Experimental
General
Unless stated otherwise, all reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) using 0.25 mm precoated silica gel F254 plates (Sigma-Aldrich). Column or flash chromatography were performed with the indicated solvents using silica gel (230-400 mesh) purchased from Dynamic Adsorbents, LLC. All melting points were obtained on a Laboratory Devices capillary melting point apparatus and are uncorrected. 1H- and 13C-NMR spectra were recorded on a Bruker VXR-300 (300 MHz) or a Bruker VXR-400 (400 MHz) spectrometer. Chemical shifts are reported relative to internal chloroform (1H, 7.26 ppm; 13C, 77.23 ppm). High resolution mass spectra were performed at the Iowa State University Mass Spectrometry Laboratory.
Plant material and extraction
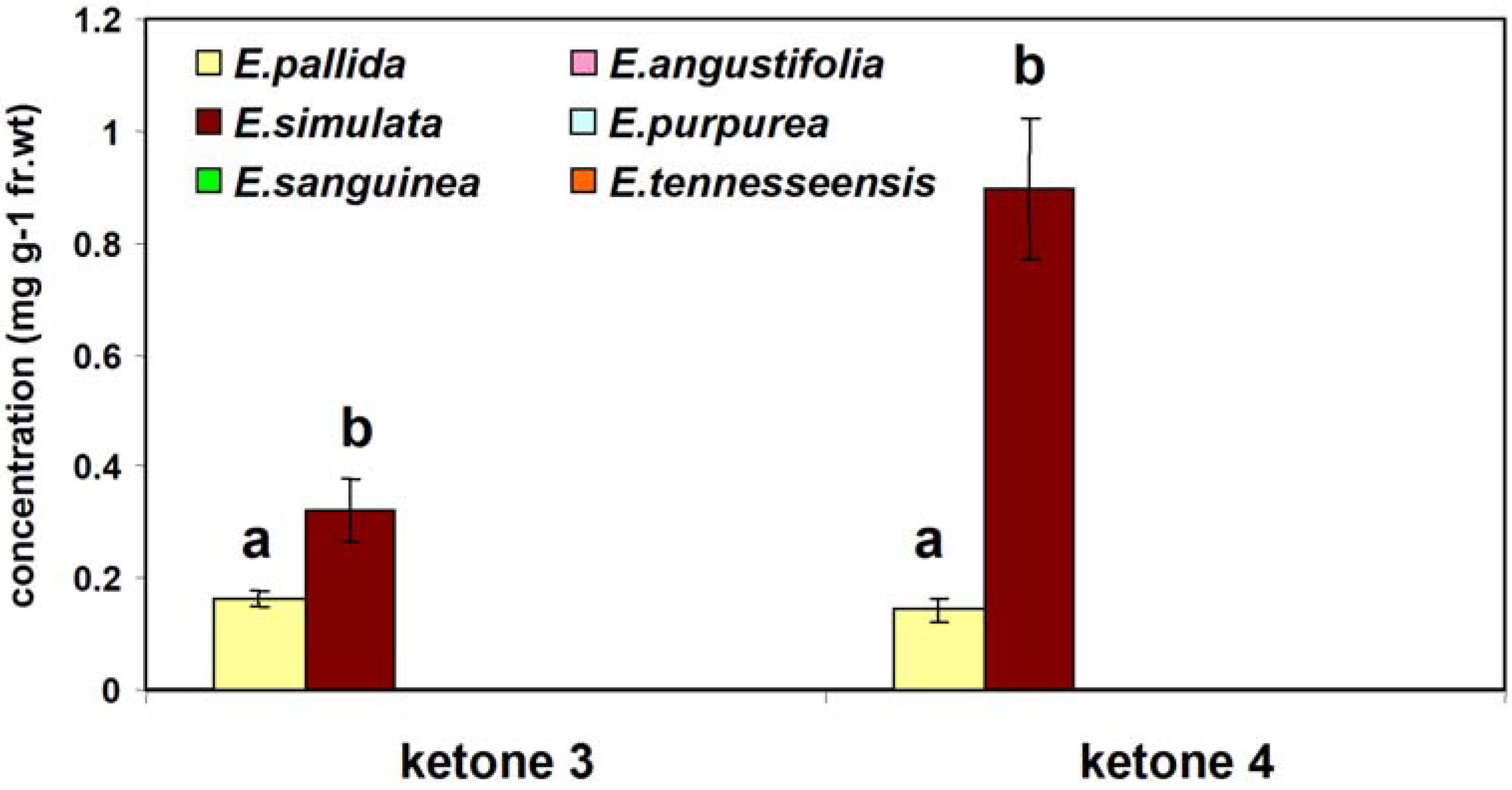
Two-year-old fresh roots of 6 species of
Echinacea,
E. angustifolia (Accession PI 631285),
E. purpurea (Accession PI 631307),
