Experimental
General
All anhydrous reactions were carried out under argon atmospheres. Solvents were dried by distillation prior to use. Solvent mixtures employed in chromatography were reported as volume to volume ratios. Starting materials were purchased from Aldrich (analytical reagent grades) and used without further purification. Analytical thin-layer chromatography (TLC) was conducted on Merck glass plates coated with silica gel 60 F254 and spots were visualized with UV light or/and an alcohol solution of anisaldehyde. Flash column chromatography was performed using Merck silica gel 60 (230-400 mesh ASTM).
Melting points were determined on a Büchi melting point apparatus and are uncorrected. 1H and 2D NMR spectra were recorded at 400 MHZ on a Bruker DRX-400 spectrometer in the indicated solvents. The coupling constants are recorded in Hertz (Hz) and the chemical shifts are reported in parts per million (δ, ppm) downfield from tetramethylsilane (TMS), which was used as an internal standard (by asterisk are indicated the overlapped peaks). For the 1H -15N GHMQC spectrum, data were acquired as 3072 x 400 data points with a total of 290 transients accumulated/t1 increment. Pulse widths were 8.55 μs for 1H and 27.7 μs for the 15N at powers of 0 and -3 dB. The F1 spectral window employed was set from 100 to 400 ppm. Pulsed field gradients, gt1-gt3, had durations of 0.8 ms. Gradient pairs were optimized as 70:30:50 for 15N.
Infrared spectra were obtained on a Nicolet Magna 750, series II spectrometer. HPLC separations were performed using a Hewlett Packard 1100 series instrument with a variable wavelength UV detector and coupled to HP Chem.-Station utilizing the manufacturer’s 5.01 software package. A Kromasil column with pore size 100 Å, internal diameter of 5μm, and a C-18 bond phase was used.
General Procedure for the Preparation of Alkylphenones
To an ice–cold solution of AlCl3 (46.8 mmol) in 1,2-dichloroethane (10 mL) an acid chloride (39.0 mmol) was added dropwise over 10 min. The resulting solution was allowed to reach the room temperature and dissolve the AlCl3. Then, the reaction mixture was cooled to 0 oC and a solution of anisole (46.8 mmol) in 1,2-dichloroethane (20 mL) was added dropwise over a period of 30 min. Upon completion, the reaction was allowed to reach the room temperature and stirred for 8-15 h. The mixture was quenched by pouring over ice (100 g) and extracted with EtOAc (3 × 25 mL). The combined organic layers were successively washed with water, NaHCO3 (satd), brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography using a 5% EtOAc/hexanes system as eluent.
1-(4-Methoxy-phenyl)-propan-1-one (1a)
Prepared according to the general procedure as outlined above affording, after flash chromatographic purification (5% EtOAc/hexanes), the title compound was obtained as a pale yellow oil (5.61 g, 73%). 1H-NMR (CDCl3) δ, 7.91 (dd, J = 2.0, 7.0 Hz, 2H, ArH), 6.89 (dd, J = 2.0, 7.0 Hz, 2H, ArH), 3.83 (s, 3H, OCH3), 2.91 (q, J = 7.4 Hz, 2H, CH2C=O), 1.18 (t, J = 7.4 Hz, 3H, CH3).
1-(4-Methoxyphenyl)-butan-one (1b)
Anisole (46.8 mmol) was reacted with butyl chloride (39.0 mmol) according to the general procedure. Flash column chromatography (5% EtOAc/hexanes) provided the title compound as a pale yellow oil (5.41 g, 65%). 1H-NMR (CDCl3) δ, 8.37 (dd, J = 2.0, 7.0 Hz, 2H, ArH), 7.34 (dd, J = 2.0, 7.0 Hz, 2H, ArH), 4.28 (s, 3H, OCH3), 3.31 (t, J = 7.3 Hz, 2H, CH2C=O), 2.18 (m, 3H, CH2), 1.41 (t, J = 7.3 Hz, 3H, CH3).
1-(4-Methoxyphenyl)-pentan-1-one (1c)
Prepared from anisole (46.8 mmol) and valeryl chloride (39.0 mmol) following the general procedure, which afforded the title compound as a yellow oil (6.56 g, 73 %). 1H-NMR (CDCl3) δ, 7.91 (dd, J = 2.0, 7.0 Hz, 2H, ArH), 6.90 (dd, J1 = 2.0, 7.0 Hz, 2H, ArH), 3.84 (s, 3H, OCH3), 2.89 (t, J = 6.2 Hz, 2H, CH2C=O), 1.68 (m, 2H, COCH2CH2), 1.37 (m, 2H, COCH2CH2CH2), 0.92 (t, J = 7.4 Hz, 3H, CH3).
General Procedure for Nitration.
To an ice–cold solution of 1a-c (6.7 mmol) in H2SO4 (12.4 mL) was added KNO3 (7.4 mmol) over a period of 30 min. The reaction was run at room temperature for 8-15 h, quenched with H2O (15-25 mL), extracted repeatedly with EtOAc (3 x 25 mL) and the combined organic layers were washed with a satd NaHCO3 solution (25 mL). The organic layer was separated, dried over anhydrous MgSO4 and concentrated under reduced pressure to afford the crude product as a white solid, which was purified by flash chromatography.
1-(4-Methoxy-3-nitro-phenyl)-propan-1-one (2a).
1a (1.1 g, 6.7 mmol) was reacted with KNO3 (0.74 g, 7.4 mmol) according to the general nitration procedure, to give a dark yellow slurry. The latter was purified by flash chromatography (20% EtOAc/hexanes) yielding a yellow solid (0.95 g, 68%), which was recrystallized from Et2O to give pale yellow fine needles. M.p. 88-89 oC; 1H-NMR (CDCl3) δ, 8.41 (d, J = 2.0 Hz, 1H, H-2’), 8.15 (dd, J = 2.0, 9.0 Hz, 1H, H-6’), 7.13 (d, J = 9.0 Hz, 1H, H-5’), 4.01 (s, 3H, OCH3), 2.97 (q, J = 7.4 Hz, 2H, CH2CH3), 1.20 (t, J = 7.4 Hz, 3H, CH2CH3); Elem. Anal.: C 57.41, H 5.30, N 6.70; C 57.59, H 5.42, N 6.52.
