High-Yielding Synthesis of Methyl Orthoformate-Protected Hydroxytyrosol and Its Use in Preparation of Hydroxytyrosyl Acetate

{kind=link}
{kind=link}
{kind=link}
Abstract
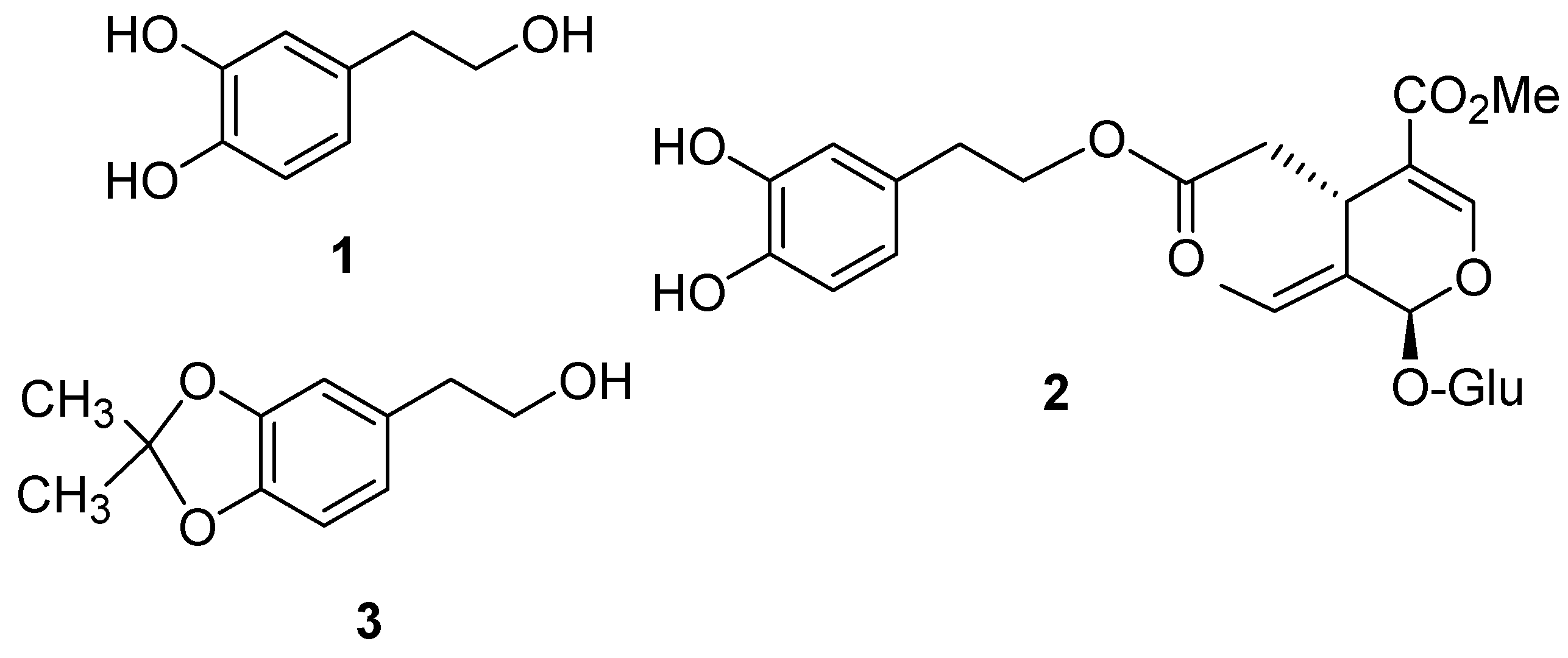
:Introduction

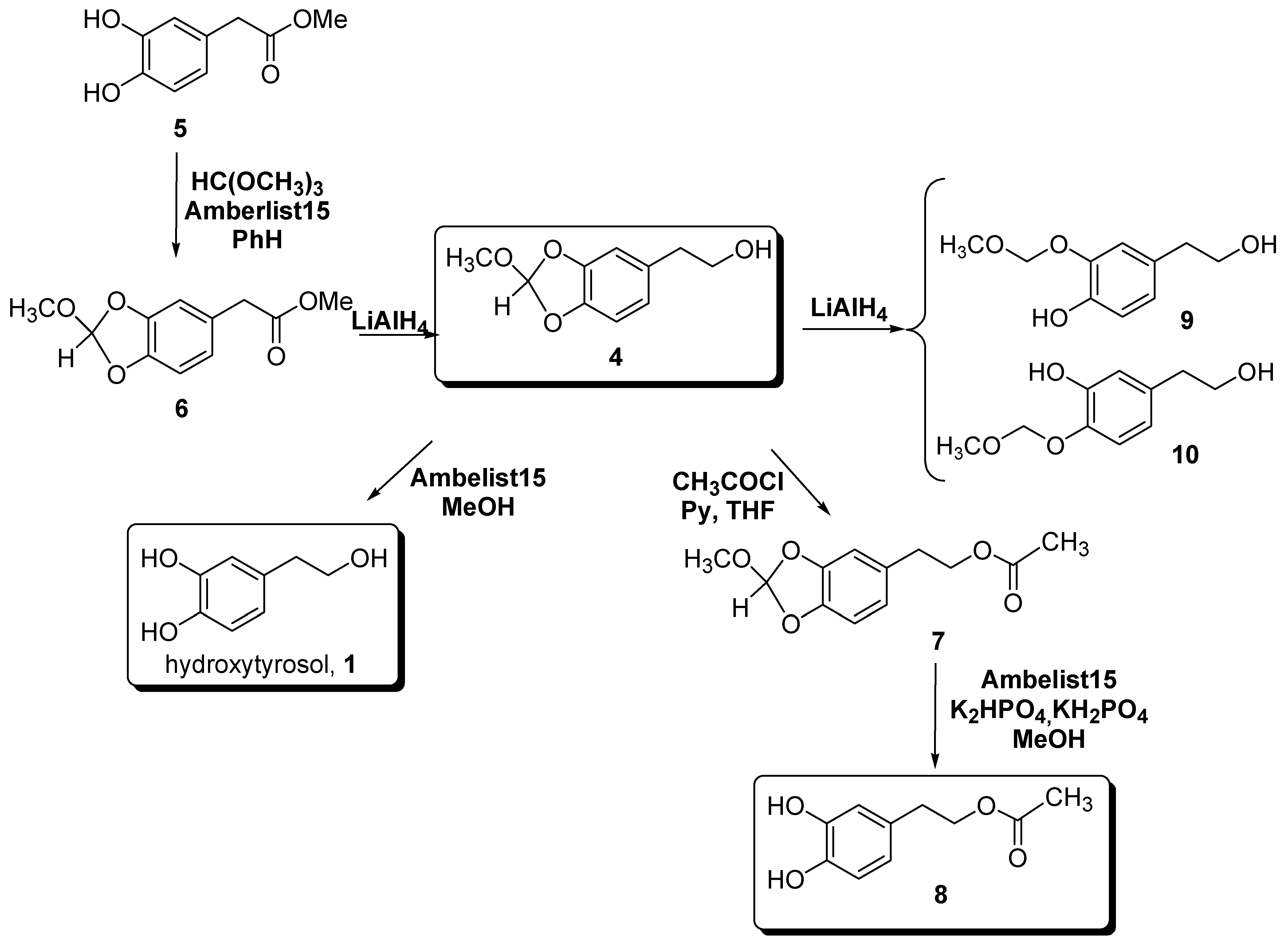
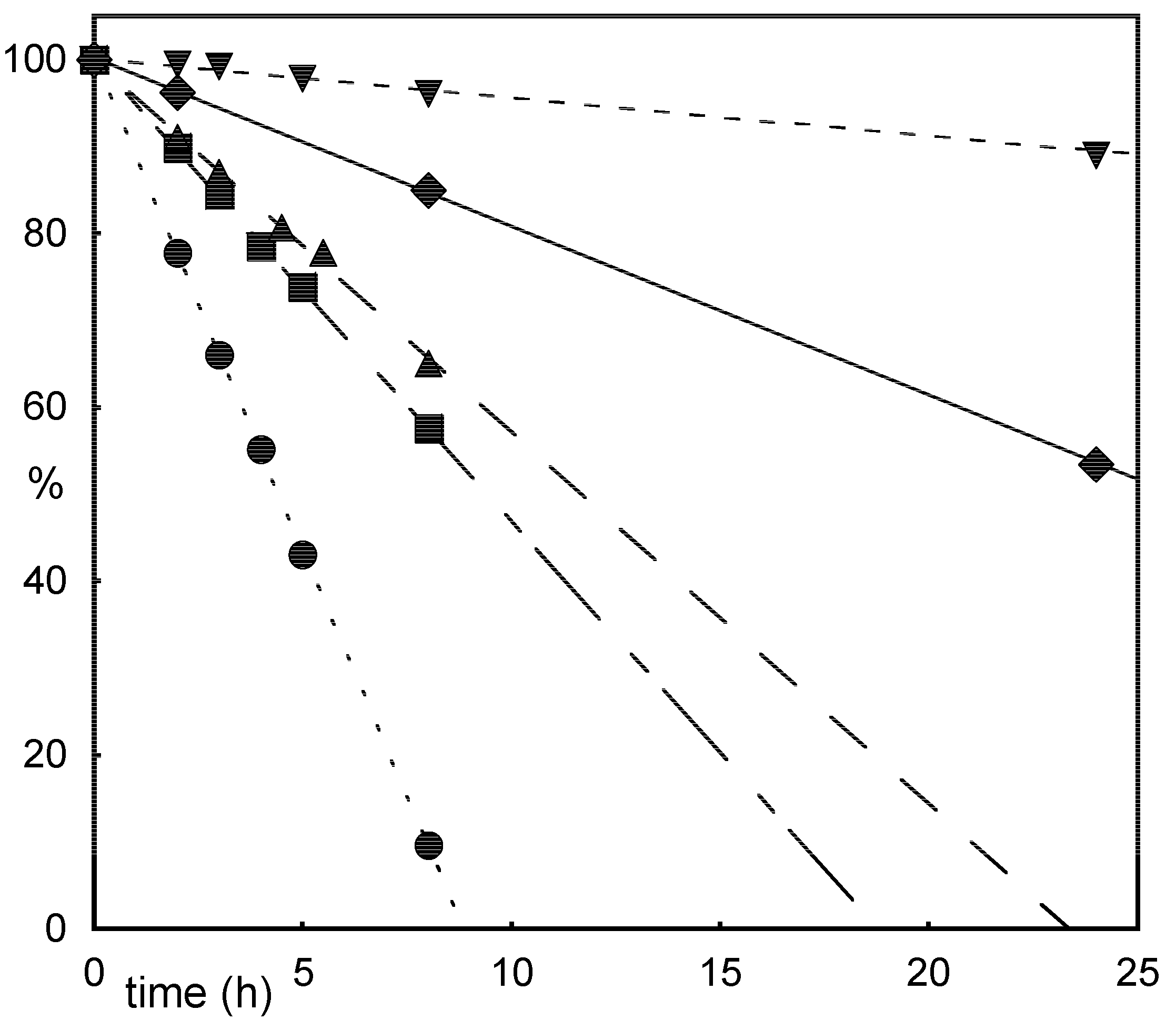
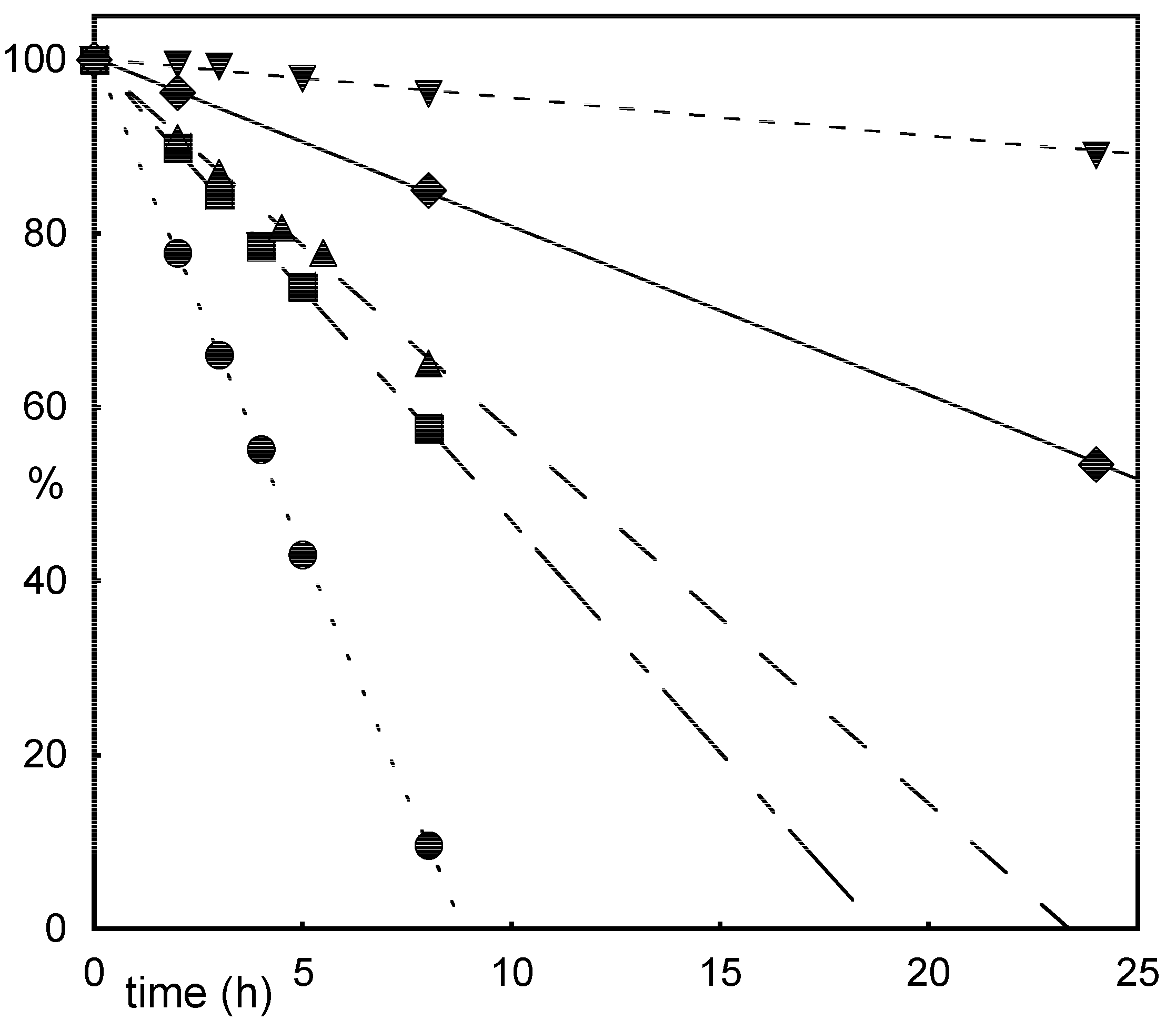
Results and Discussion
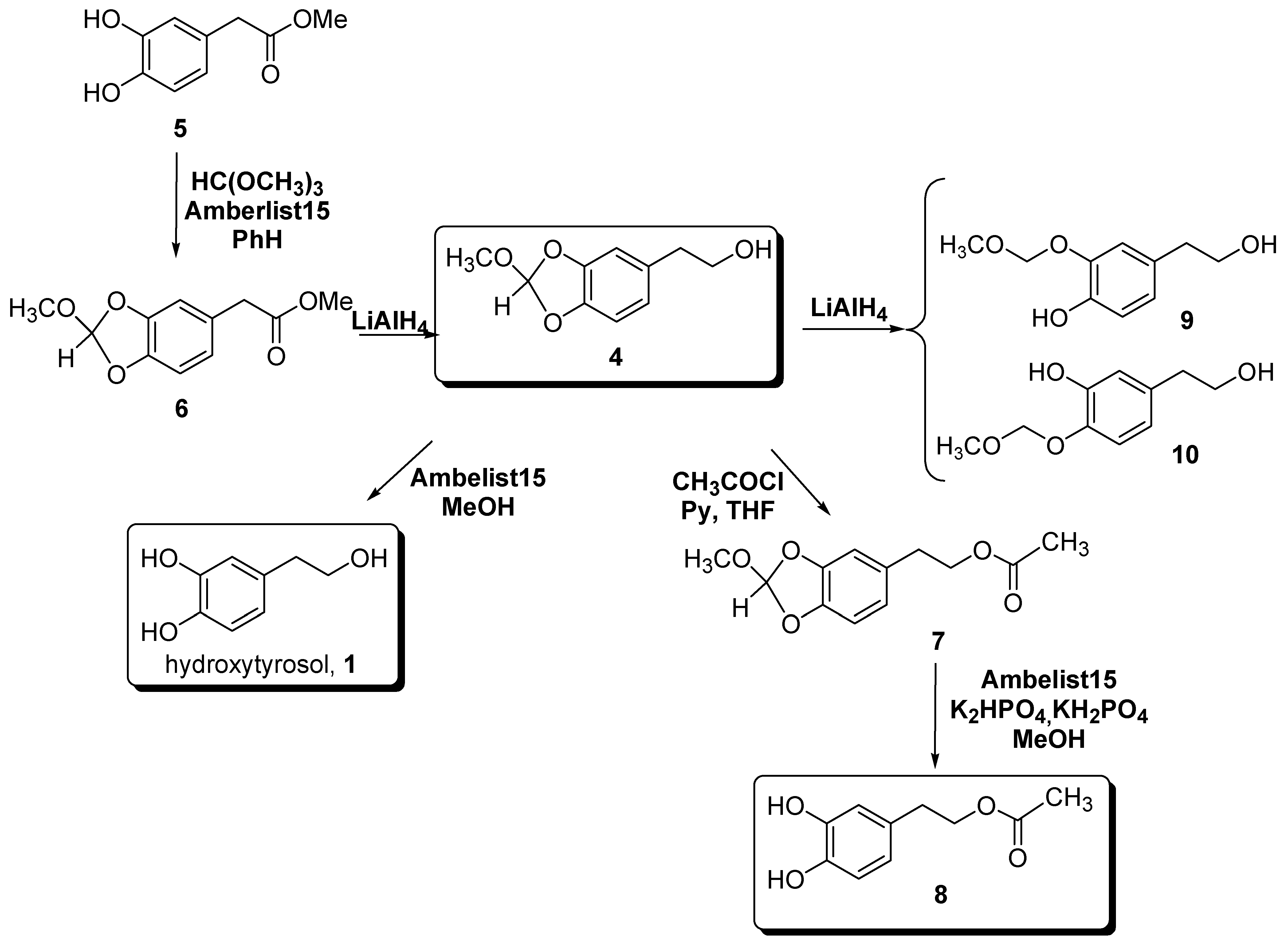
Experimental Section
General
Acknowledgements
References and Notes
- Silva, S.; Gomes, L.; Leitão, F.; Coelho, A. V.; Boas, L. V. Phenolic compounds and antioxidant activity of olea europaea L. fruits and leaves. Food Sci. Technol. Int. 2006, 12, 385–396. [Google Scholar]
- Lo Scalzo, R.; Scarpati, M.L.; Verzegnassi, B.; Vita, G. Olea Europaea chemicals repellent to Dacus Oleae females. J. Chem. Ecol. 1994, 20, 1813–1823. [Google Scholar]
- Le Tutor, B.; Guedon, D. Antioxidative activities of Olea europaea leaves and related phenolic compounds. Phytochemistry 1992, 31, 1173–1178. [Google Scholar]
