-495. https://doi.org/10.3390/11060486
APA StyleWalsh, A. J., Davis, M. L., & Fraser, W. (2006). Solid Phase Synthesis of a Metronidazole Oligonucleotide Conjugate. Molecules, 11(6), 486-495. https://doi.org/10.3390/11060486