1-(4-Methoxy-3-nitro-phenyl)-butan-1-one (2b)
Prepared according to general procedure for nitration from 1b (1.19 g, 6.7 mmol) and purified by flash chromatography (20% EtOAc/hexanes) to afford a white solid (0.91 g, 61%). M.p. 69-70 oC; 1H- NMR (CDCl3) δ, 8.46 (d, J = 2.2 Hz, 1H, H-2’), 8.19 (dd, J = 2.2, 8.8 Hz, H-6’), 7.18 (d, J = 8.8 Hz, H-5’), 4.06 (s, 3H, OCH3), 2.94 (t, J = 7.3 Hz, 2H, CH2C=O), 2.63 (m, 2H, CH2), 1.03 (t, J = 7.4 Hz, 3H, CH3); Elem. Anal.: C 59.19, H 5.87, N 6.27; C 59.39, H 5.72, N 6.41.
1-(4-Methoxy-3-nitro-phenyl)-pentan-1-one (2c)
Prepared according to general procedure for nitration as described above from 1c and purified by flash chromatography (20% EtOAc/hexanes) to afford a white crystalline solid (1.0 g, 63%). M.p. 81-82 oC; 1H-NMR (CDCl3) δ, 8.40 (d, J = 2.5 Hz, 1H, H-2’), 8.14 (dd, J = 2.5, 9.4 Hz, 1H, H-6’), 7.12 (d, J = 9.4 Hz, 1H, H-5’), 4.01 (s, 3H, OCH3), 2.91 (t, J = 7.4 Hz, 2H, CH2CH2CH2CH3), 1.68 (m, 2H, CH2CH2CH2CH3), 1.37 (m, 2H, CH2CH2CH2CH3), 0.92 (t, J = 7.4 Hz, 3H, CH2CH2CH2CH3); Elem. Anal.: C 60.75, H 6.37, N 5.90; C 60.61, H 6.45, N 5.82.
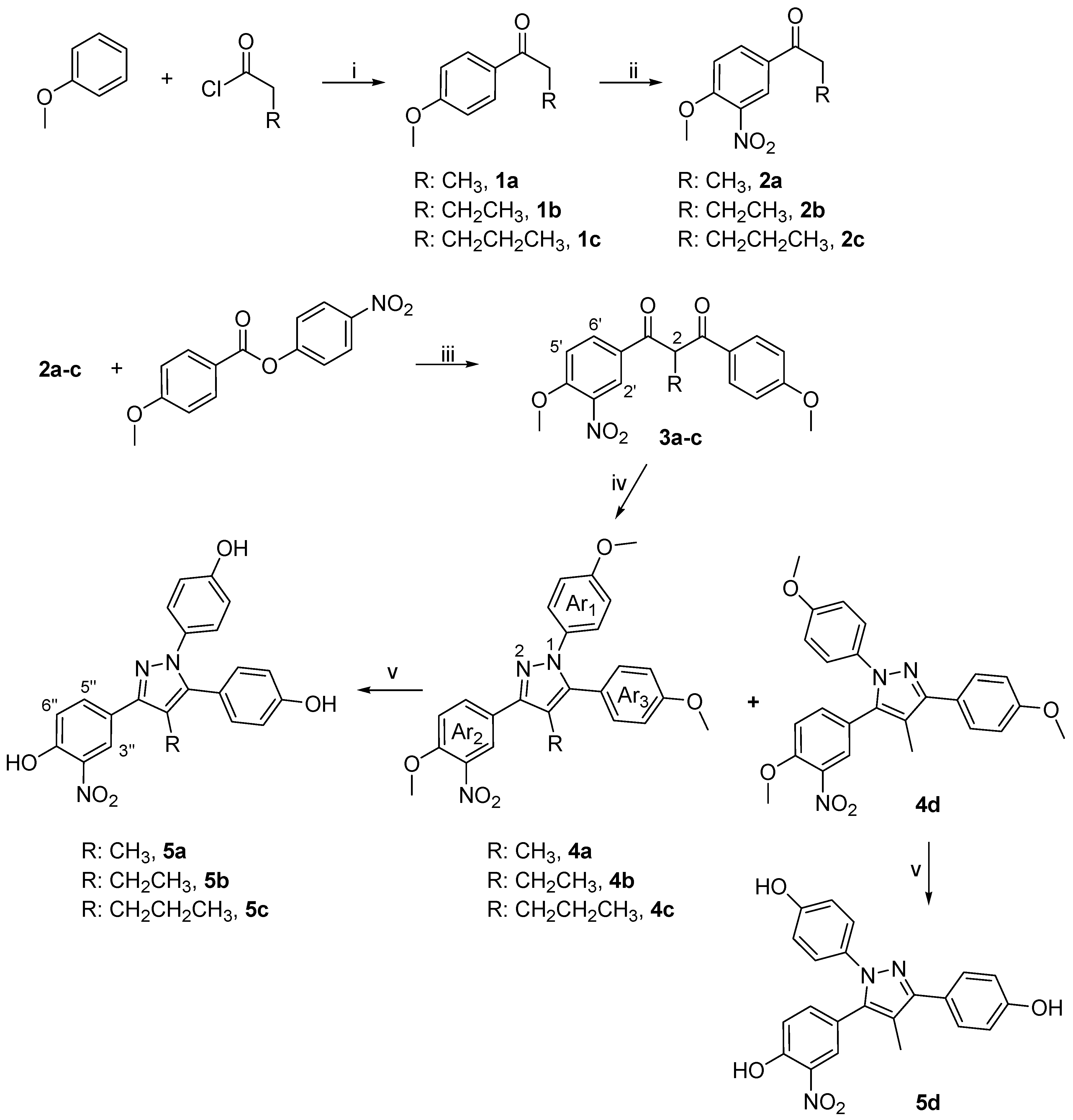
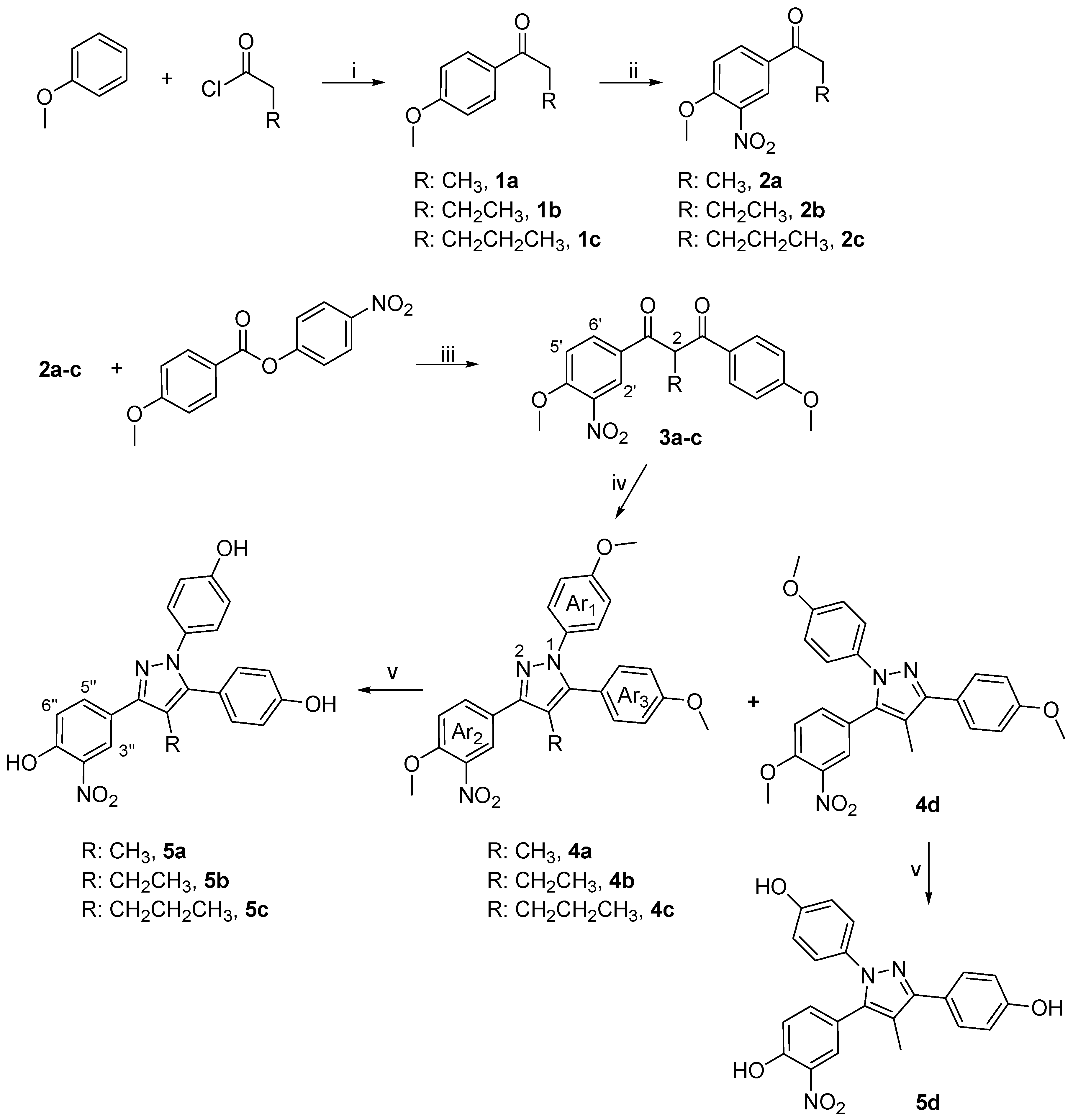
General Procedure for the Preparation of Diketones 3a-c
To an ice–cold solution of 2a-c (0.97 mmol) and 4-nitrophenyl-4-methoxy benzoate (0.97 mmol) [prepared from p-nitrophenol and 4-methoxybenzoic acid using diisopropylcarbodiimide and 4-dimethylaminopyridine] in THF (35 mL), was added a 1.0 M solution of lithium hexamethyldisilamide (3.03 mmol) dropwise over a period of 5 min. The reaction was allowed to reach the room temperature and stirred for additional 1.5 h and quenched with H2O (25 mL). Then, the mixture was repeatedly extracted with Et2O and the combined