- Medina, E.; De Castro, A.; Romero, C.; Brenes, M. Comparison of the concentrations of phenolic compounds in olive oils and other plant oils: correlation with antimicrobial activity. J. Agric. Food Chem. 2006, 54, 4954–4961. [Google Scholar] Bisignano, G.; Tomaino, A.; Lo Cascio, R.; Crisafi, G.; Uccella, N.; Saija, A. On the in-vitro antimicrobial activity of oleuropein and hydroxytyrosol. J. Pharm. Pharmacol. 1999, 51, 971–974. [Google Scholar]
- Fabiani, R.; De Bartolomeo, A.; Rosignoli, P.; Servili, M.; Selvaggini, R.; Montedoro, G. F.; Di Saverio, C.; Morozzi, G. Virgin olive oil phenols inhibit proliferation of human promyelocytic leukemia cells (HL60) by inducing apoptosis and differentiation. J. Nutr. 2006, 136, 614–619. [Google Scholar] Gill, C.I.R.; Boyd, A.; McDermot, E.; McCann, M.; Servili, M.; Selvaggini, R.; Taticchi, A.; Esposto, S.; Montedoro, G.F.; McGlynn, H.; Rowland, I. Potential anti-cancer effects of virgin olive oil phenols on colorectal carcinogenesis models in vitro. Int. J. Cancer 2005, 117, 1–7. [Google Scholar]
