Facile Synthesis of Optically Active Imidazole Derivatives

Department of Organic Chemistry, Faculty of Chemical Technology, University of Pardubice, nam. Cs. legii 565, Pardubice, 53210, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(5), 1183-1190; https://doi.org/10.3390/12051183
Submission received: 11 May 2007
/
Revised: 22 May 2007
/
Accepted: 23 May 2007
/
Published: 30 May 2007
(This article belongs to the Special Issue Heterocycles)
Abstract
:Five optically active imidazole derivatives have been synthesized via a facile 4-step reaction sequence starting from commercially available and inexpensive N-Cbz amino acids. While microwave assisted condensation was unsuccessful, the condensation of the corresponding α-bromoketones with formamidine acetate in liquid ammonia was revealed to be a useful method for the synthesis of such imidazole derivatives. The derivatives thus prepared are structurally-related to histamine.
Introduction
From time immemorial, organic chemists have been attempting to synthesize, isolate and characterize heterocyclic molecules for their unique chemical and physical properties. Despite the fact that many convenient synthetic methods have been utilized for preparing the basic heterocycles [1], a facile synthesis of optically active analogues still remains a challenge. Among the many possible targets enantiopure five-membered heterocyclic derivatives such as imidazole are of particularly interest. For instance, naturally occurring 4-substituted imidazoles such as histidine or histamine and their significance as an essential amino acid or its decarboxylation product, are well known. Imidazole chemistry currently attracts considerable attention, where the imidazole derivatives are widely applied as N-ligands coordinating transition metals [2,3]. The application of imidazoles in medicinal chemistry [4] or chemistry of natural products/alkaloids [5,6] or of 1,3-disubstituted imidazole salts as ionic liquids [7,8] are also well known. Although a few examples of the synthesis and applications of optically active imidazole derivatives have been published [9,10,11,12,13], development of an efficient synthesis of such derivatives still requires more attention. Recently, we have published the synthesis and application of enantiopure 2-phenylimidazoles starting from the commercially available α-amino acids as a source of chirality [14,15]. It would be challenging to synthesize 2,5-unsubstituted imidazoles bearing similar chains with a chiral amine (depending on the starting amino acids used). In addition, optically active imidazole derivatives formed upon cross-coupling reactions at the 2 position with another heterocycles (pyridine, phenanthroline, etc.), in order to further support the ability to bind transition metal ions, might provide interesting enantiopure nitrogen ligands. A possible application of such materials in asymmetric catalysis or as histamine-related products is apparent. Hence, we describe herein the synthesis of the proposed optically active 4-substituted imidazole derivatives as well as their detailed structural analysis.
Results and Discussion
One of the synthetic pathways used for the construction of the N(1)-C(5) and N(3)-C(4) bonds of the imidazole ring involves reaction of 1,3-bifunctional electrophiles with various amidines. In order to prepare a 2-unsubstituted imidazole, formamide must be employed, while the 1,3-bifunctional electrophiles could be represented by various haloketones [16,17,18] or hydroxyketones [20]. Sugar based hydroxyketones or hydroxyaldehydes and their application as suitable 1,3-bifunctional electrophiles are also well known [20,21]. The condensation may be enhanced by microwave irradiation [22] as well. Nevertheless, the most used method for imidazole ring construction involves a condensation of the α-bromoketones with formamidine in liquid ammonia [16,17,18,19,20,21].
During our investigation in the field of enantiopure imidazole synthesis, we have developed an efficient method utilizing N-Cbz protected α-amino acids 1 as a suitable chiral starting material. Activation of the carboxylic function of the α-amino acid via mixed anhydrides 2, followed by its reaction with diazomethane afforded α-diazoketones 3. Treatment of the α-diazoketones with hydrobromic acid smoothly provided α-bromoketones 4, while nitrogen was liberated. In this way prepared α-bromoketones may serve as suitable 1,3-bifunctional electrophiles and, using the known condition for condensation in liquid ammonia as described above, we have prepared five optically active imidazole derivatives 5 bearing substituents at position 4 (Scheme 1).
(S)-Cbz-Alanine, (S)-Cbz-valine, (S)-Cbz-leucine, (S)-Cbz-isoleucine and (S)-Cbz-phenylalanine were used as the starting N-Cbz protected α-amino acids. The condensation reactions of the α-bromoketones with formamidine acetate were carried out in a pressure vessel containing liquefied ammonia at 70 °C. The reaction mixture was stirred overnight, extracted and purified by column chromatography to afford pure imidazoles 5a-e. Yields and selected physical properties of the compounds prepared are summarized in the Table 1.
Scheme 1.