organic layers were washed with H2O, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford a crude solid. The unreacted ester was removed by the addition of a solution of 40% EtOAc/ hexanes and the filtration of the insoluble ester. The filtrate was concentrated in vacuo and purified by flash chromatography using various proportions of EtOAc/hexanes as eluent system.
1-(4-Methoxy-3-nitrophenyl)-3-(4-methoxyphenyl)-2-methyl-propane-1,3-dione (3a)
Prepared according to general procedure for preparation of diketones outlined above from 2a (0.6 g, 2.9 mmol) and purified by flash chromatography (20% EtOAc/hexanes) to yield the title product as an orange oil (0.74 g, 74%). 1H-NMR (CDCl3) δ, 8.39 (d, J = 2.2 Hz, 1H, H-2’), 8.09 (dd, J = 2.2, 8.4 Hz, 1H, H-6’), 7.08 (d, J = 8.4 Hz, 1H, H-5’), 7.92 (d , J = 8.9 Hz, 2H, ArH), 6.93 (d, J= 8.9 Hz, 2H, ArH), 5.17 (m, 1H, H-2), 3.97 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 1.52 (d, J = 7.0 Hz, 3H, CH3); Elem. Anal.: C 62.97, H 4.99, N 4.08; C 62.71, H 4.81, N 4.19.
2-Ethyl-1-(4-methoxy-3-nitrophenyl)-3-(4-methoxyphenyl)-propane-1,3-dione (3b)
Ketone 2b (0.4 g, 1.8 mmol) reacted with 4-nitrophenyl 4-methoxybenzoate (1.8 mmol) as above to afford after flash chromatographic purification (40% EtOAc/hexanes) compound 3b as a dark yellow oil (0.26g, 68%), while 0.15 g of the ketone 2b remained unreacted. 1H-NMR (CDCl3) δ, 8.46 (d, J = 2.2 Hz, 1H, H-2’), 8.19 (dd, J = 2.2, 8.9 Hz, 1H, H-6’), 7.99 (d, J = 8.9 Hz, 1H, H-5’), 7.74 (d , J = 8.9 Hz, 2H, Ar2H), 6.97 (d, J= 8.9 Hz, 2H, Ar2H), 4.97 (t, J = 6.7 Hz, 1H, H-2), 4.02 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 2.18 (m, 2H, CH2), 1.05 (t, J = 7.4 Hz, 3H, CH3); Elem. Anal.: C 63.86, H 5.36, N 3.92; C 63.61, H 5.45, N 3.82.