- See for example: Nousis, L.; Doulias, P.; Aligiannis, N.; Bazios, D.; Agalias, A.; Galaris, D.; Mitakou, S. DNA protecting and genotoxic effects of olive oil related components in cells exposed to hydrogen peroxide. Free Radical Res. 2005, 39, 787–795. [Google Scholar] Hashimoto, T.; Ibi, M.; Matsuno, K.; Nakashima, S.; Tanigawa, T.; Yoshikawa, T.; Yabe-Nishimura, C. An endogenous metabolite of dopamine, 3,4-dihydroxyphenylethanol, acts as a unique cytoprotective agent against oxidative stress-induced injury. Free Radical Bio. Med. 2004, 36, 555–564. [Google Scholar]
- González-Santiago, M.; Martín-Bautista, E.; Carrero, J. J.; Fonollá, J.; Baró, L.; Bartolomé, M. V.; Gil-Loyzaga, P.; Loper-Huertas, E. One-month administration of hydroxytyrosol, a phenolic antioxidant present in olive oil, to hyperlipemic rabbits improves blood lipid profile, antioxidant status and reduces atherosclerosis development. Atherosclerosis 2006, 188, 35–42. [Google Scholar]
- Tabera, G. J. J.; Ruiz, R. A.; Senorans, R. F. J.; Ibanez, E. E.; Reglero, R. G. J.; Albi, V. T.; Lanzon, R. A.; Perez, C. C.; Guinda, G. A.; Rada, R. M. Method of obtaining high-value-added compounds from olive leaves. PCT WO2005075614, 2005. [Google Scholar] Fernandez-Bolaños, G.J.; Moreno, A.H. Method for obtaining purified hydroxytyrosol from products and by-products derived from the olive tree. PCT US2004102657, 2004. [Google Scholar] Crea, R. Hydroxytyrosol-rich composition from olive vegetation water and method of use thereof. PCT WO2004005228, 2004. [Google Scholar]
- Capasso, R.; Evidente, A.; Avolio, S.; Solla, F. A higly convenient synthesis of hydroxytyrosol and its recovery from agricultural waste waters. J. Agric. Food Chem. 1999, 47, 1745–1748. [Google Scholar] Tuck, K.L.; Tan, H.; Hayball, P.J. Synthesis of tritium-labeled hydroxytyrosol, a phenolic compound found in olive