E. pallida (Accession PI 631279),
E. sanguinea (Accession PI 633672),
E. simulata (Accession PI 631251) and
E. tennesseensis (Accession PI 631350), provided by Dr. Mark P. Widrlechner at the USDA-ARS North Central Regional Plant Introduction Station, were studied to evaluate the natural distribution of ketones
3 and
4 in
Echinacea species. The plant extraction method is the same as that in our previously published work [
16]. Similarly, 7-hydroxy-(
E)-
N-isobutylundeca-2-ene-8,10-diynamide (C
15H
21O
2) was added as an internal standard prior to extraction for quantification purposes. All experiments were performed in triplicate on independently extracted plant samples from three individual plants.
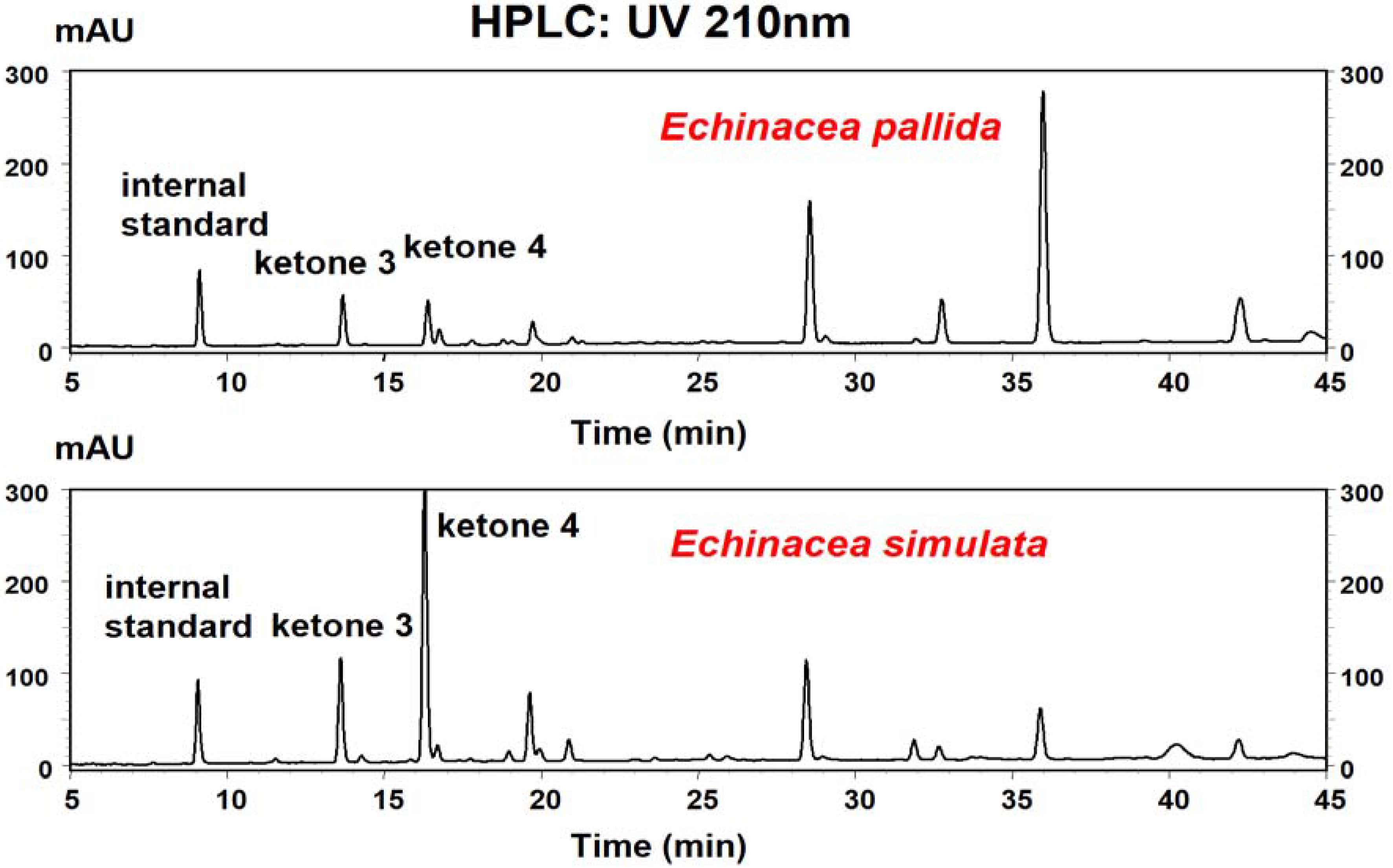
HPLC analysis
HPLC procedures were as previously described [
20]. Ketones
3 and