1-(4-Methoxy-3-nitrophenyl)-3-(4-methoxyphenyl)-2-propylpropane-1,3-dione (3c)
Prepared according to general procedure for preparation of 1,3-diketones outlined above from 2c (0.71 g, 3.0 mmol) and purified by flash chromatography (50% EtOAc/hexanes) to provide a red oil (0.44 g, 67%) and 0.29 g of the unreacted ketone 2c. 1H NMR (CDCl3) δ, 8.47 (d, J = 2.2 Hz, 1H, H-2’), 8.20 (dd, J = 2.2, 8.8 Hz, 1H, H-6’), 7.97 (d, J = 8.8 Hz, 2H, Ar2H), 7.14 (d , J = 8.8 Hz, 1H, H-5’), 6.98 (d, J= 8.8 Hz, 2H, Ar2H), 5.05 (t, J = 6.6 Hz, 1H, H-2), 4.03 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 2.12 (m, 2H, CH2), 1.46 (m, 2H, CH2), 0.99 (t, J = 7.3 Hz, 3H, CH3). Elem. Anal.: C 64.68, H 5.70, N 3.77; C 64.80, H 5.65, N 3.82.
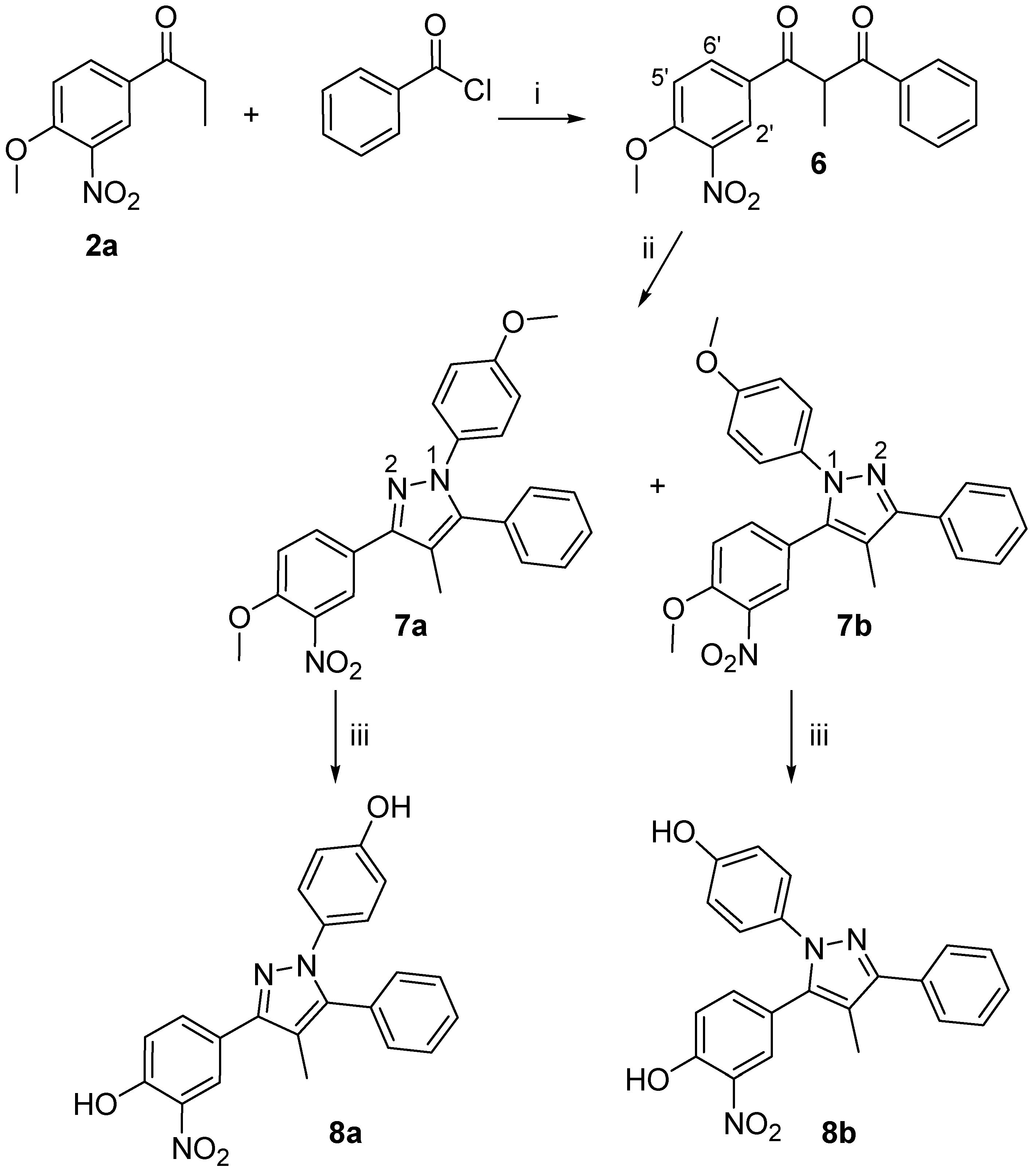
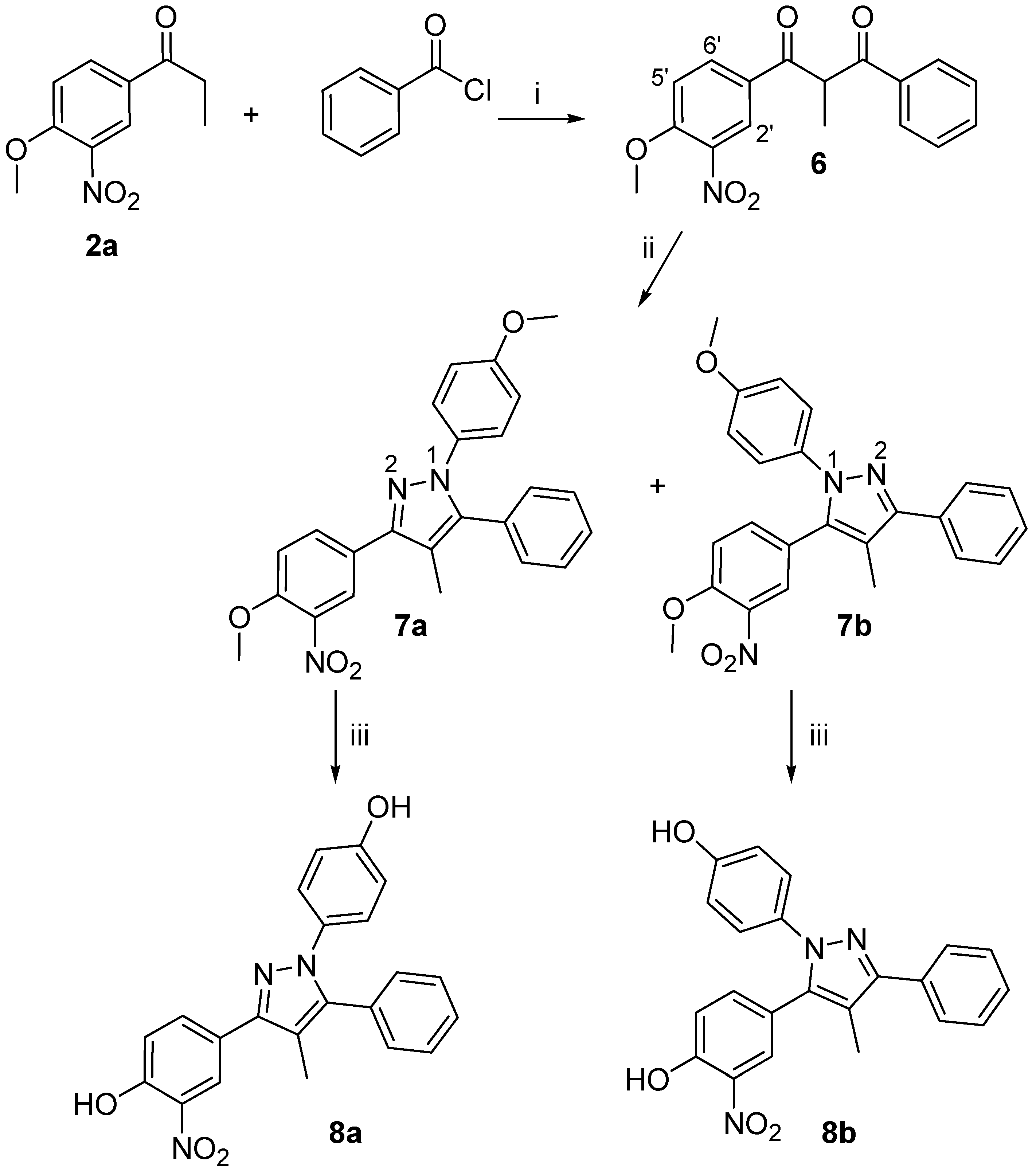
1-(4-Methoxy-3-nitrophenyl)-2-methyl-3-phenylpropane-1,3-dione (6)
To a solution of 2a (1.21 g, 5.8 mmol) in THF (15 mL), a solution of LHMDS (5.8 mmol, 1M in THF) was added dropwise over 15 min. Upon complete addition of LHMDS, the reaction was stirred for additional 15 min. Then, benzoyl chloride (5.8 mmol, in 3 mL THF) was added dropwise and the reaction was run under stirring for 12 h. The reaction mixture was repeatedly extracted with Et2O and the combined organic layers were