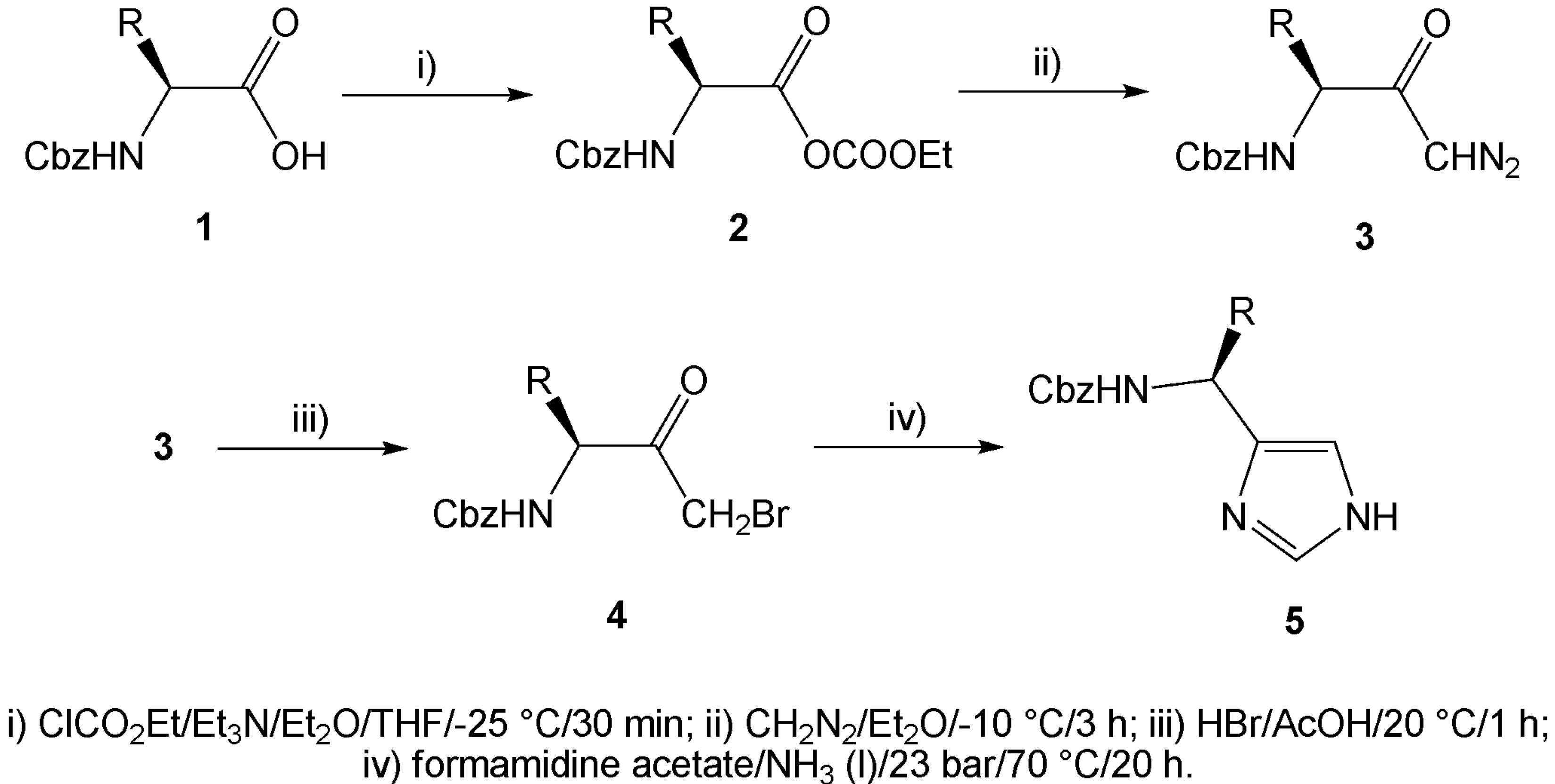

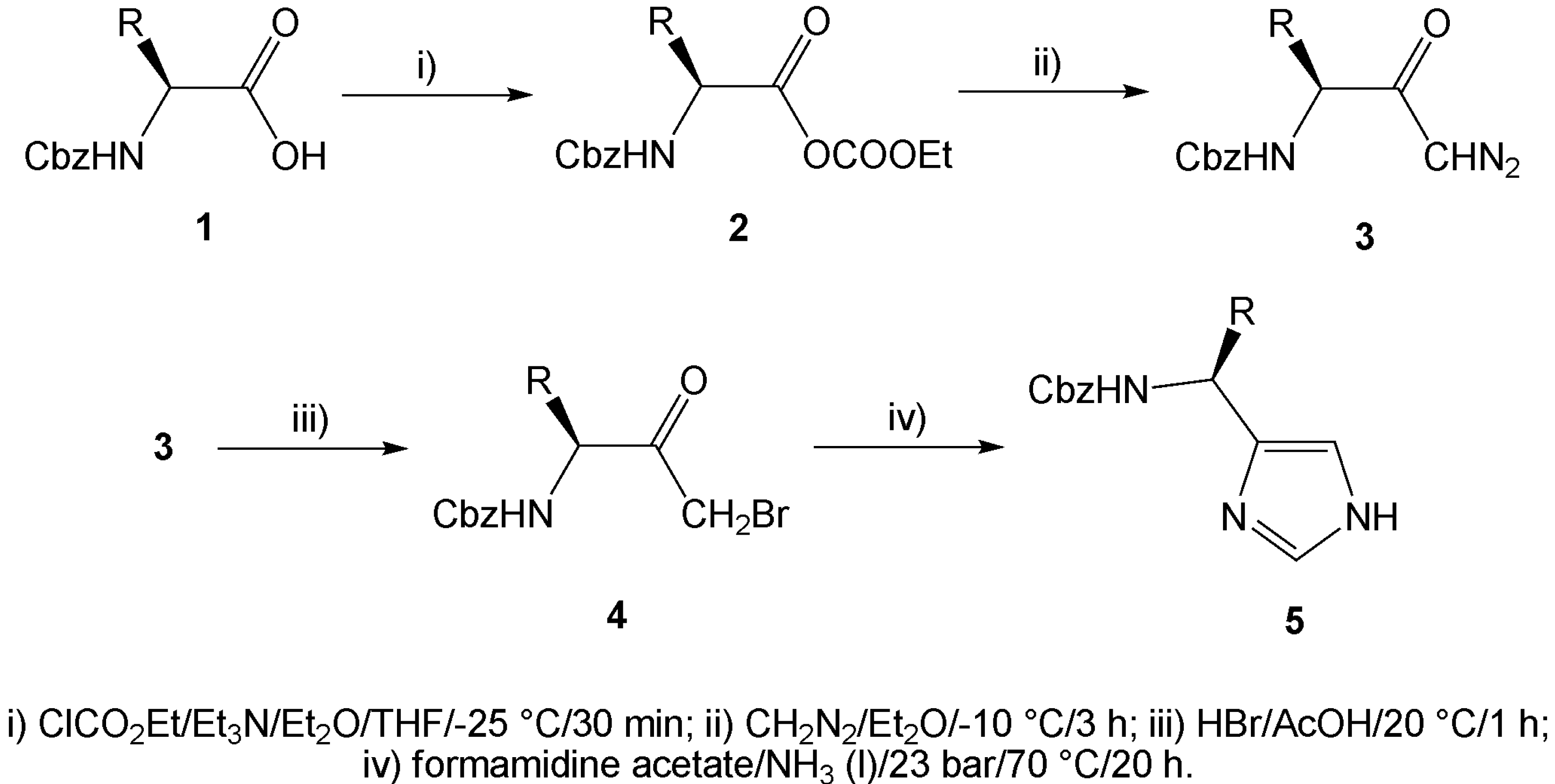{kind=link}
{kind=link}
| Comp. | R / Starting amino acid | Yield[a] [%] | e.e. [%] | [α]D20 |
|---|---|---|---|---|
| 5a | CH3 / (S)-Alanine | 81/69 | 99 | -15.8 (c 1, MeOH) |
| 5b | (CH3)2CH / (S)-Valine | 49/42 | 99 | -31.7 (c 0.52, MeOH) |
| 5c | (CH3)2CHCH2 / (S)-Leucine | 69/57 | 99 | -28.5 (c 0.46, MeOH) |
| 5d | CH3CH2(CH3)CH / (S)-Isoleucine | 72/56 | 99 | -35.6 (c 1, MeOH) |
| 5e | PhCH2 / (S)-Phenylalanine | 40/37 | 99 | -29.0 (c 1, MeOH) |
[a] GC/isolated yields of the final condensation reaction
The enantiomeric excesses (e.e.) were determined by 1H-NMR spectroscopy using (R)-Mosher‘s acid. The resulting spectra were compared with the spectra obtained from the corresponding racemate. No racemization was observed throughout the entire reaction sequence. All of the synthesized imidazoles were further characterized by 1H- and 13C-NMR spectroscopy, EI-MS and elemental analysis (see Experimental section). The NMR structural analyses of the ligands showed the presence of strong hydrogen bonds in a common CDCl3 solution, which resulted in hindered imidazole tautomerism and hindered rotation in the protecting carbamate function used. 1H- and 13C-NMR spectra then showed broad signals or two set of signals, without expected spin–spin interactions. The hindered imidazole tautomerism and rotation in the carbamate function was suppressed if measured in deuteromethanol. Hence, the 1H- and 13C-NMR spectra listed in the Experimental were measured in CD3OD.
