Discovery of a Novel CCR5 Antagonist Lead Compound Through Fragment Assembly

Abstract
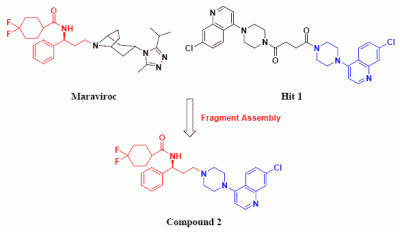
:
Introduction

Results and Discussion
Identification of Compound 1 by Virtual Screening and Calcium Mobilization Assay

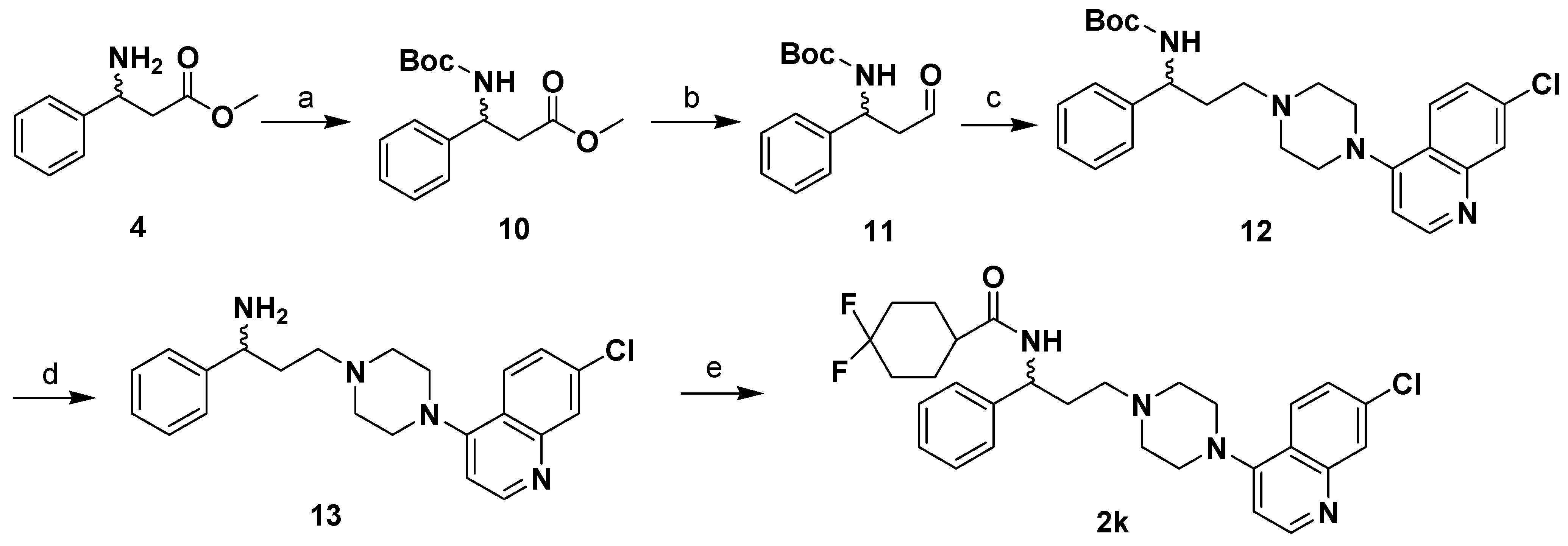
Design and Synthesis of Compounds 2 and 2a-l


