1,2-Diaryl(3-pyridyl)ethanone Oximes. Intermolecular Hydrogen Bonding Networks Revealed by X-ray Diffraction

Abstract
:Introduction

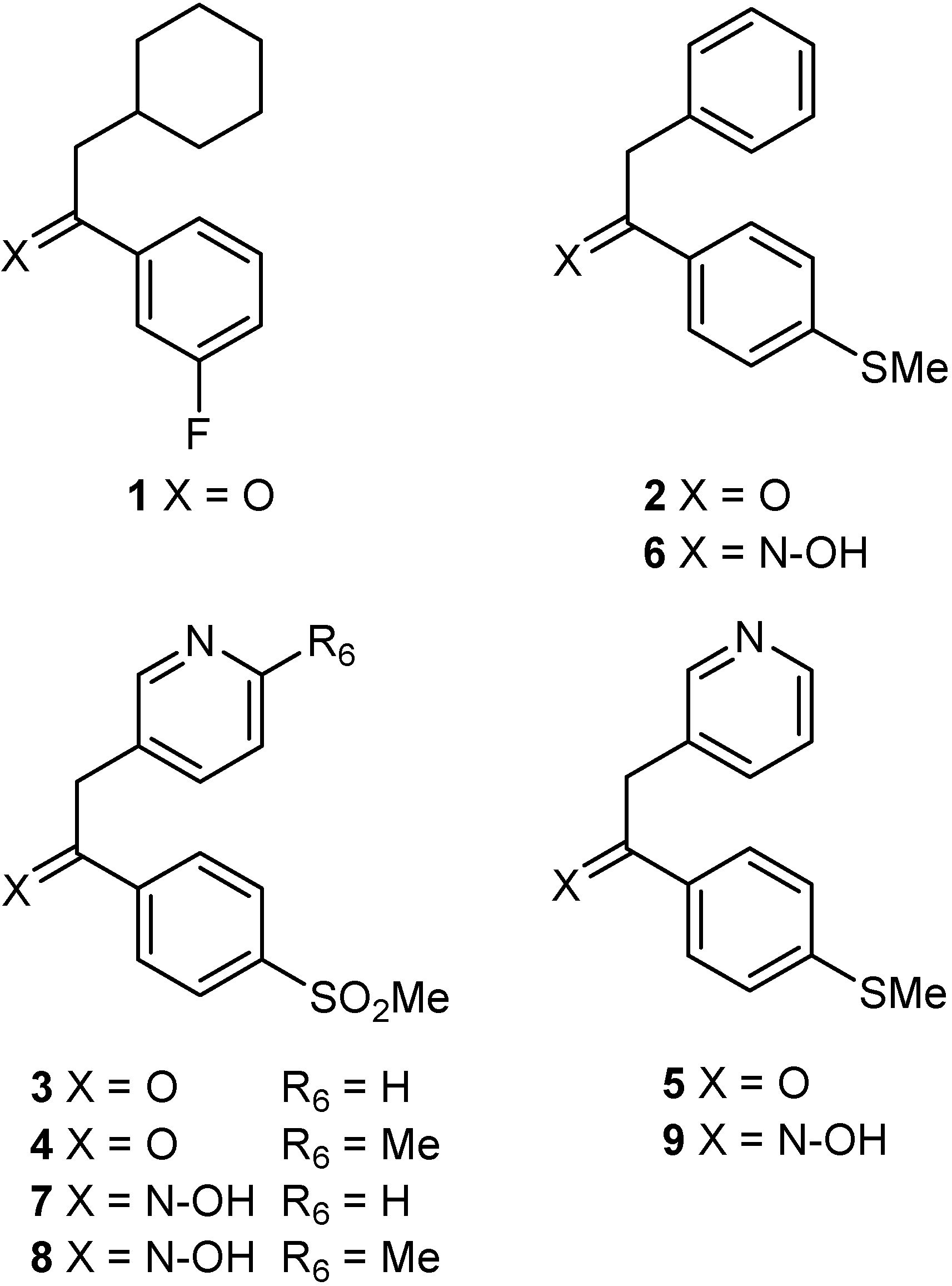