washed with H2O, dried over MgSO4 and concentrated under reduced pressure to afford a crude solid, which was purified by flash chromatography eluting using 5% EtOAc/hexanes as solvent eluting system (1.34 g, 74%). 1H-NMR (CDCl3) δ, 8.41 (d, J = 2.0 Hz, 1H, H-2’), 8.25 (d, J = 9.4 Hz, 1H, H-5’), 8.11 (dd, J = 2.0, 9.4 Hz, 1H, H-6’), 7.93 (t , J = 9.0 Hz, 2H, ArH), 7.46 (m, J = 7.8 Hz, 1H, ArH), 7.13 (d, J = 9.0 Hz, 2H, ArH), 5.19 (q, J = 7.0 Hz, 1H, CHCH3), 3.99 (s, 3H, OCH3), 1.24 (d, J = 7.0 Hz, 3H, CH3); Elem. Anal.: C 68.17, H 4.83, N 4.47; C 67.35, H 4.71, N 4.35.
General Procedure for Pyrazole Synthesis
To a solution of diketone (1.0 mmol) in DMF (30 mL) and THF (10 mL), 4-methoxy-phenylhydrazine hydrochloride (4-5 equiv) was added. The mixture was brought to reflux and the reaction progress was monitored by TLC analysis (10-20 h). Then the reaction mixture was allowed to cool to room temperature and diluted with H2O (30 mL). The product was repeatedly extracted with EtOAc (3 x 25 mL) and the combined organic layers and sequentially washed with a satd LiCl solution (25 mL), satd NaHSO3 (25 mL), and brine (25 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure to afford a crude oil, which was purified by flash chromatography using EtOAc/hexanes as eluting solvent system.