Calcium Mobilization Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (μM) |
| 1 |  | 2.00 |
| 2 |  | 0.692 |
| 2a | 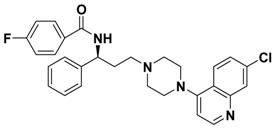 | -- |
| 2b |  | 2.66 |
| 2c | 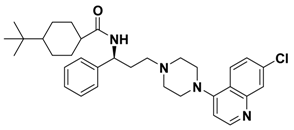 | -- |
| 2d | 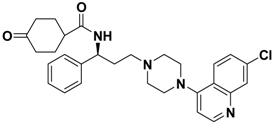 | > 1.00 |
| 2e | 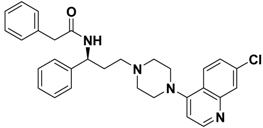 | > 10.0 |
| 2f | 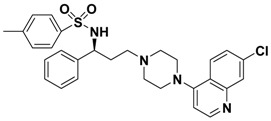 | > 10.0 |
| 2g | 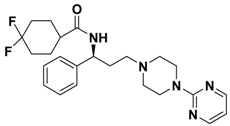 | > 1.00 |
| 2h | 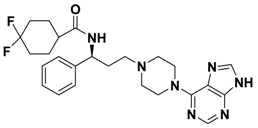 | > 10.0 |
| 2i | 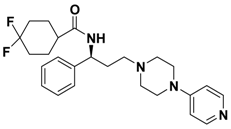 | > 10.0 |
| 2j | 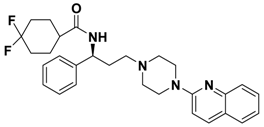 | 0.233 |
| 2k | 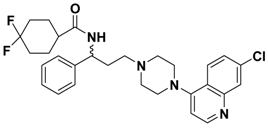 | 0.669 |
| 2l | 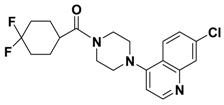 | > 1.00 |
| Maraviroc |  | 0.00261 |
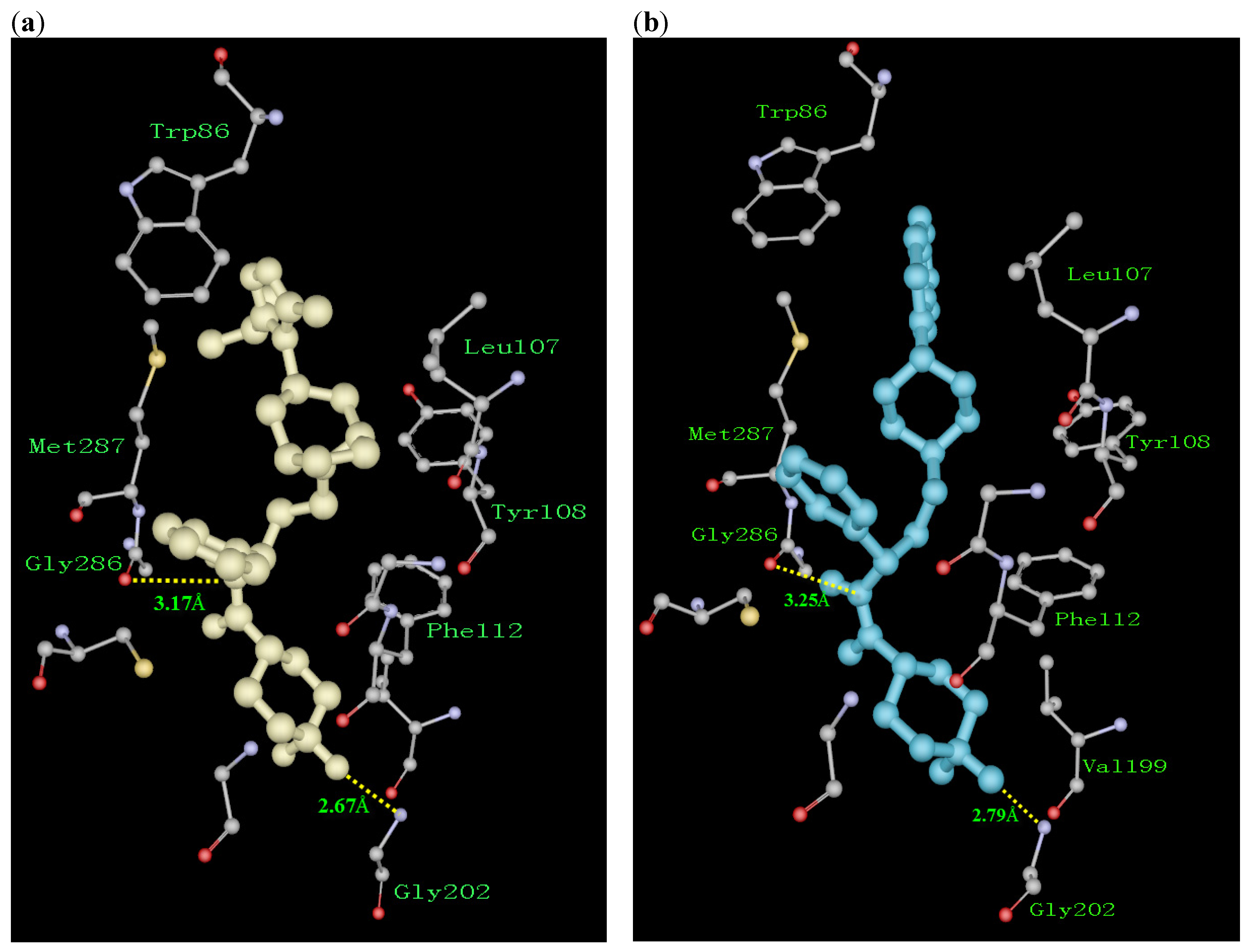
Structure and Activity Relationship Correction with the Binding Models