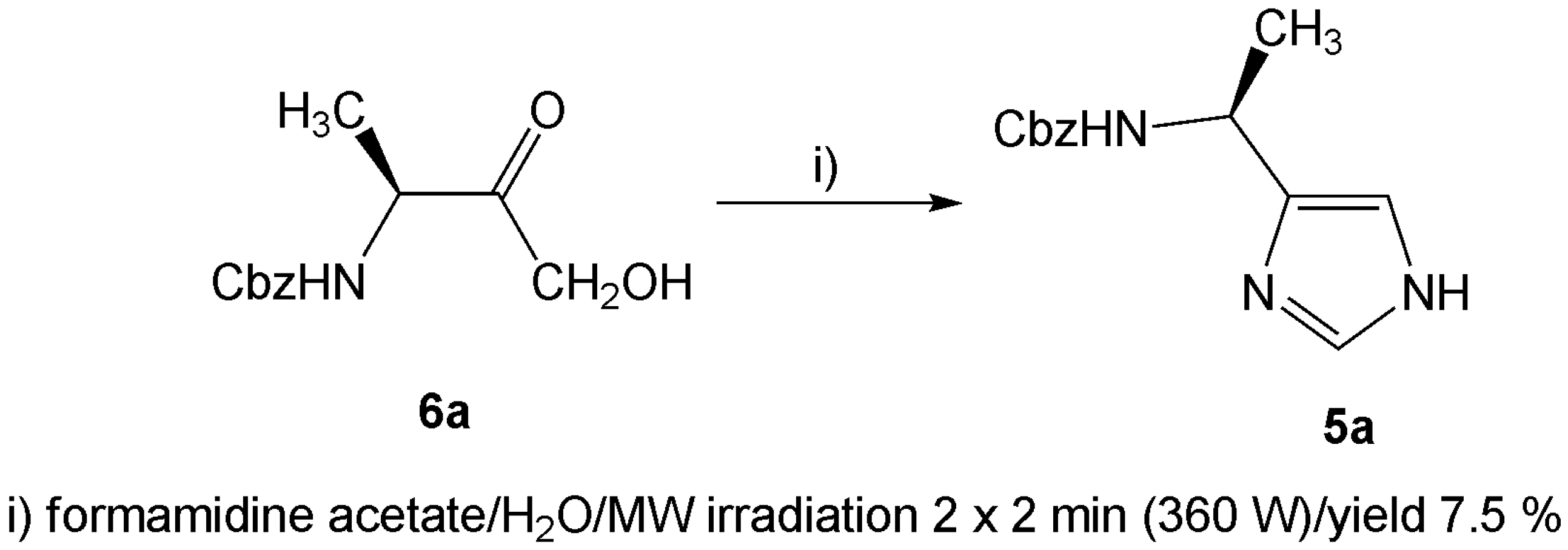
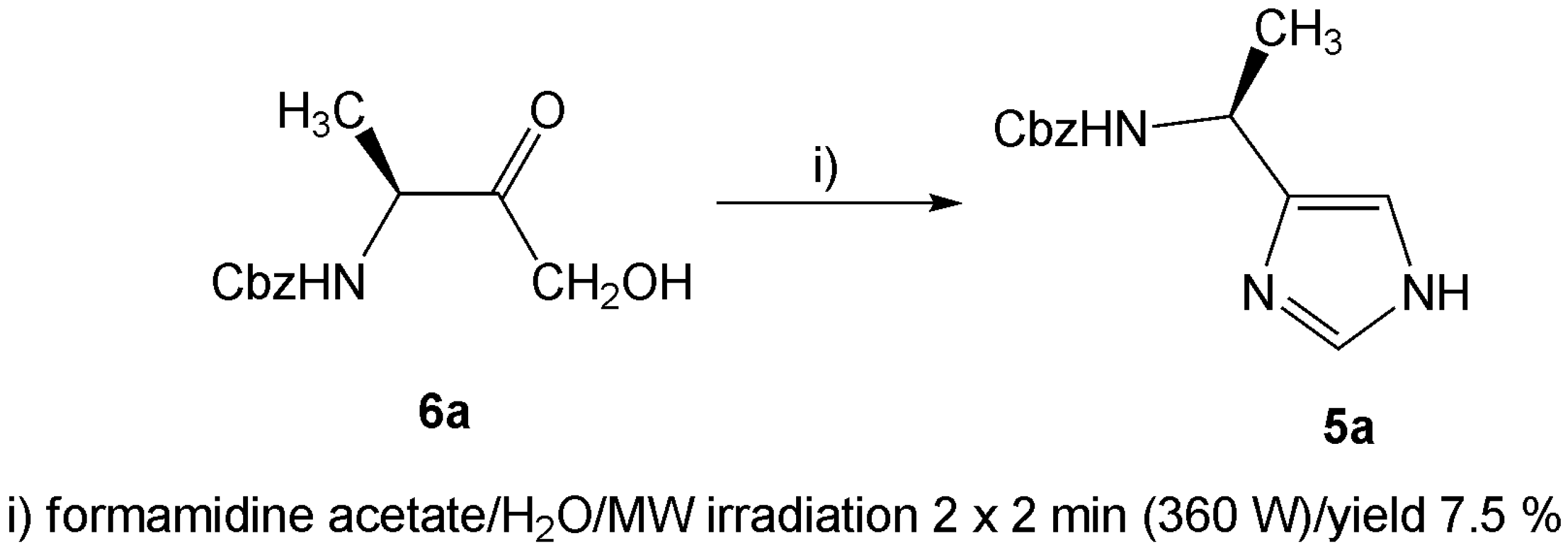
In addition to the condensation reaction carried out in liquid ammonia, microwave assisted reaction [22] was attempted as well. The α-hydroxyketones 6, obtained from the conversion of the α-diazoketone 3 using hydrochloric acid [23], were treated with formamidine acetate and irradiated with microwaves (Scheme 2). Only traces of one corresponding imidazole 5a could be isolated, even after we tried to optimize the reaction conditions. It is also noteworthy that no formation of the corresponding imidazole was observed if another α-hydroxy- or α-bromoketone used. Hence, the microwave assisted reaction was shown to be unsuitable for our derivatives.