3-(4-Methoxy-3-nitrophenyl)-1,5-bis-(4-methoxyphenyl)-4-methyl-1H-pyrazole (4a) and 5-(4-methoxy-3-nitrophenyl)-1,3-bis-(4-methoxyphenyl)-4-methyl-1H-pyrazole (4d)
Diketone 3a was reacted with 4-methoxyphenylhydrazine hydrochlorides as described above. Flash chromatographic purification (20% EtOAc/hexanes) provided an inseparable mixture of the title compounds (68%). The latter were separated by semipreparative HPLC separation using H2O/CH3CN (15:85) as eluent in a flow rate of 1.6 ml/min, furnishing the two regioisomers in a 4:1 ratio for 4a and 4d respectively (UV detector at 254 nm, tR4a= 12.2 min, tR4d= 15.4 min). 4a: 1H-NMR (CDCl3) δ, 8.32 (d, J = 2.2 Hz, 1H, H-2’), 8.03 (dd, J = 2.2, 8.7 Hz, 1H, H-6’), 7.71 (d, J = 8.7 Hz, 1H, H-5’), 7.26 (d, J = 8.5 Hz, 2H, Ar3H), 7.14 (d, J = 8.9 Hz, 2H, Ar1H), 6.92 (d, J = 8.5 Hz, 2H, Ar3H), 6.82 (d, J = 8.9 Hz, 2H, Ar1H), 4.02 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.24 (s, 3H, CH3); 1H-15N NMR (CDCl3) δ, N-1 205.6, N-2 301.6; Elem. Anal.: C 67.41, H 5.20, N 9.43; C 67.25, H 5.41, N 9.32; 4d: 1H-NMR (CDCl3) δ, 8.34 (d, J = 2.1 Hz, 1H, H-2’), 8.03 (dd, J = 2.1, 8.8 Hz, 1H, H-6’), 7.60 (d, J = 8.8 Hz, 1H, H-5’), 7.22 (d, J = 8.5 Hz, 2H, Ar3H), 7.08 (d, J = 8.7 Hz, 2H, Ar1H), 6.92 (d, J = 8.5 Hz, 2H, Ar3H), 6.80 (d, J = 8.7 Hz, 2H, Ar1H), 4.04 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.16 (s, 3H, CH3); 1H-15N NMR (CDCl3) δ, N-1 197.4, N-2 294.7; Elem. Anal.: C 67.41, H 5.20, N 9.43; C 67.54, H 5.32, N 9.30.
4-Ethyl-3-(4-methoxy-3-nitrophenyl)-1,5-bis-(4-methoxyphenyl)-1H-pyrazole (4b)
Diketone 3b (0.23 g, 0.6 mmol) was reacted with 4-methoxyphenylhydrazine hydrochloride (0.53 g, 3.0 mmol) according to the general procedure. Upon purification by flash chromatography (40% EtOAc/hexanes) the title compound was obtained as a tan solid (0.21 g, 75%). M.p. 121-122 oC; 1H- NMR (CDCl3) δ, 8.32 (d, J = 2.2 Hz, 1H, H-2’), 8.02 (dd, J = 2.2, 8.7 Hz, 1H, H-6’), 7.68 (d, J = 8.7 Hz, 1H, H-5’), 7.21 (dd, J = 2.0, 8.8 Hz, 2H, Ar3H), 7.17 (dd, J = 2.0, 8.7 Hz, 2H, Ar1H), 6.92 (dd, J = 2.0, 8.8 Hz, 2H, Ar3H), 6.82 (dd, J = 2.0, 8.7 Hz, 2H, Ar1H), 4.03 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.67 (q, J = 7.5 Hz, 2H, CH2), 1.09 (t, J = 7.5 Hz, 3H, CH3); 1H-15N-NMR (CDCl3) δ, N-1 201.8, N-2 299.2; Elem. Anal. : C 67.96, H 5.48, N 9.14; C 68.11, H 5.53, N 9.22.
3-(4-Methoxy-3-nitrophenyl)-1,5-bis-(4-methoxyphenyl)-4-propyl-1H-pyrazole (4c)
Diketone 3c (0.2 g, 0.5mmol) was reacted with 4-methoxyphenylhydrazine hydrochloride (0.48 g, 2.5 mmol) for 20 h, according to the general procedure. The resulting dark brown residue was purified by flash column chromatography (40% EtOAc/hexanes) to provide the title product 4c as a pale brown solid (0.1 g, 85%) and 0.1 g of the substrate 3c. M.p. 131-133 oC; 1H-NMR (CDCl3) δ, 8.31 (d, J = 2.0 Hz, 1H, H-2’), 8.01 (dd, J = 2.0, 8.1 Hz, 1H, H-6’), 7.68 (d, J = 8.1 Hz, 1H, H-5’), 7.21 (d, J = 7.1 Hz, 2H, Ar3H), 7.16 (d, J = 8.5 Hz, 2H, Ar1H), 6.93 (d, J = 7.1 Hz, 2H, Ar3H), 6.83 (d, J = 8.5 Hz, 2H, Ar1H), 4.04 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.60 (t, J = 7.7 Hz, 2H, CH2), 1.45 (m, 2H, CH2), 0.84 (t, J = 7.2 Hz, 3H, CH3); 1H-15N-NMR (CDCl3) δ, N-1 202.9, N-2 300.1; Elem. Anal.: C 68.48, H 5.75, N 8.87; C 68.61, H 5.59, N 8.75.
3-(4-Methoxy-3-nitro-phenyl)-1-(4-methoxy-phenyl)-4-methyl-5-phenyl-1H-pyrazole (7a) and 5-(4-Methoxy-3-nitro-phenyl)-1-(4-methoxy-phenyl)-4-methyl-3-phenyl-1H-pyrazole (7b)
A stirred solution of compound 6 (0.33 g, 1.05 mmol) in DMF (30mL) and THF (10mL), was reacted with 4-methoxyphenylhydrazine hydrochloride (0.92 g, 5.25 mmol) according to the general procedure for pyrazole synthesis. Upon purification by flash chromatography (10% EtOAc/hexanes), an inseparable mixture of the title compounds was obtained as a dark brown solid (0.16g, 69%), while 0.15 g of the starting compound 6 was recovered. Semipreparative HPLC separation using H2O/CH3CN (20:80) as eluent and flow rate of 1.6 ml/min, provided equal amounts of the two regioisomers (UV detector at 254 nm, tR7a= 11.2 min, tR7b= 13.7 min). 7a: M.p. 127-128 oC; 1H-NMR (CDCl3) δ, 8.42 (d, J = 2.2 Hz, 1H, H-2’), 8.11 (dd, J = 2.2, 8.7 Hz, H-6’), 7.45 (d, J = 8.7 Hz, 1H, H-5’), 7.28-7.07 (m, 4H, ArH), 7.01-6.97 (m, 5H, ArH), 4.35 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.28 (s, 3H, CH3). 1H-15N-NMR (CDCl3) δ, N-1 203.7, N-2 299.6; Elem. Anal.: C 69.39, H 5.10, N 10.11; C 69.51, H 5.02, N 10.30; 7b: M.p. 141-143 oC; 1H-NMR (CDCl3) δ, 8.30 (d, J = 2.2 Hz, 1H, H-2’), 8.09 (dd, J = 2.2, 8.7 Hz, 1Η, H-6’), 7.40 (d, J = 8.7 Hz, 1H, H-5’), 7.37-7.14 (m, 4H, ArH), 7.08-6.89 (m, 5H, ArH), 4.04 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.25 (s, 3H, CH3); 1H-15N-NMR (CDCl3) δ, N-1 199.8, N-2 295.7; Elem. Anal.: C 69.39, H 5.10, N 10.11; C 69.67, H 4.93, N 9.94.