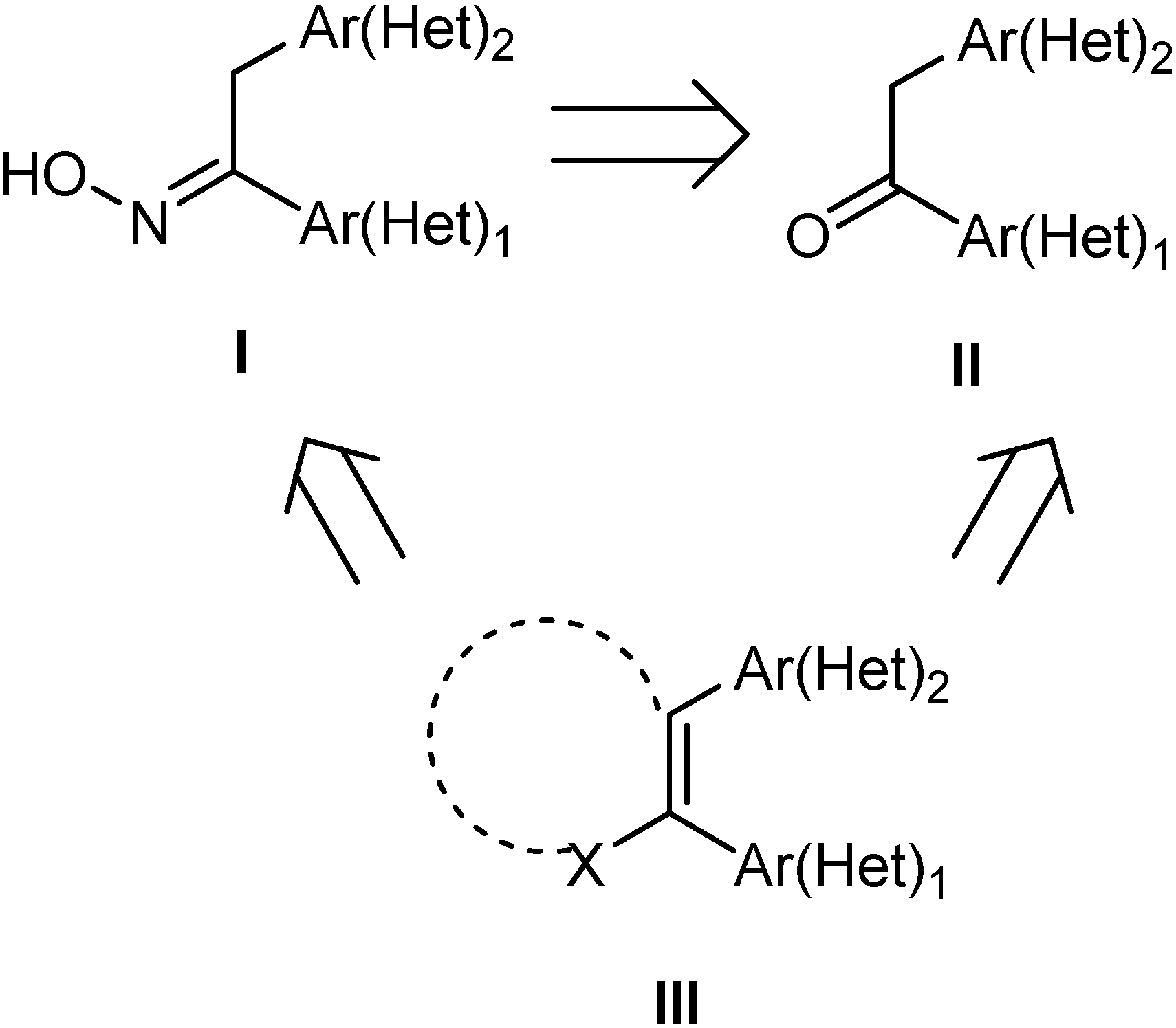
Results and Discussion