Scheme 2.
Conclusions
Five novel imidazole derivatives have been synthesized and isolated in satisfactory yields (69-37%). The crucial step of the reaction sequence was shown to be the condensation of the corresponding α-bromoketones with formamidine acetate in liquid ammonia. In CDCl3 solutions the products showed hindered imidazole tautomerism and also hindered carbamate function rotation, which makes the NMR analysis difficult and, therefore, the spectra were measured in deuteromethanol. Microwave assisted reaction of the α-hydroxyketones was revealed to be incompatible with the above discussed intermediates. Overall, we have developed a facile 4-step synthesis of histamine-related imidazole derivatives starting from the commercially available and inexpensive N-Cbz amino acids. Cross-couplings of the synthesized imidazoles with other heterocycles, in order to produce even larger nitrogen ligands with stronger coordinating ability, are now under investigation.
Experimental
General
Reagents and solvents (reagent grade) were purchased from Aldrich or Fluka and used as received. The α-bromoketones 4 and α-hydroxyketones 6 were synthesized according to the literature procedures [14,23]. Evaporation and concentration in vacuo were performed at water aspirator pressure. The condensation reactions in liquid ammonia were carried out in a ROTH pressure vessel. The microwave assisted reactions were conducted in a MILESTONE MLS ETHOS 1600 URM oven (300 W; 2.45 GHz). Column chromatography (CC) was carried out with SiO2 60 (particle size 0.040-0.063 mm, 230-400 mesh; Merck) and commercially available solvents. Thin-layer chromatography (TLC) was conducted on aluminium sheets coated with SiO2 60 F254 obtained from Merck, with visualization by UV lamp (254 or 360 nm). Melting points (M.p.) were measured on a Buchi B-540 melting-point apparatus in open capillaries and are uncorrected. 1H- and 13C-NMR spectra were recorded in CD3OD at 360 MHz or 90 MHz, respectively, with Bruker AMX 360 instrument at 20 °C. Chemical shifts are reported in ppm relative to the signal of Me4Si. Residual solvent signals in the 1H- and 13C-NMR spectra were used as an internal reference (CD3OD – 3.31 and 49.15 ppm for 1H- and 13C-NMR, respectively). Coupling constants (J) are given in Hz. The apparent resonance multiplicity is described as s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet) and m (multiplet). Optical rotation values were measured on a Perkin Elmer 341 instrument, concentration c is given in g/100 mL CH3OH. The mass spectra were measured on GC/MS configuration comprised of an Agilent Technologies – 6890N gas chromatograph (HP-5MS column, length 30 m, I.D. 0.25 mm, film 0.25 μm) equipped with a 5973 Network MS detector (EI 70 eV, mass range 33-550 Da).
General method for the condensations in liquid ammonia
Into a cooled (-78 °C) pressure vessel equipped with a magnetic stirrer, ammonia (100 mL) was liquefied, followed by addition of the appropriate α-bromoketone (6.3 mmol) and formamidine acetate (0.65 g; 6.3 mmol). The reaction mixture was stirred 20 h at 70 °C at 23-26 bar, the residual ammonia evaporated, the residue taken up in CHCl3 (100 mL) and the organic extract washed successively with sat. aq. potassium carbonate (3 × 100 mL) and water (200 mL). The organic layer was dried (Na2SO4) and evaporated in vacuo. CC (SiO2; EtOAc/hexane 1:1 to CHCl3/CH3OH/NH4OH 5:1:0.1) afforded the pure products.
(S)-Benzyl 1-(1H-imidazol-4-yl)ethylcarbamate (5a). Synthesized from α-bromoketone 4a (69 % yield) as a white solid; Rf = 0.55 (SiO2; CHCl3/CH3OH/NH4OH 5:1:0.1); M.p. 119-121 °C; [α]D20 = -15.8 (c 1, CH3OH); 1H-NMR: δ = 1.46 (3H, d, J = 6.9, CH3), 4.81 (1H, q, J = 6.9, CH), 5.04-5.11 (2H, m, PhCH2), 6.90 (1H, s, 4-Him), 7.27-7.34 (5H, m, Ar), 7.59 (1H, d, J = 1.2, 2-Him); 13C-NMR: δ = 21.32 (CH3), 45.92 (CH), 67.53 (PhCH2), 116.27 (5-Cim), 128.95 (Ar), 129.09 (Ar), 129.57 (Ar), 136.48 (2-Cim), 138.50 (Ar), 141.9 (4-Cim), 158.53 (CO); EI-MS (70 eV) m/z (%): 245 (1), 154 (10), 137 (10), 122 (50), 108 (90), 94 (60), 79 (100), 67 (10), 51 (20), 44 (70); Elemental analysis (%) calcd. for C13H15N3O2 (245.12): C 63.66, H 6.16, N 17.13; found: C 63.66, H 6.15, N 17.06.