General Demethylation Procedure
To a stirred solution of methyl-protected pyrazole (1 equiv) in CH2Cl2 at –78 oC was added dropwise a 1M BBr3 solution in CH2Cl2 (3-5 equiv). Upon completion of BBr3 addition, the reaction was stirred at –78 oC for 1 h and then allowed to reach room temperature. Stirring was continued for additional 16 h, then the mixture was cooled to 0 oC, and carefully quenched with H2O (15-25 mL) and extracted repeatedly with EtOAc. The combined organic layers were dried over Na2SO4 and evaporated under reduced pressure. Flash chromatographic purification using a 5% MeOH/CH2Cl2 mixture as the eluting solvent system provided the desired demethylation products.
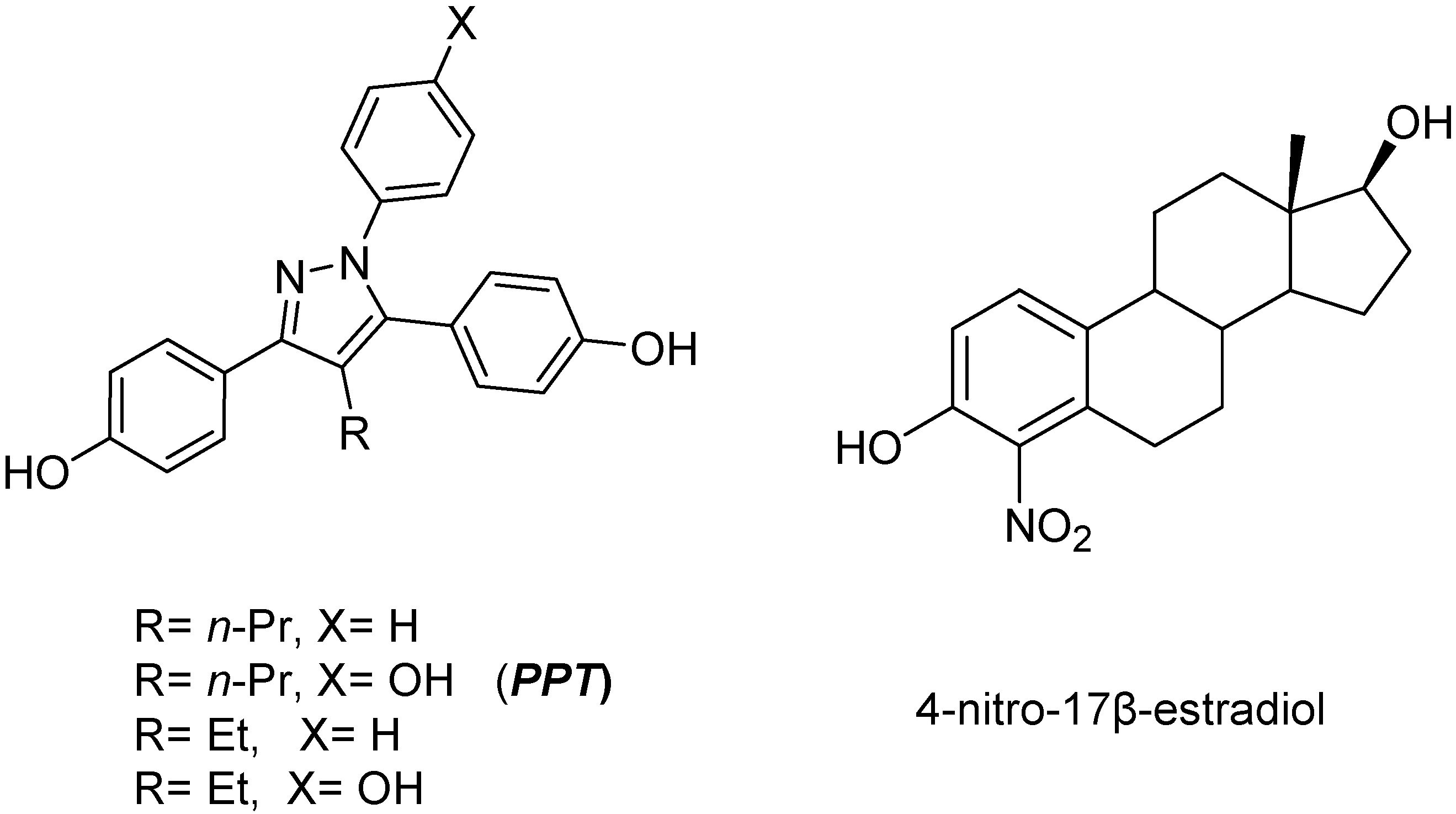
4-[1,5-Bis-(4-hydroxyphenyl)-4-methyl-1H-pyrazol-3-yl]-2-nitrophenol (5a)
A stirred solution of 4a in CH2Cl2 was deprotected using BBr3 according to the general demethylation procedure. Purification by flash chromatography afforded the title compound as a tan solid (87%). M.p. 155-156 oC; 1H-NMR (acetone-D6) δ, 8.87 (s, 3H, OH), 8.52 (d, J = 2.2 Hz, 1H, H-3’’), 8.19 (dd, J = 2.2, 8.7 Hz, 1H, H-5’’), 7.32 (d, J = 8.7 Hz, 1H, H-6’’), 7.16 (d, J = 8.6 Hz, 2H, Ar3H), 7.11 (d, J = 8.6 Hz, 2H, Ar1H), 6.91 (d, J = 8.6 Hz, 2H, Ar3H), 6.81 (d, J = 8.6 Hz, 2H, Ar1H), 2.26 (s, 3H, CH3). ΙR (ZnSe) v = 3303 cm-1, (OH); Elem. Anal.: C 65.50, H 4.25, N 10.42; C 65.31, H 4.11, N 10.58.