oil. J. Agric. Food Chem. 2000, 48, 4087–4090. [Google Scholar] Bai, C.; Yan, X.; Takenaka, M.; Sekyya, S.; Nagata, T. Determination of synthetic hydroxytyrosol in rat plasma by GC-MS. J. Agric. Food Chem. 1998, 46, 3998–4001. [Google Scholar] Fernández-Bolaños, J.; Rodríguez, G.; Gómez, E.; Guillén, R.; Jiménez, A.; Heredia, A.; Rodriguez, R. Total recovery of the waste of two-phase olive oil processing: isolation of added-value compounds. J. Agric. Food Chem. 2004, 52, 5849–5855. [Google Scholar] Briante, R.; Patumi, M.; Febbraio, F.; Nucci, R. Production of highly purified hydroxytyrosol from olea europaea leaf extract biotransformed by hyperthermophilic β-glycosidase. J. Biotechnol. 2004, 111, 67–77. [Google Scholar]
- Alcudia, G. F.; Cert, V. A.; Espartero Sanchez, J. L.; Mateo, B. R.; Trujillo Perez-Lanzac, M. Method of preparing Hydroxytyrosol esters, esters thus obtained and use of same. PCT 2004/005237, 2004. [Google Scholar] Trujillo, M.; Mateos, R.; Collantes De