(S)-Benzyl 1-(1H-imidazol-4-yl)-2-methylpropylcarbamate (5b). Synthesized from α-bromoketone 4b (42 % yield) as an amorphous solid; Rf = 0.61 (SiO2; CHCl3/CH3OH/NH4OH 5:1:0.1); M.p. 95-100 °C; [α]D20 = -31.7 (c 0.52, CH3OH); 1H-NMR: δ = 0.86 (3H, d, J = 6.7, CH3), 0.92 (3H, d, J = 6.7, CH3), 2.07-2.16 (1H, m, CH), 4.50 (1H, d, J = 7.2, NCH), 5.03-5.11 (2H, m, PhCH2), 6.93 (1H, s, 4-Him), 7.25-7.35 (5h, m, Ar), 7.64 (1H, d, J = 1.1, 2-Him); 13C-NMR: δ = 18.94 (CH3), 20.15 (CH3), 33.92 (CH), 56.26 (NCH), 67.61 (PhCH2), 117.19 (5-Cim), 128.90 (Ar), 129.09 (Ar), 129.58 (Ar), 136.25 (2-Cim), 138.53 (Ar), 140.04 (4-Cim), 158.68 (CO); EI-MS (70 eV) m/z (%): 274 (20), 230 (20), 166 (100), 136 (10), 122 (10), 108 (30), 91 (30), 79 (30), 53 (10), 45 (70); Elemental analysis (%) calcd. for C15H19N3O2 (273.15): C 65.91, H 7.01, N 15.37; found: C 65.91, H 7.01, N 15.21.
(S)-Benzyl 1-(1H-imidazol-4-yl)-3-methylbutylcarbamate (5c). Synthesized from α-bromoketone 4c (57 % yield) as an amorphous solid; Rf = 0.63 (SiO2; CHCl3/CH3OH/NH4OH 5:1:0.1); M.p. 32-36 °C; [α]D20 = - 28.5 (c 1, CH3OH); 1H-NMR: δ = 0.93 (6H, d, J = 6.1, 2 × CH3), 1.61-1.70 (3H, m, CH + CH2), 4.78 (1H, t, J = 7.9, NCH), 5.05-5.11 (2H, m, PhCH2), 6.90 (1H, s, 4-Him), 7.24-7.33 (5H, m, Ar), 7.58 (1H, d, J = 1.2, 2-Him); 13C-NMR: δ = 22.55 (CH3), 23.38 (CH3), 26.17 (CH2), 45.42 (CH), 48.44 (CH), 67.53 (PhCH2), 116.60 (5-Cim), 128.89 (Ar), 129.07 (Ar), 129.57 (Ar), 136.43 (2-Cim), 138.55 (Ar), 141.44 (4-Cim), 158.42 (CO); EI-MS (70 eV) m/z (%): 287 (1), 230 (20), 196 (10), 186 (20), 136 (10), 122 (100), 108 (50), 91 (60), 79 (60), 67 (10), 51 (10), 39 (10); Elemental analysis (%) calcd. for C16H21N3O2 (287.16): C 66.88, H 7.37, N 14.62; found: C 66.64, H 7.34, N 14.55.
(1S,2R)-Benzyl-1-(1H-imidazol-4-yl)-2-methylbutylcarbamate (5d). Synthesized from α-bromoketone 4d (56 % yield) as an amorphous solid; Rf = 0.60 (SiO2; CHCl3/CH3OH/NH4OH 5:1:0.1); M.p. 45-49 °C; [α]D20 = - 35.6 (c 0.46, CH3OH); 1H-NMR: δ = 0.81 (3H, d, J = 6.7, CH3), 0.90 (3H, t, J = 7.3, CH3), 1.15 + 1.54 (2H, 2 × m, CH2), 1.87-1.94 (1H, m, CH), 4.57 (1H, d, J = 7.3, NCH), 5.00-5.10 (2H, m, PhCH2), 6.91 (1H, s, 4-Him), 7.25-7.32 (5H, m, Ar), 7.60 (1H, d, J = 0.8, 2-Him); 13C-NMR: δ = 11.79 (CH3), 16.41 (CH3), 26.28 (CH2), 40.33 (CH), 55.08 (CH), 67.57 (PhCH2), 117.31 (5-Cim), 128.87 (Ar), 129.06 (Ar), 129.55 (Ar), 136.35 (2-Cim), 138.50 (Ar), 139.93 (4-Cim), 158.54 (CO); EI-MS (70 eV) m/z (%): 287 (1), 230 (20), 186 (20), 136 (20), 122 (100), 108 (70), 91 (60), 79 (70), 67 (20), 51 (20), 39 (20); Elemental analysis (%) calcd. for C16H21N3O2 (287.16): C 66.88, H 7.37, N 14.62; found: C 66.70, H 7.25, N 14.62.