4 were quantified at UV 210 nm based on the internal standard because they have the same UV absorption at 210 nm. The internal quantification method used here can account for variations in extraction efficiencies in different extracts. The limit of HPLC detection for each of the ketones is approximately 0.04 μg mL
−1.
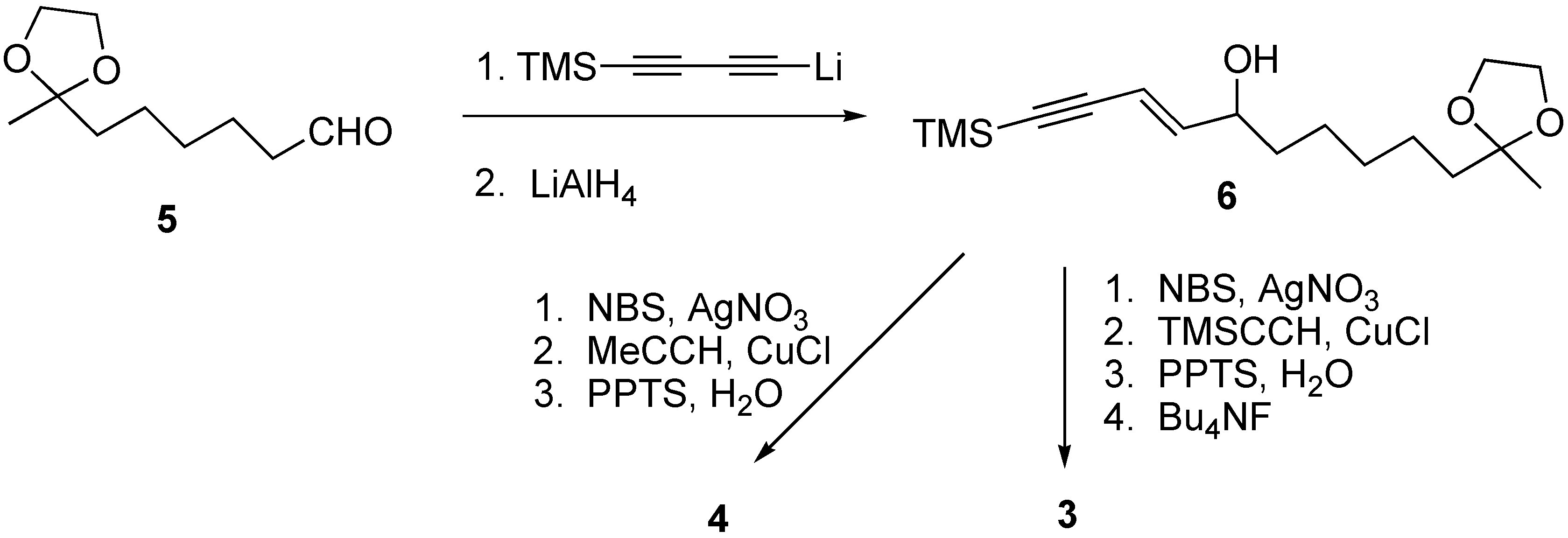
1-Trimethylsilyl-11-oxodocec-3-en-1-yn-5-ol ethylene ketal (6)
MeLi-LiBr complex (5 mL, 1.5 M soln in ether) was added to a solution of 1,4-bis(trimethylsilyl)-1,3-butadiyne (1.46 g, 7.5 mmol) in THF (10 mL) at 0 oC under Ar. The mixture was warmed to room temperature and stirred for 4 h. The mixture was then cooled down to -78 oC and aldehyde 5 (0.700 g, 3.76 mmol) in THF (4 mL) was added via cannula. The mixture was stirred for 1 h while warmed to rt. The reaction was quenched with sat NH4Cl (aq) and the aqueous layer was extracted with ethyl ether. The combined organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the diynol (0.890 g, 77 % yield); 1H-NMR (400 MHz, CDCl3) δ: 4.45-4.38 (m, 1H), 3.97-3.89 (m, 4H), 1.89-1.80 (brs, 1H), 1.74-1.60 (m, 4H), 1.48-1.32 (m, 6H), 1.31 (s, 3H), 0.19 (s, 9H).
To a solution of above diynol (0.600 g, 1.94 mmol) in diethyl ether (20 mL) was added LAH (0.088 g, 2.33 mmol) at 0 oC and the mixture was stirred for 2 h while warming to rt. The mixture was poured into ice cold water (5 mL) then extracted with diethyl ether. The organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give compound 6 (0.590 g, 98 % yield). 1H-NMR (300 MHz, CDCl3) δ: 6.19 (dd, J= 15.9, 6.0 Hz, 1H), 5.72 (d, J= 15.9 Hz, 1H), 4.28-4.10 (m, 1H), 3.96-3.87 (m, 4H), 1.68-1.47 (m, 6H), 1.42-1.32 (m, 5H), 1.30 (s, 3H), 0.19 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 147.0, 110.3, 110.0, 72.4, 66.1, 64.8, 39.3, 36.9, 29.8, 25.4, 24.2, 23.9, 15.5, 0.1; HRMS m/e (EI) for C17H30O3Si (M)+ calcd 310.1964, measured 310.1921.
