Synthesis and Activity of a New Series of(Z)-3-Phenyl-2-benzoylpropenoic Acid Derivatives as Aldose Reductase Inhibitors

Abstract
:Introduction
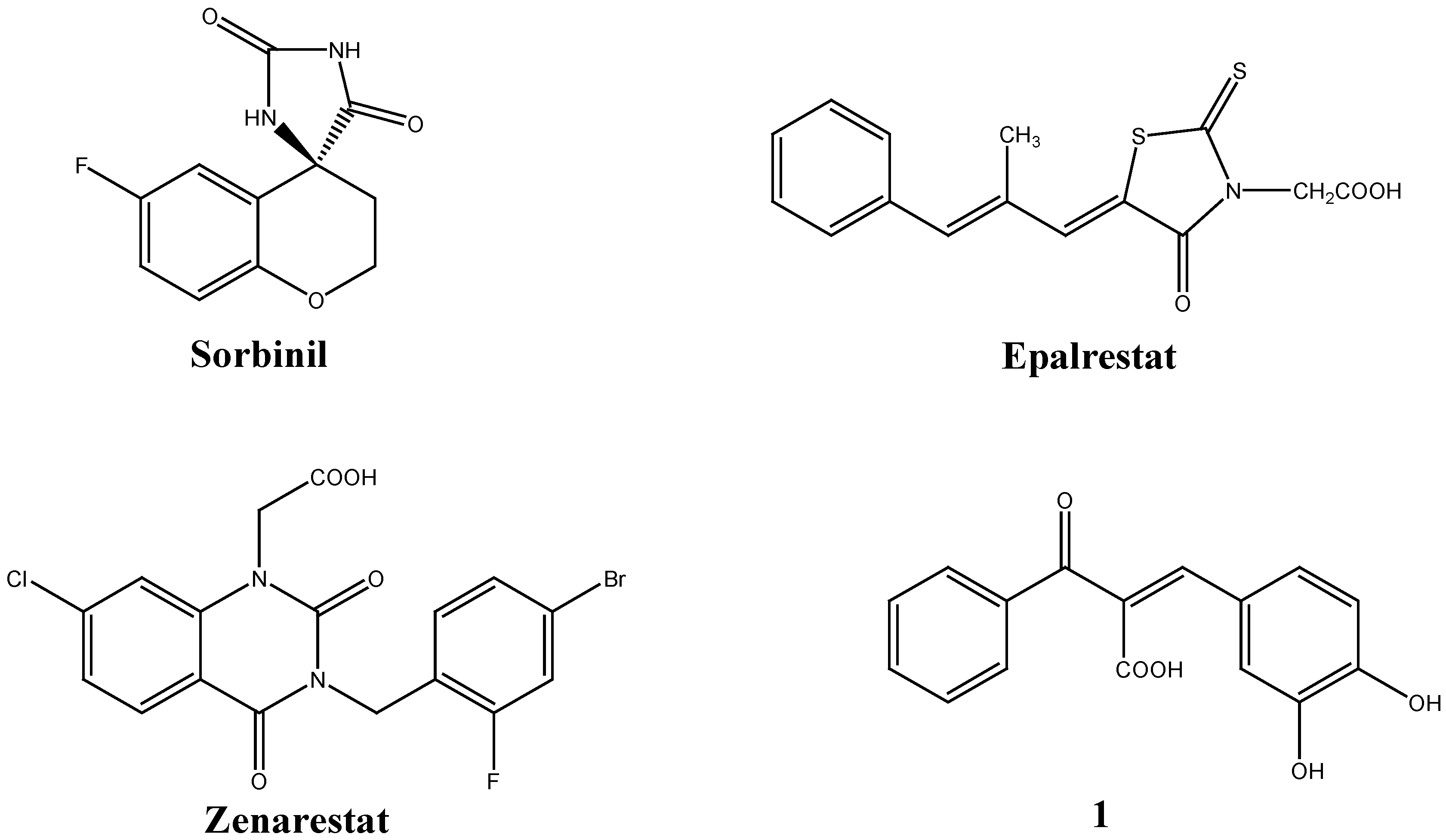
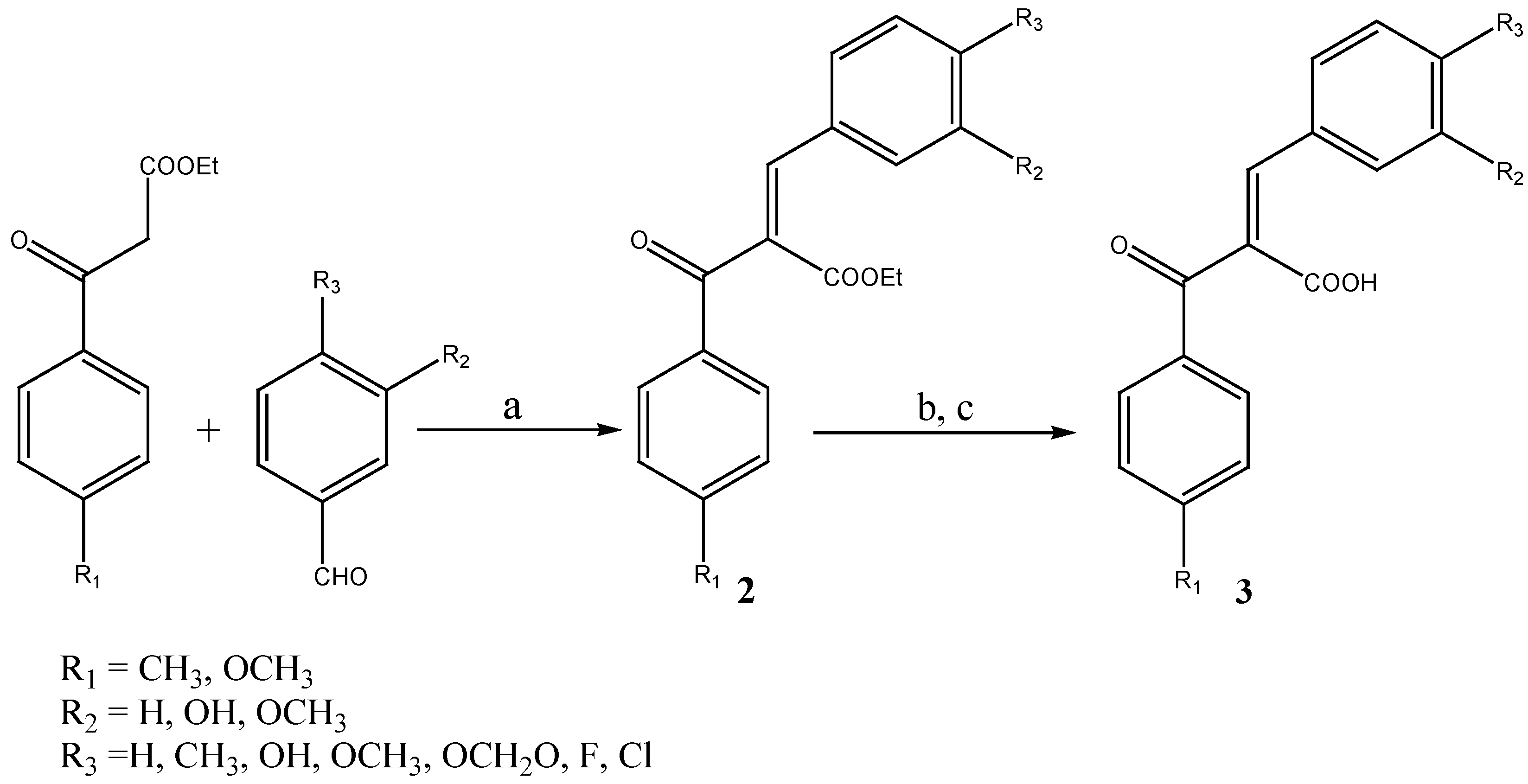
Results and Discussion
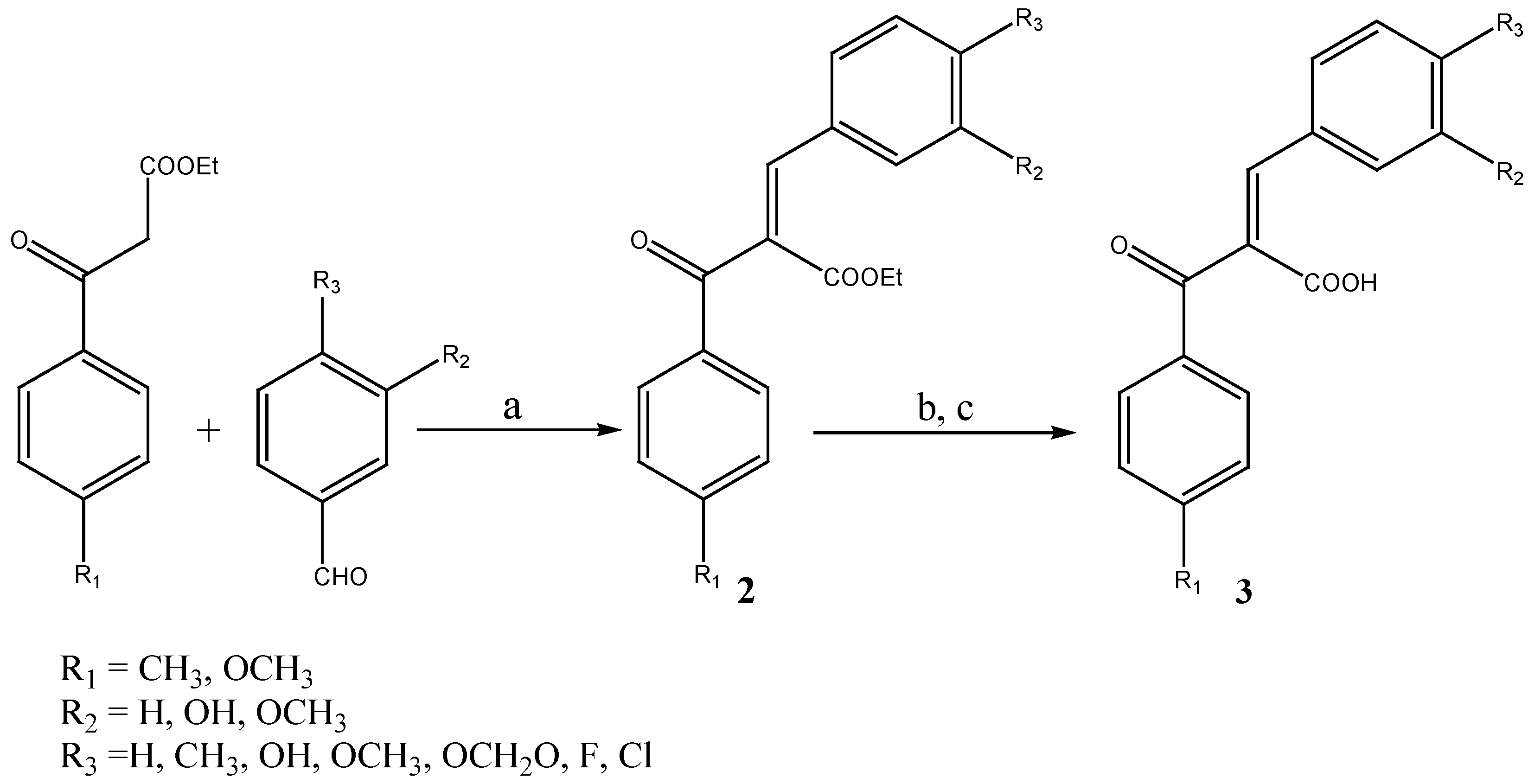
Aldose reductase inhibitory activity

{kind=link}
{kind=link}
{kind=link}

| Compds | R1 | R2 | R3 | IC50 (μM)a |
|---|---|---|---|---|
| 3a | CH3 | H | H | n.a.b |
| 3b | CH3 | H | OCH3 | n.a. |
| 3c | CH3 | H | F | n.a. |
| 3d | CH3 | H | Cl | n.a. |
| 3e | CH3 | OCH2O | n.a. | |
| 3f | CH3 | H | OH | n.a. |
| 3g | CH3 | OH | H | n.a. |
| 3h | CH3 | H | CH3 | n.a. |
| 3i | CH3 | OCH3 | H | 7.95 |
| 3j | CH3 | OCH3 | OH | 15.69 |
| 3k | CH3 | OH | OH | 0.49 |
| 3l | OCH3 | OH | OH | 30.84 |
| 3m | OCH3 | OCH2O | n.a. | |
| 3n | OCH3 | H | Cl | n.a. |