(S)-Benzyl 1-(1H-imidazol-4-yl)-2-phenylethylcarbamate (5e). Synthesized from α-bromoketone 4e (37 % yield) as a brown solid; Rf = 0.65 (SiO2; CHCl3/CH3OH/NH4OH 5:1:0.1); M.p. 138-140 °C; [α]D20 = -29.0 (c 1, CH3OH); 1H-NMR: δ = 2.99 (1H, dd, 2J (H,H) = 13.6, 3J (H,H) = 8.8, CH2), 3.20 (1H, dd, 2J (H,H) = 13.6, 3J (H,H) = 6.1, CH2), 3.92-5.05 (4H, m, CH + NH + CH2 - merged with residual CH3OH), 6.85 (1H, s, 4-Him), 7.11-7.32 (10H, m, 2 × Ar), 7.62 (1H, d, J = 1.1, 2-Him); 13C-NMR: δ = 42.50 (CH2), 52.21 (NCH), 67.41 (PhCH2), 116.42 (5-Cim), 127. 48 (Ar), 128.74 (Ar), 128.99 (Ar), 129.35 (Ar), 129.55 (Ar), 130.53 (Ar), 136.52 (2-Cim), 138.52 (Ar), 139.77 (4-Cim), 141.59 (Ar), 158.26 (CO); EI-MS (70 eV) m/z (%): 230 (10), 213 (20), 169 (80), 142 (30), 122 (100), 115 (30), 108 (60), 91 (60), 79 (60), 65 (20), 51 (20). Elemental analysis (%) calcd. for C19H21N3O2 (321.15): C 71.01, H 5.96, N 13.08; found: C 70. 97, H 5.96, N 13.08.
Microwave assisted condensation
A few drops of water were added to a mixture of 6a (0.2 g; 0.84 mmol) and formamidine acetate (0.17 g; 1.68 mmol), which was then treated for 2 × 2 min in a 360 W microwave oven. The resulting mass was taken up in CHCl3 (10 mL), washed successively with water (2 × 10 mL) and concentrated in vacuo. The crude product was purified in the same way as described above for the condensation in liquid ammonia. Yield 7.5 %.
Acknowledgments
This research was supported by the Ministry of Education, Youth and Sport (MSM 0021627501) and by the Czech Science Foundation (203/07/P013).
References
- Katritzky, A. R.; Pozharskii, A. F. Handbook of Heterocyclic Chemistry, 2nd Edition ed; Pergamon: New York, 2000. [Google Scholar]
- Kamaraj, K.; Kim, E.; Galliker, B.; Zakharov, L. N.; Rheingold, A. R.; Zuberbuhler, A. D.; Karlin, K. D. Copper(I) and copper(II) complexes possessing cross-linked imidazole-phenol ligands: Structures and dioxygen reactivity. J. Am. Chem. Soc. 2003, 15, 6028–6029. [Google Scholar]
- Moore, L. R.; Cooks, S. M.; Anderson, M. S.; Schanz, H.-J.; Griffin, S. T.; Rogers, R. D.; Kirk, M. C.; Shaughnessy, K. H. Synthesis and characterization of water-soluble silver and palladium imidazol-2-ylidene complexes with noncoordinating anionic substituents. Organometallics 2006, 25, 5151–5158. [Google Scholar]
- Wiglenda, T.; Gust, R. Structure-Activity Relationship study to understand the estrogen receptor-dependent gene activation of aryl- and alkyl-substituted 1H-imidazoles. J. Med. Chem. 2007, 50, 1475–1484. [Google Scholar]
- Baran, P. S.; O’Malley, D. P.; Zografos, A. L. Sceptrin as a potential biosynthetic precursor to complex pyrrole–imidazole alkaloids: The total synthesis of ageliferin. Angew. Chem. Int. Ed. 2004, 43, 2674–2677. [Google Scholar]
- O’Malley, D. P.; Li, K.; Maue, M.; Zografos, A. L.; Baran, P. S. Total synthesis of dimeric pyrrole-imidazole alkaloids: Sceptrin, ageliferin, nagelamide E, oxysceptrin, nakamuric acid, and the axinellamine carbon skeleton. J. Am. Chem. Soc. 2007, 129, 4762–4775. [Google Scholar]
- Wang, R.; Xiao, J.-C.; Twamley, B.; Shreeve, J. M. Efficient Heck reactions catalyzed by a highly recyclable palladium(II) complex of a pyridyl-functionalized imidazolium-based ionic liquid. Org. Biomol. Chem. 2007, 5, 671–678. [Google Scholar]
- Kan, H.-C.; Tseng, M.-C.; Chu, Y.-H. Bicyclic imidazolium-based ionic liquids: Synthesis and characterization. Tetrahedron 2007, 63, 1644–1653. [Google Scholar]
- Yang, C.-G.; Wang, J.; Jiang, B. Enantioselective synthesis of the aminoimidazole segment of Dragmacidin D. Tetrahedron Lett. 2002, 43, 1063–1066. [Google Scholar]