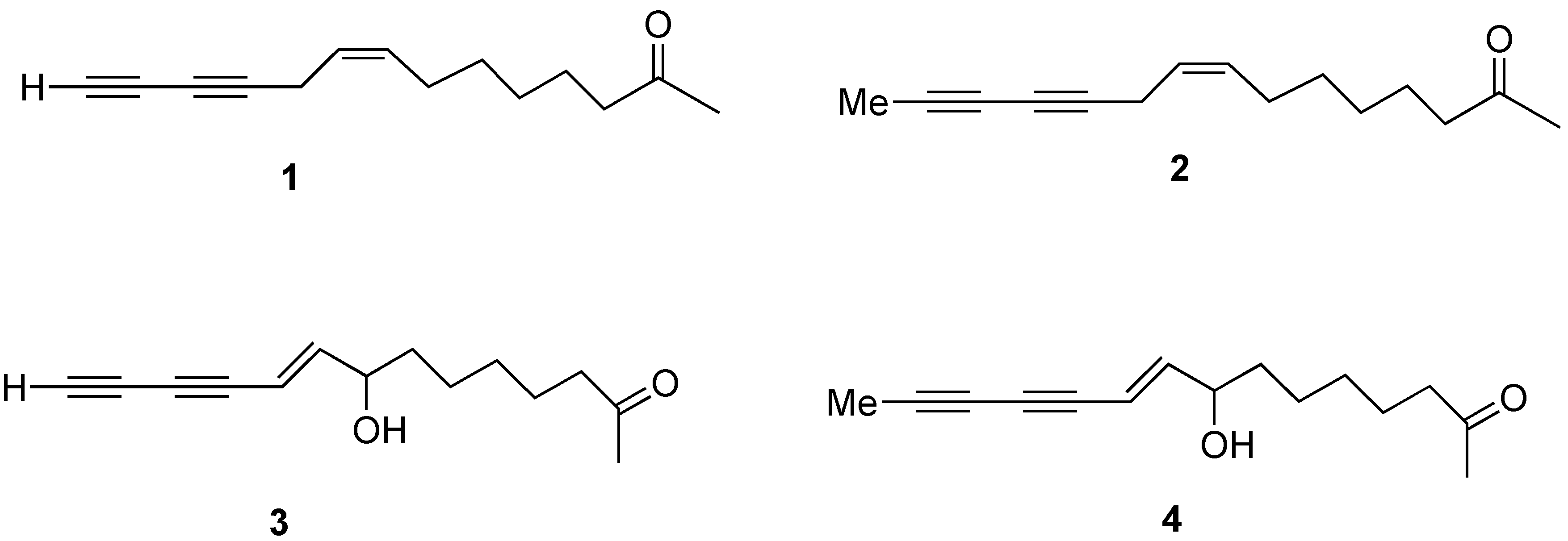
8-Hydroxytetradeca-(9E)-ene-11,13-diyn-2-one (3)
To a solution of compound 6 (0.125 g, 0.40 mmol) in acetone (10 mL) was added N-bromo-succinimide (0.086 g, 0.48 mmol) and AgNO3 (0.004 g, 0.02 mmol) at rt. After stirring for 1 h at this temperature, the mixture was cooled to 0 oC and cold water (5 mL) was added. The aqueous layer was extracted with Et2O, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (hexanes-EtOAc) to give the corresponding bromoacetylene (0.055 g, 43% yield).
To a solution of trimethylsilylacetylene (0.052 mL, 0.37 mmol) and bromoacetylene (0.040 g, 0.126 mmol) in degassed piperidine (1 mL) was added CuCl (0.002 g, 0.013 mmol) at 0 oC. The mixture was stirred at rt for 0.5 h. The reaction was quenched with sat. NH4Cl(aq) (1 mL) and extracted with ethyl ether (3 x 10 mL). The organic phase was washed with brine (2 x 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash chromatography to give the corresponding alcohol (0.035g, 83% yield).
To a solution of the above alcohol (0.030 g, 0.08 mmol) in water/acetone (1 mL/1 mL) was added PPTS (0.002 g, 0.008 mmol) at rt. The mixture was heated at 40 oC for 3 h and extracted with diethyl ether. The organic layer was washed with brine (2 x 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the expected hydroxyketone (0.022 g, 95% yield).
To a solution of hydroxyketone (0.022 g, 0.076 mmol) in THF (5 mL) was added TBAF (0.114 mL, 1M soln in THF) at 0 oC. The mixture was stirred for 1h at rt and solvent was removed. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 3 (0.015 g, 89 % yield). 1H-NMR (400 MHz, CDCl3) δ: 6.34 (dd, J= 16.5, 5.6 Hz, 1H), 5.73 (d, J= 16Hz, 1H), 4.22-4.17 (m, 1H), 2.42 (t, J= 7.6 Hz, 2H), 2.41 (s, 1H), 1.60 (brs, 1H), 1.62-1.50 (m, 6H), 1.40-1.30 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 209.4, 150.6, 108.0, 74.3, 73.9, 72.1, 71.3, 68.3, 43.8, 36.8, 30.2, 29.2, 25.2, 23.8; HRMS m/e (EI) for C14H18O2 (M)+ calcd. 218.1307, measured 218.1229.