| 3o | OCH3 | OH | H | n.a. |
| Epalrestat | 0.075 | |||
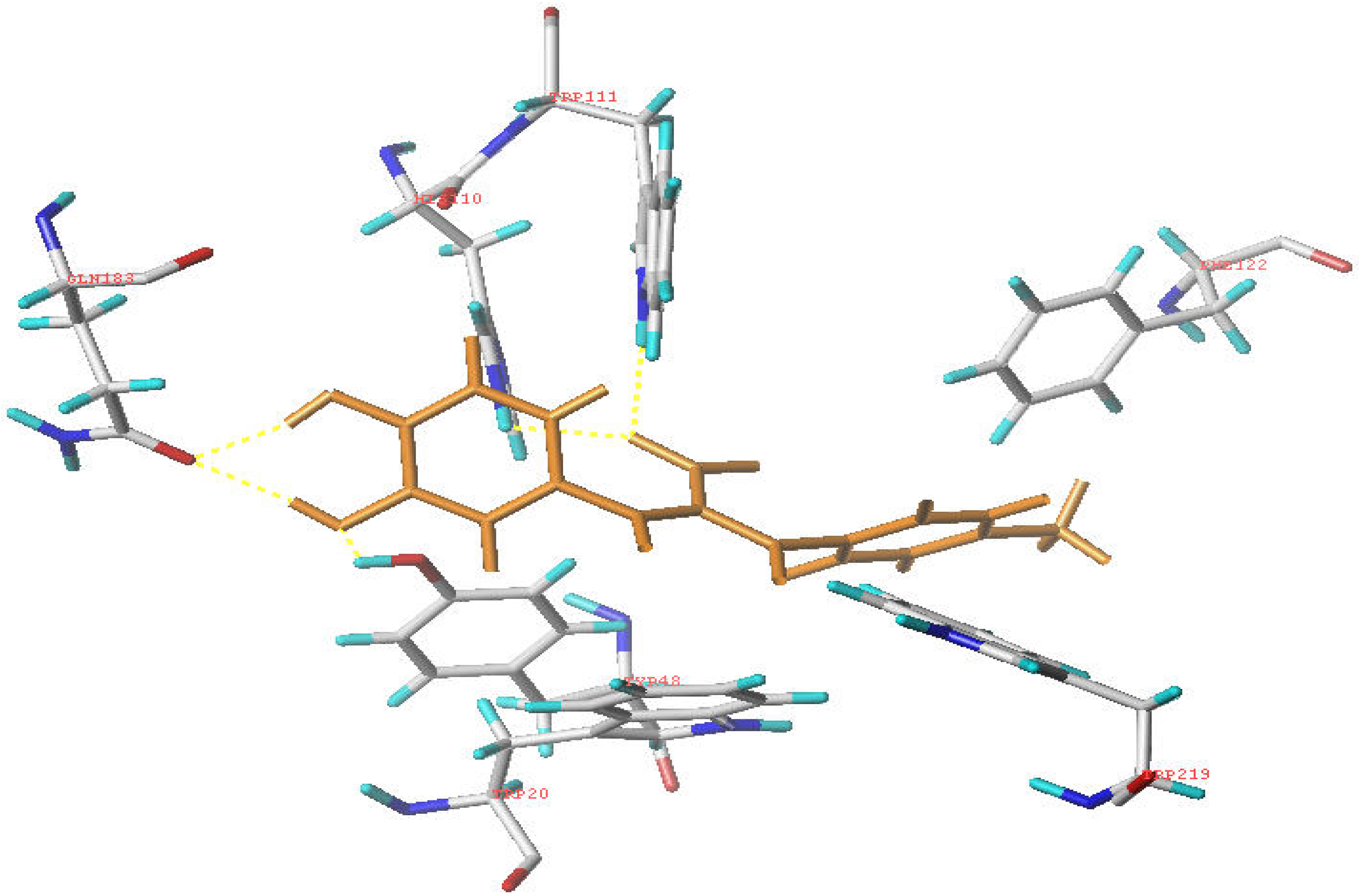
Molecular modeling
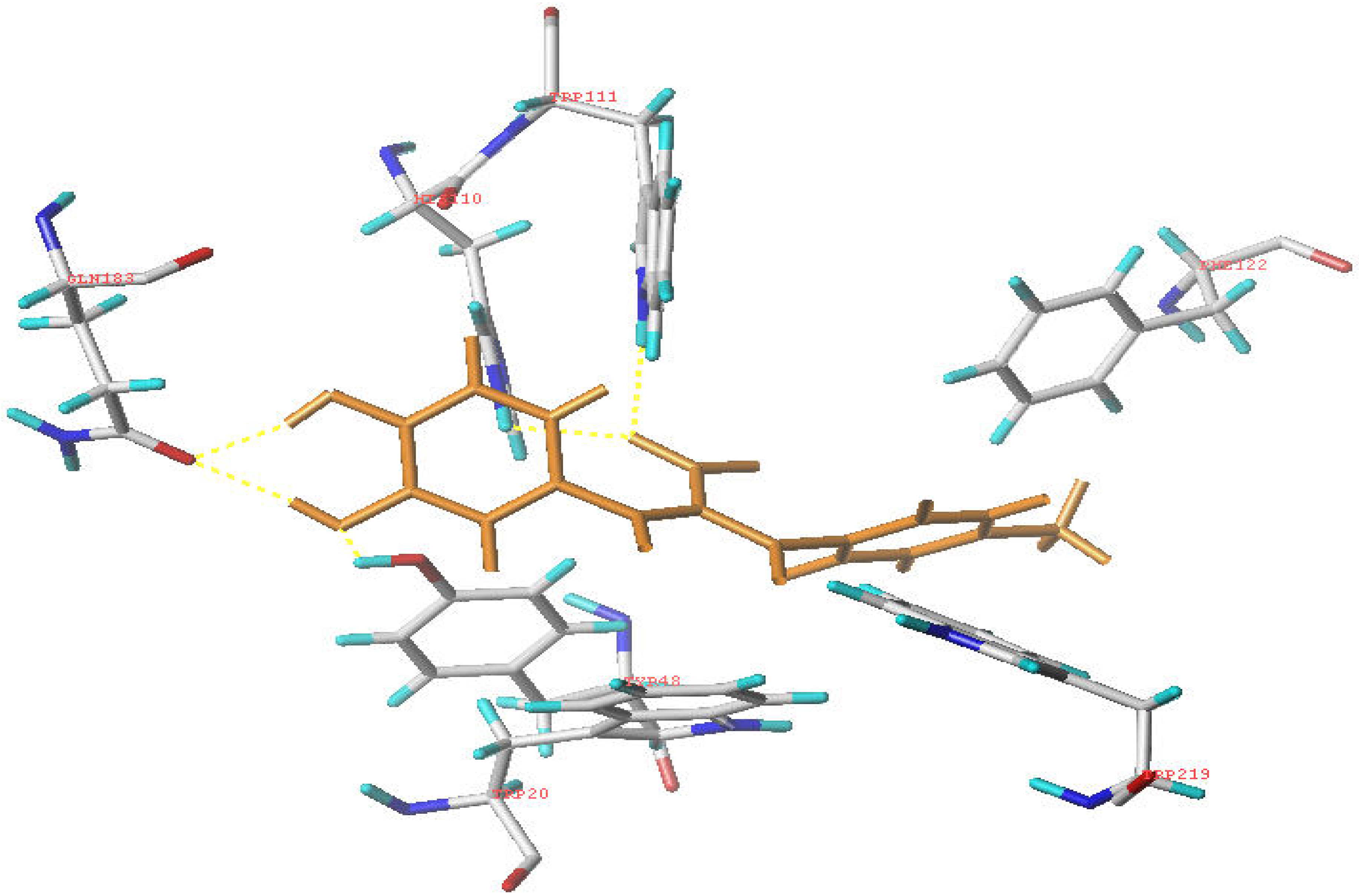
Conclusions
Experimental
General
General procedure for the preparation of (Z)-3-phenyl-2-benzoylpropenoic acids 3a-o
Aldose reductase inhibitory assay
Computational methods
Acknowledgments
References
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol. Rev. 1998, 50, 21–33. [Google Scholar]
- Settimo, F. D.; Primofiore, G.; Motta, C. L.; Sartini, S.; Taliani, S.; Simorini, F.; Marini, A. M.; Lavecchia, A.; Novellino, E.; Boldrini, E. Naphtho[1,2-d]isothiazole acetic acid derivatives as a novel class of selective aldose reductase inhibitors. J. Med. Chem. 2005, 48, 6897–6907. [Google Scholar]