- Santagostini, L.; Gullotti, M.; Pagliarin, R.; Bianchi, E.; Casella, L.; Monzani, E. Functional mimics of copper enzymes. Synthesis and stereochemical properties of the copper(II) complexes of a trinucleating ligand derived from L-Histidine. Tetrahedron: Asymmetry 1999, 10, 281–295. [Google Scholar]
- You, J.-S.; Yu, X.-Q.; Zhang, G.-L.; Xiang, Q.-X.; Lan, J.-B.; Xie, R.-G. Novel chiral imidazole cyclophane receptors: synthesis and enantioselective recognition for amino acid derivatives. Chem. Commun. 2001, 1816–1817. [Google Scholar]
- Suwinski, J.; Szczepankiewicz, W.; Swierczek, K.; Walczak, K. Synthesis of chiral imidazole derivatives as purine precursors. Eur. J. Org. Chem. 2003, 1080–1084. [Google Scholar]
- Jiang, H.-Y.; Zhou, C.-H.; Luo, K.; Chen, H.; Lan, J.-B.; Xie, R.-G. Chiral imidazole metalloenzyme models: Synthesis and enantioselective hydrolysis for α-amino acid esters. J. Mol. Catal. A – Chem. 2006, 260, 288–294. [Google Scholar]
- Bures, F.; Kulhanek, J. Chiral imidazole derivatives synthesis from enantiopure N-protected α-amino acids. Tetrahedron: Asymmetry 2005, 16, 1347–1354. [Google Scholar]
- Bures, F.; Szotkowski, T.; Kulhanek, J.; Pytela, O.; Ludwig, M.; Holcapek, M. Novel nitrogen ligands based on imidazole derivatives and their application in asymmetric catalysis. Tetrahedron: Asymmetry 2006, 17, 900–907. [Google Scholar]
- Sellier, C.; Buschauer, A.; Elz, S.; Schunack, W. Synthesis of (Z)- and (E)-3-(1H)-imidazol-4-yl)-2-propenamine and some 3-(1H-imidazol-4-yl)propanamines. Liebigs Ann. Chem. 1992, 317–323. [Google Scholar]
- Leschke, C.; Altman, J.; Schunack, W. Bis[1H-imidazol-4(5)-yl]ethane and bis(1-tritylimidazol-4-yl)alkanes. Synthesis 1993, 197–198. [Google Scholar]
- Elz, S.; Schunack, W. An alternative synthesis of Homohistamine and structurally related (imidazol-4-yl)alkylamines. Z. Naturforsch. 1987, 42b, 238–242. [Google Scholar]
- Griffith, R. K.; DiPietro, R. A. An improved preparation of imidazole-4(5)-methanol hydrochloride. Synthesis 1983, 576. [Google Scholar]
- Siendt, H.; Tschamber, T.; Streith, J. Improved double epimerisation of (D)-glucose into (D)-gulose and the synthesis of (D)-xylo-imidazolopiperidinose. Tetrahedron Lett. 1999, 40, 5191–5192. [Google Scholar]
- Streith, J.; Boiron, A.; Frankowski, A.; Le Nouen, D.; Rudyk, H.; Tschamber, T. On the way to glycoprocessing inhibotors: A general one-pot synthesis of imidazolosugars. Synthesis 1995, 944–946. [Google Scholar]
- Tschamber, T.; Rudyk, H.; Le Nouen, D. Expeditious syntheses of imidazole C-nucleosides (=C-glycosylimidazoles) from carbohydrates and formamidine acetate. Helv. Chim. Acta 1999, 82, 2015–2019. [Google Scholar]
- Ramtohul, Y. K.; James, M. N. G.; Vederas, J. C. Synthesis and evaluation of keto-glutamine analogues as inhibitors of Hepatitis A virus 3C proteinase. J. Org. Chem. 2002, 67, 3169–3178. [Google Scholar]
- Sample Availability: Samples of the compounds 5a-e are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Marek, A.; Kulhanek, J.; Ludwig, M.; Bures, F. Facile Synthesis of Optically Active Imidazole Derivatives. Molecules 2007, 12, 1183-1190. https://doi.org/10.3390/12051183
AMA Style
Marek A, Kulhanek J, Ludwig M, Bures F. Facile Synthesis of Optically Active Imidazole Derivatives. Molecules. 2007; 12(5):1183-1190. https://doi.org/10.3390/12051183
Chicago/Turabian StyleMarek, Ales, Jiri Kulhanek, Miroslav Ludwig, and Filip Bures. 2007. "Facile Synthesis of Optically Active Imidazole Derivatives" Molecules 12, no. 5: 1183-1190. https://doi.org/10.3390/12051183