8-Hydroxy-pentadeca-(9E)-ene-11,13-diyn-2-one (4)
To a solution of compound 6 (0.125 g, 0.40 mmol) in acetone (10 mL) was added N-bromo-succinimide (0.086 g, 0.48 mmol) and AgNO3 (0.004 g, 0.02 mmol) at rt. After stirring for 1 hr at rt, the mixture was cooled to 0 oC and cold water (5 mL) was added. The aqueous layer was extracted with Et2O, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (hexane-EtOAc) to give the bromoacetylene (0.055 g, 43 % yield).
Degassed piperidine (2 mL), bromoacetylene (0.060 g, 0.189 mmol) and CuCl (0.003 g, 0.019 mmol) were mixed in a sealed tube. The mixture was cooled to -78 oC and excess propyne gas (condensed to liquid, 2 mL) was added by blowing along the wall of the tube. The mixture was slowly warmed to rt. After stirring for 2h at rt, the mixture was cooled to -78 oC and the sealed tube was opened then slowly warmed to room temperature while excess propyne was evaporated. Sat. NH4Cl (aq) was added to the mixture then extracted with ethyl ether. The organic layer was washed with 10% HCl (aq), brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was dissolved in 1:1 water/acetone (2 mL) and treated with PPTS (0.003 g, 0.012 mmol) at rt. The mixture was heated at 40 oC for 3 h and extracted with diethyl ether. The organic layer was washed with brine (2 x 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 4 (0.027 g, 63 % yield). 1H-NMR (300 MHz, CDCl3) δ: 6.26 (dd, J= 15.9, 6.0 Hz, 1H), 5.71 (d, J= 15.9 Hz, 1H), 4.20-4.12 (m, 1H), 2.42 (t, J= 7.5 Hz, 2H), 2.13 (s, 3H), 1.98 (s, 3H), 1.62-1.49 (m, 4H), 1.39-1.24 (m, 4H); 13C-NMR (75 MHz, CDCl3) δ 209.4, 148.7, 109.1, 81.8, 80.4, 76.8, 72.2, 64.5, 43.8, 36.8, 30.1, 29.2, 25.2, 23.8, 4.8; HRMS m/e (EI) for C15H20O2 (M)+ calcd 232.1463, measured 232.1497.
3,5-Heptadiyn-1-ol (8)
Degassed piperidine (5.5 mL), 4-iodo-3-butynol (1.22 g, 6.2 mmol) and CuCl (0.061 g, 0.62 mmol) were mixedIn a tube. The mixture was cooled to -78 oC and excess propyne gas was added by blowing along the wall of the tube. The propyne was condensed to liquid (2 mL) in tube and the tube was sealed. The mixture was slowly warmed to rt. After stirring for 2 h at rt, the mixture was cooled to -78 oC and the sealed tube was opened. The mixture was warmed to rt slowly to evaporate excess propyne. NH4Cl (aq) (20 mL) was added to the mixture then extracted with Et2O (3 x 20 mL). Organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate= 2:1) to give compound 8 (538 mg, 81 % yield). 1H-NMR (300 MHz, CDCl3) δ: 3.74-3.71 (m, 2H), 2.51 (t, J = 6.8 Hz, 2H), 1.91 (s, 3H).