- Van Zandt, M. C.; Jones, M. L.; Gunn, D. E.; Geraci, L. S.; Jones, J. H. Discovery of 3-[(4,5,7-trifluorobenzothiazol-2-yl)methyl]indole-N-acetic acid and congeners as highly potent and selective inhibitors of aldose reductase for treatment of chronic diabetic complications. J. Med. Chem. 2005, 48, 3141–3152. [Google Scholar]
- Stevens, J. F.; Taylor, A. V.; Nickerson, G. B.; Ivancic, M. Prenylflavonoid variation in (Humulus lupulus): distribution and taxonomic significance of xanthogalenol and 4′-O-methylxanthohumol. Phytochemistry 2000, 53, 759–775. [Google Scholar]
- Liu, M.; Wilairat, P.; Go, M. L. Antimalarial alkoxylated and hydroxylated chalones: structure-activity relationship analysis. J. Med. Chem. 2001, 44, 4443–4452. [Google Scholar]
- Babu, M. A.; Shakya, N.; Prathipati, P.; Kaskhedikar, S. G.; Saxena, A. K. Development of 3D-QSAR models for 5-Lipoxygenase antagonists: chalcones. Bioorg. Med. Chem. 2003, 10, 4035–4041. [Google Scholar]
- Kumar, S. K.; Hager, E.; Pettit, C.; Gurulingappa, H.; Davidson, N. E. Design, Synthesis, and Evaluation of Novel Boronic-Chalcone Derivatives as Antitumor Agents. J. Med. Chem. 2003, 46, 2813–2815. [Google Scholar]
- Satyanarayana, M.; Tiwari, P.; Tripathi, B. K.; Srivastava, A. K.; Pratap, R. Synthesis and antihyperglycemic activity of chalcone based aryloxypropanolamines. Bioorg. Med. Chem. 2004, 12, 883–889. [Google Scholar]
- Matsuda, H.; Morikawa, T.; Toguchida, I.; Yoshikawa, M. Structural requirements of flavonoids and related compounds for aldose reductase inhibitory activity. Chem. Pharm. Bull. 2002, 50, 788–795. [Google Scholar]
- Aida, K.; Tawata, M.; Shindo, H.; Onaya, T.; Sasaki, H.; Yamaguchi, T.; Chin, M.; Mitsuhashi, H. Isoliquiritigenin: a new aldose reductase inhibitor from glycyrrhizae radix. Planta Med. 1990, 56, 254–258. [Google Scholar]