4-[2,5-Bis-(4-hydroxyphenyl)-4-methyl-1H-pyrazol-3-yl]-3-nitrophenol (5d)
A stirred solution of 4d in CH2Cl2 was deprotected using BBr3 according to the general demethylation procedure. Purification by flash chromatography afforded the title compound as a pale yellow solid (88%). M.p. 161-162 oC;1H-NMR (acetone-D6) δ, 8.86 (s, 3H, OH), 8.05 (d, J = 1.9 Hz, 1H, H-3’’), 7.66 (dd, J = 1.9, 8.9 Hz, 1H, H-5’’), 7.23 (d, J = 8.9 Hz, 1H, H-6’’), 7.19 (d, J = 8.8 Hz, 2H, Ar3H), 7.02 (d, J = 8.8 Hz, 2H, Ar1H), 6.95 (d, J = 8.8 Hz, 2H, Ar3H), 6.83 (d, J = 8.8 Hz, 2H, Ar1H), 2.25 (s, 3H, CH3). ΙR (ZnSe) v = 3313 cm-1 (OH); Elem. Anal.: C 65.50, H 4.25, N 10.42; C 65.20, H 4.14, N 10.53.
4-[4-Ethyl-1,5-bis-(4-hydroxyphenyl)-1H-pyrazol-3-yl]-2-nitrophenol (5b)
A stirred solution of 4b in CH2Cl2 was deprotected using BBr3 according to the general demethylation procedure. Purification by flash chromatography afforded the title compound as a tan solid (74%). 1H-NMR (acetone–D6) δ, 10.52 (s, 1H, OH), 8.73 (s, 1H, OH), 8.64 (s, 1H, OH), 8.53 (d, J = 2.2 Hz, 1H, H-3’’), 8.19 (dd, J = 2.2, 8.7 Hz, 1H, H-5’’), 7.33 (d, J = 8.7 Hz, 1H, H-6’’), 7.16 (d, J = 8.6 Hz, 2H, Ar3H), 7.13 (d, J = 8.6 Hz, 2H, Ar1H), 6.92 (d, J = 8.6 Hz, 2H, Ar3H), 6.80 (d, J = 8.6 Hz, 2H, Ar1H), 2.69 (q, J = 7.5, 12.5 Hz, 2H, CH2), 1.11 (t, J = 7.5 Hz, 3H, CH3). ΙR (ZnSe) v = 3283 cm-1, (OH); Elem. Anal. : C 66.18, H 4.59, N 10.07; C 66.33, H 4.74, N 9.88.
4-[1,5-Bis-(4-hydroxyphenyl)-4-propyl-1H-pyrazol-3-yl]-2-nitrophenol (5c)
A stirred solution of 4c in CH2Cl2 was deprotected using BBr3 according to the general demethylation procedure. Purification by flash chromatography afforded the title compound as a tan solid (60%). 1H-NMR (acetone–D6) δ, 10.52 (s, 1H, OH), 8.78 (s, 1H, OH), 8.68 (s, 1H, OH), 8.50 (d, J = 2.2 Hz, 1H, H-3’’), 8.20 (dd, J = 2.1, 8.7 Hz, 1H, H-5’’), 7.38 (d, J = 8.7 Hz, 1H, H-6’’), 7.16 (d, J = 8.3 Hz, 2H, Ar3H), 7.12 (d, J = 7.7 Hz, 2H, Ar1H), 6.89 (d, J = 8.3 Hz, 2H, Ar3H), 6.81 (d, J = 7.7 Hz, 2H, Ar1H), 2.65 (t, J = 7.9 Hz, 2H, CH2), 1.20 (m, 2H, CH2), 0.85 (t, J = 7.9 Hz, 2H, CH3); IR (ZnSe) v = 3283 cm-1 (OH); Elem. Anal. : C 66.81, H 4.91, N 9.74; C 67.02, H 5.10, N 9.88.
4-[1-(4-Hydroxyphenyl)-4-methyl-5-phenyl-1H-pyrazol-3-yl]-2-nitrophenol (8a)
A stirred solution of 7a in CH2Cl2 was deprotected using BBr3 according to the general desmethylation procedure. Purification by flash chromatography afforded the title compound as a tan solid (70%). 1H-NMR (acetone–D6) δ, 9.30 (s, 1H, OH), 8.70 (s, 1H, OH), 8.40 (d, J = 2.2 Hz, 1H, H-3”), 8.07 (dd, J = 2.2, 8.7 Hz, H-5”), 7.35 (d, J = 8.7 Hz, 1H, H-6”), 7.19-7.11 (m, 5H, ArH), 7.08-6.98 (m, 4H, ArH), 4.35 (s, 3H, CH3O), 3.80 (s, 3H, CH3O), 2.28 (s, 3H, CH3); IR (ZnSe) v = 3193 cm-1 (OH); Elem. Anal.: C 68.21, H 4.42, N 10.85; C 68.44, H 4.29, N 10.71.
4-[2-(4-Hydroxyphenyl)-4-methyl-5-phenyl-2H-pyrazol-3-yl]-2-nitrophenol (8b)
A stirred solution of 7b in CH2Cl2 was deprotected using BBr3 according to the general desmethylation procedure. Purification by flash chromatography afforded the title compound as a tan solid (78%). 1H-NMR (acetone–D6) δ, 9.28 (s, 1H, OH), 8.69 (s, 1H, OH), 8.25 (d, J = 2.2 Hz, 1H, H-3”), 8.02 (dd, J = 2.2, 8.7 Hz, 1Η, H-5”), 7.47-7.24 (m, 4H, ArH), 7.26 (d, J = 8.7 Hz, 1H, H-6”), 6.98-6.78 (m, 5H, ArH), 4.04 (s, 3H, CH3O), 3.80 (s, 3H, CH3O), 2.25 (s, 3H, CH3); IR (ZnSe) v = 3199 cm-1 (OH); Elem. Anal.: C 68.21, H 4.42, N 10.85; C 68.39, H 4.49, N 10.71.
Binding to isolated human ERα and ERβ
The relative binding affinity (RBA) values were assessed as previously described [
22]. Briefly, the concentrations of
5a-d, 8a,b that inhibited ES2 (a fluorescein-labelled estrogen from Invitrogen) binding to isolated human ERα or ERβ (Invitrogen) by 50% (IC
50), as assessed using a Beacon 2000 Fluorescence Polarization Reader (Invitrogen), were used to derive the RBA values of
Table 1, as described in the legend to the Table.


{kind=link}
{kind=link}
{kind=link}