Triphenyl(hepta-3,5-diynyl)phosphonium Iodide (9)
To a solution of imidazole (374 mg, 5.5 mmol) and Ph3P (1.44 g, 5.5 mmol) in Et2O–MeCN (12 mL/4 mL) was slowly added iodine (1.40 g, 5.5 mmol) at 0 °C. The resulting slurry was warmed to room temperature and then stirred for 20 min. The slurry was cooled to 0 °C and the alcohol 8 (538 mg, 4.9 mmol) was added in Et2O (10 mL) at 0 °C. The solution was slowly warmed to room temperature and then stirred for 1 h. The reaction was quenched by adding hexane (30 mL). The organic layer was washed with aq NaHCO3, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes–EtOAc, 2:1) to give the iodide (1.04 g, 98 % yield). 1H-NMR (300 MHz, CDCl3) δ: 3.13 (t, J = 7.2 Hz, 2H), 2.77 (t, J = 7.2 Hz, 2H), 1.83 (s, 3H). To a solution of PPh3 (0.793 g, 3.03 mmol) in acetonitrile was added the above iodide (0.60 g, 2.75 mmol) and the mixture was boiled for 24 h, then cooled to rt and the solvent was removed to give 9 as a yellowish oil. (1.02 g, 78% yield). 1H-NMR (400 MHz, CDCl3) δ: 7.76-7.53 (m, 15H), 3.86-3.80 (m, 2H), 2.79 (dt, J = 20.8, 6.4 Hz, 2H), 1.87 (s, 3H).
Pentadec-8Z-ene-11,13-diyn-2-one (2)
To a solution of compound 9 (612 mg, 1.19 mmol) in THF (10 mL) was added NaHMDS (1.19 mL, 1M in THF) at -78 oC. The mixture was stirred for 20 min at -78 oC, then aldehyde 5 (201 mg, 1.08 mmol) in THF (3 mL) was added by cannula. The mixture was slowly warmed to rt then stirred for 12 h. The reaction was quenched with NH4Cl (5 mL) and extracted with Et2O (20 mL). The organic layer was washed with water, brine, dried (MgSO4) and concentrated. The residue was purified via column chromatography (hexanes/ethyl acetate= 2:1) to give an enediyne (173 mg, 54 % yield). 1H-NMR (400 MHz, CDCl3) δ: 5.46 (dt, J = 10.4, 7.2 Hz, 1H), 5.36 (dt, J = 10.4, 7.2 Hz, 1H), 3.96-3.88 (m, 4H), 2.98 (d, J = 7.2 Hz, 2H), 2.01 (td, J = 7.2, 6.0 Hz, 2H), 1.89 (s, 3H), 1.63-1.59 (m, 2H), 1.41-1.29 (m, 6H), 1.30 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 132.7, 122.9, 110.4, 75.0, 73.6, 65.3, 64.8, 64.7, 39.4, 29.7, 29.5, 27.3, 24.2, 24.0, 17.8, 4.4.
To a solution of the above compound (56 mg, 0.22 mmol) in 1:1 acetone/water (2 mL) was added PPTS (4.6 mg, 0.022 mmol) at rt. The mixture was heated at 40 oC for 3 h and extracted with ether. The organic layer was washed with brine (2 x 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 2 (43 mg, 93% yield). 1H-NMR (400 MHz, CDCl3) δ: 5.48-5.34 (m, 2H), 2.96 (d, J = 7.2 Hz, 2H), 2.41 (t, J = 7.2 Hz, 2H), 2.12 (s, 3H), 2.01 (td, J = 7.2, 6.0 Hz, 2H), 1.88 (s, 3H), 1.60-1.51 (m, 2H), 1.42-1.22 (m, 6H); 13C-NMR (100 MHz, CDCl3) δ: 209.4, 132.5, 123.1, 74.9, 73.6, 65.3, 64.8, 43.9, 30.1, 29.2, 28.9, 27.2, 23.9, 17.8, 4.4; HRMS (EI): m/z calcd. for C15H20O : 216.1514; found: 216.1510.
Statistical analysis
Statistical analyses were performed using SAS software version 8.02 (SAS Institute Inc., Cary, NC). One-way analysis of variance followed by the Tukey test was used to compare means. Significance of difference was defined at p < 0.01.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