- Severi, F.; Benvenuti, S.; Costantino, L.; Vampa, G.; Michele, M.; Antolini, L. Synthesis and activity of a new series of chalcones as aldose reductase inhibitors. Eur. J. Med. Chem. 1998, 33, 859–866. [Google Scholar]
- Wang, S. J.; Zhang, Z. Y.; Wu, J.; Zhou, W. F.; Cheng, M. S. Synthesis and aldose reductase inhibitory activity of α-(phenylmethylene)-β-oxo-benzenepropanoic acid derivatives. Chin. J. Med. Chem. 2006, 16, 1–5. [Google Scholar]
- Milcent, R.; Malanda, J. C.; Barbier, G.; Vaissermann, J. Synthesis of ethyl 2-aminodihydro-5- pyrimidincarboxylate derivatives and 3,7-diethoxycarbonyl-4,6-dihydro-2,4,6,8-tetraaryl-1H-pyrimido [1,2-a]pyrimidines. J. Heterocyclic Chem. 1997, 34, 329–336. [Google Scholar] [CrossRef]
- Halder, A. B.; Crabbe, M. J. Bovine lens aldehyde reductase (aldose reductase). Purification, kinetics and mechanism. Biochem. J. 1984, 219, 33–39. [Google Scholar]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar]
- Chung, S.; LaMendola, J. Cloning and sequence determination of human placental aldose reductase gene. J. Biol. Chem. 1989, 264, 14775–14777. [Google Scholar]
- Urzhumtsev, A.; Tête-Favier, F.; Mitschler, A.; Barbanton, J.; Barth, P.; Urzhumtseva, L.; Biellmann, J. F.; Podjarny, A. D.; Moras, D. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997, 5, 601–612. [Google Scholar]
- Podjarny, A.; Cachau, R. E.; Schneider, T.; Van Zandt, M.; Joachimiak, A. Subatomic and atomic crystallographic studies of aldose reductase: implications for inhibitor binding. Cell. Mol. Life Sci. 2004, 61, 763–773. [Google Scholar]
- SYBYL Molecular Modelling System, version 6.9.1; Tripos Inc.: St. Louis, MO, 2003.
- Sample Availability: Available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Wang, S.-J.; Yan, J.-F.; Hao, D.; Niu, X.-W.; Cheng, M.-S. Synthesis and Activity of a New Series of(Z)-3-Phenyl-2-benzoylpropenoic Acid Derivatives as Aldose Reductase Inhibitors. Molecules 2007, 12, 885-895. https://doi.org/10.3390/12040885
Wang S-J, Yan J-F, Hao D, Niu X-W, Cheng M-S. Synthesis and Activity of a New Series of(Z)-3-Phenyl-2-benzoylpropenoic Acid Derivatives as Aldose Reductase Inhibitors. Molecules. 2007; 12(4):885-895. https://doi.org/10.3390/12040885
Chicago/Turabian StyleWang, Shao-Jie, Ju-Fang Yan, Dong Hao, Xin-Wen Niu, and Mao-Sheng Cheng. 2007. "Synthesis and Activity of a New Series of(Z)-3-Phenyl-2-benzoylpropenoic Acid Derivatives as Aldose Reductase Inhibitors" Molecules 12, no. 4: 885-895. https://doi.org/10.3390/12040885