Teran, L.; Espartero, J.L.; Cert, R.; Jover, M.; Alcudia, F.; Bautista, J.; Cert, A.; Parrado, J. Lipophilic hydroxytyrosol esters. Antioxidant activity in lipid matrices and biological systems. J. Agric. Food Chem. 2006, 54, 3779–3785. [Google Scholar] Gordon, M.H.; Pavia-Martins, F.; Almeida, M. Antioxidant activity of hydroxytyrosol acetate compared with that of other olive oil polyphenols. J. Agric. Food Chem. 2001, 49, 2480–2485. [Google Scholar]
- Torregiani, E.; Seu, G.; Minassi, A.; Appendino, G. Cerium-(III) chloride-promoted chemoselective esterification of phenolic alcohols. Tetrahedron Lett. 2005, 46, 2193–2196. [Google Scholar]
- Gambacorta, A.; Tofani, D.; Bernini, R.; Migliorini, A. High yielding preparation of a stable precursor of hydroxytyrosol by total synthesis and from natural glycoside oleuropein. J. Agric. Food Chem. 2007, 55, 3386–3391. [Google Scholar]
- Narayanan, J.; Hayakawa, Y.; Fan, J.; Kirk, K.L. Convenient syntheses of biogenic aldehydes, 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde. Bioorg. Chem. 2003, 31, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals, 4th ed.; Butterworth-Heinemann: Oxford, 1997. [Google Scholar]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gambacorta, A.; Tofani, D.; Migliorini, A. High-Yielding Synthesis of Methyl Orthoformate-Protected Hydroxytyrosol and Its Use in Preparation of Hydroxytyrosyl Acetate. Molecules 2007, 12, 1762-1770. https://doi.org/10.3390/12081762
Gambacorta A, Tofani D, Migliorini A. High-Yielding Synthesis of Methyl Orthoformate-Protected Hydroxytyrosol and Its Use in Preparation of Hydroxytyrosyl Acetate. Molecules. 2007; 12(8):1762-1770. https://doi.org/10.3390/12081762
Chicago/Turabian StyleGambacorta, Augusto, Daniela Tofani, and Antonella Migliorini. 2007. "High-Yielding Synthesis of Methyl Orthoformate-Protected Hydroxytyrosol and Its Use in Preparation of Hydroxytyrosyl Acetate" Molecules 12, no. 8: 1762-1770. https://doi.org/10.3390/12081762