Conclusions
Experimental
General
Virtual Screening by Molecular Docking
Calcium Mobilization Assay
Acknowledgements
References
- Doranz, B. J.; Berson, J. F.; Rucker, J.; Doms, R. W. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1). Immunol. Res. 1997, 16, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C. C.; Kennedy, P. E.; Berger, E. A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G proteincoupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C. C.; Feng, Y.; Kennedy, P. E.; Murphy, P. M.; Berger, E. CC-CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage- tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Marzio, P.; Marmon, S.; Sutton, R. E.; Hill, C. M.; Davis, C. B.; Peiper, S. C.; Schall, T. J.; Littman, D. R.; Landau, N. R. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G. P.; Martin, S. R.; Huang, Y.; Nagashima, K. A.; Cayanan, C.; Maddon, P. J.; Koup, R. A.; Moore, J. P.; Paxton, W. A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Gerard, N. P.; Wyatt, R.; Choe, H.; Parolin, C.; Ruffing, N.; Borsetti, A.; Cardoso, A. A.; Desjardin, E.; Newman, W.; Gerard, C.; Sodroski, J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P. D.; Wyatt, R.; Robinson, J.; Sweet, R. W.; Sodroski, J.; Hendricks, W. A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [PubMed]
- Cocchi, F.; Devico, A. L.; Garzinodemo, A.; Arya, S. K.; Gallo, R. C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Clapham, P. R.; Picard, L.; Offord, R. E.; Rosenkilde, M. M.; Schwartz, T. W.; Buser, R.; Wells, T. N. C.; Proudfoot, A. E. I. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 1997, 276, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Heveker, N.; Montes, M.; Germeroth, L.; Amara, A.; Trautmann, A.; Alizon, M.; Schneider- Mergener, J. Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr. Biol. 1998, 8, 369–376. [Google Scholar] [CrossRef]
- Olson, W. C.; Rabut, G. E.; Nagashima, K. A.; Tran, D. N.; Anselma, D. J.; Monard, S. P.; Segal, J. P.; Thompson, D. A.; Kajumo, F.; Guo, Y.; Moore, J. P.; Maddon, P. J.; Dragic, T. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J. Virol. 1999, 73, 4145–4155. [Google Scholar] [PubMed]
- Liu, R.; Paxton, W. A.; Choe, S.; Ceradini, D.; Martin, S. R.; Horuk, R.; MacDonald, M. E.; Stuhlmann, H.; Koup, R. A.; Landau, N. R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Palani, A.; Tagat, J. R. Discovery and Development of Small-Molecule Chemokine Coreceptor CCR5 Antagonists. J. Med. Chem. 2006, 49, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; Webster, R.; Armour, D.; Price, D.; Stammen, B.; Wood, A.; Perros, M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Dunning, L.; Jaroch, S.; Kochanny, M. J.; Lee, W.; Lian, X.; Liang, M.; Lu, S.; Qnuffer, J.; Phillips, G.; Wei, G.; Ye, B. WO Patent 2004002960, 2004.
- Xu, Y.; Liu, H.; Niu, C.; Luo, C.; Luo, X.; Shen, J.; Chen, K.; Jiang, H. Molecular docking and 3D QSAR studies on 1-amino-2-phenyl-4-(piperidin-1-yl)-butanes based on the structural modeling of human CCR5 receptor. Bioorg. Med. Chem. 2004, 12, 6193–6208. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C. A.; Motoshima, H.; Fox, B. A.; Le Trong, I.; Teller, D. C.; Okada, T.; Stenkamp, R. E.; Yamamoto, M.; Miyano, M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Gui, C.; Zhang, L.; Qin, Y.; Xu, Q.; Zhang, J.; Liu, H.; Shen, X.; Jiang, H. Discovering novel chemical inhibitors of human cyclophilin A:Virtual screening, synthesis, and bioassay. Bioorg. Med. Chem. 2006, 14, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T. J.; Makino, S.; Skillman, A. G.; Kuntz, I. D. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Morris, G. M.; Goodsell, D. S.; Halliday, R.S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Weiner, S. J.; Kollman, P. A.; Nguyen, D. T.; Case, D. A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef]
- Baba, M.; Takashima, K.; Miyake, H.; Kanzaki, N.; Teshima, K.; Wang, X.; Shiraishi, M.; Iizawa, Y. TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans. Antimicrob. Agents Chemother. 2005, 49, 4584–4591. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Kuehne, P.; Linden, A.; Hesse, M. Asymmetric Synthesis of the Alkaloids Mayfoline and N(1)- Acetyl-N(1)-Deoxymayfoline. Helv. Chim. Acta 1996, 79, 1085–1094. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds reported in this paper are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Zhou, E.; Yu, K.; Zhu, J.; Zhang, Y.; Xie, X.; Li, J.; Jiang, H. Discovery of a Novel CCR5 Antagonist Lead Compound Through Fragment Assembly. Molecules 2008, 13, 2426-2441. https://doi.org/10.3390/molecules13102426
Liu Y, Zhou E, Yu K, Zhu J, Zhang Y, Xie X, Li J, Jiang H. Discovery of a Novel CCR5 Antagonist Lead Compound Through Fragment Assembly. Molecules. 2008; 13(10):2426-2441. https://doi.org/10.3390/molecules13102426
Chicago/Turabian StyleLiu, Yanqing, Enkun Zhou, Kunqian Yu, Jin Zhu, Yu Zhang, Xin Xie, Jian Li, and Hualiang Jiang. 2008. "Discovery of a Novel CCR5 Antagonist Lead Compound Through Fragment Assembly" Molecules 13, no. 10: 2426-2441. https://doi.org/10.3390/molecules13102426