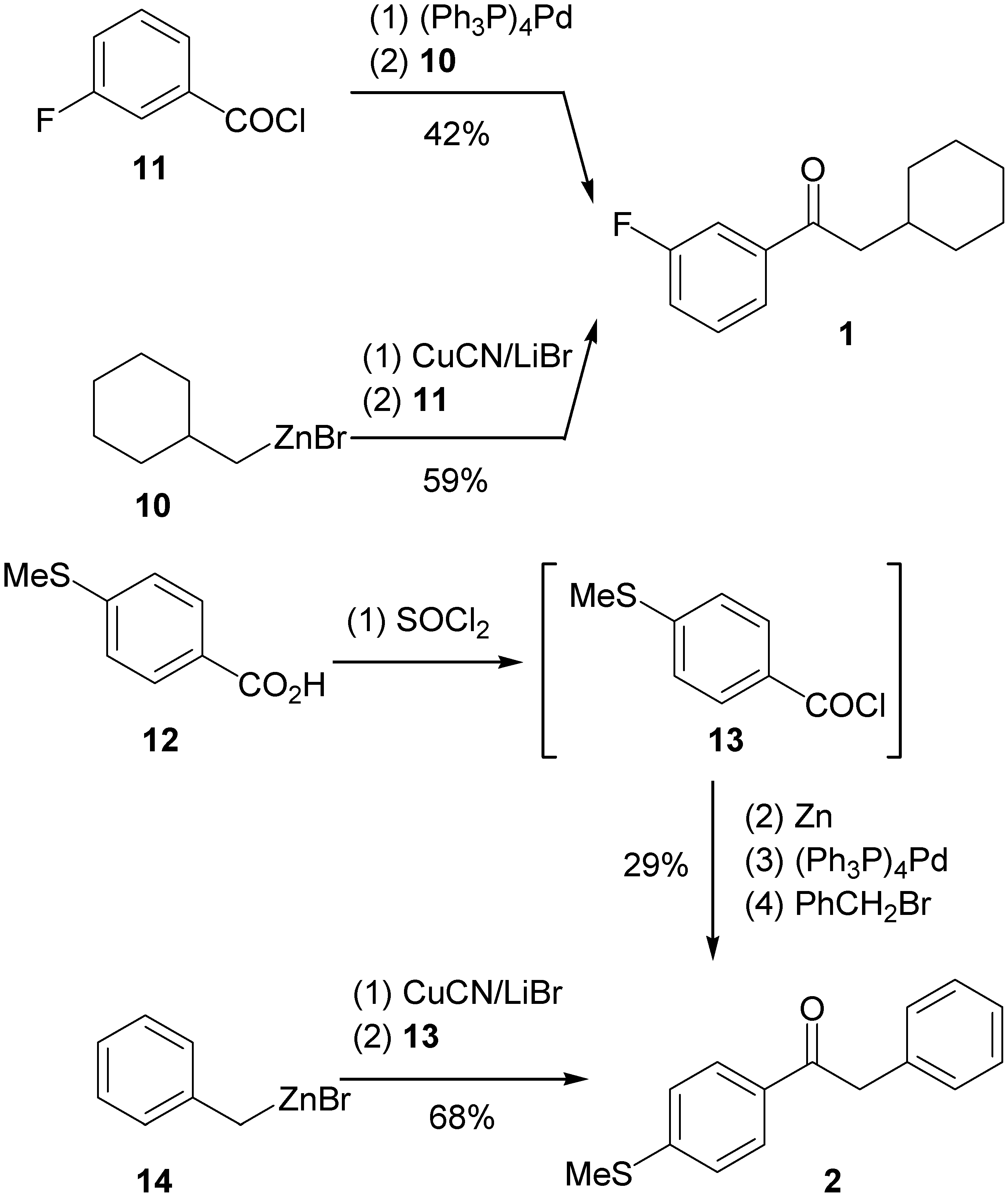
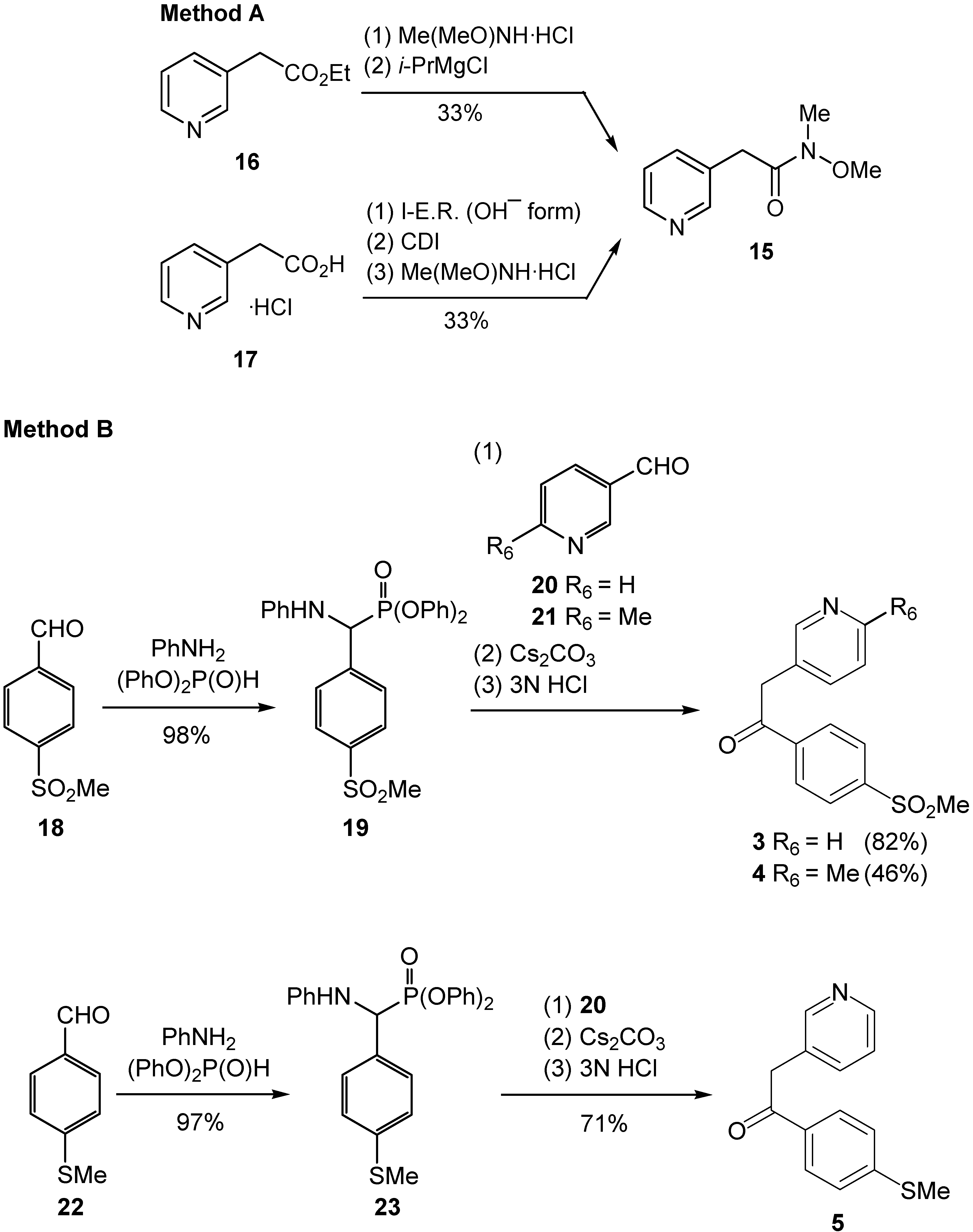
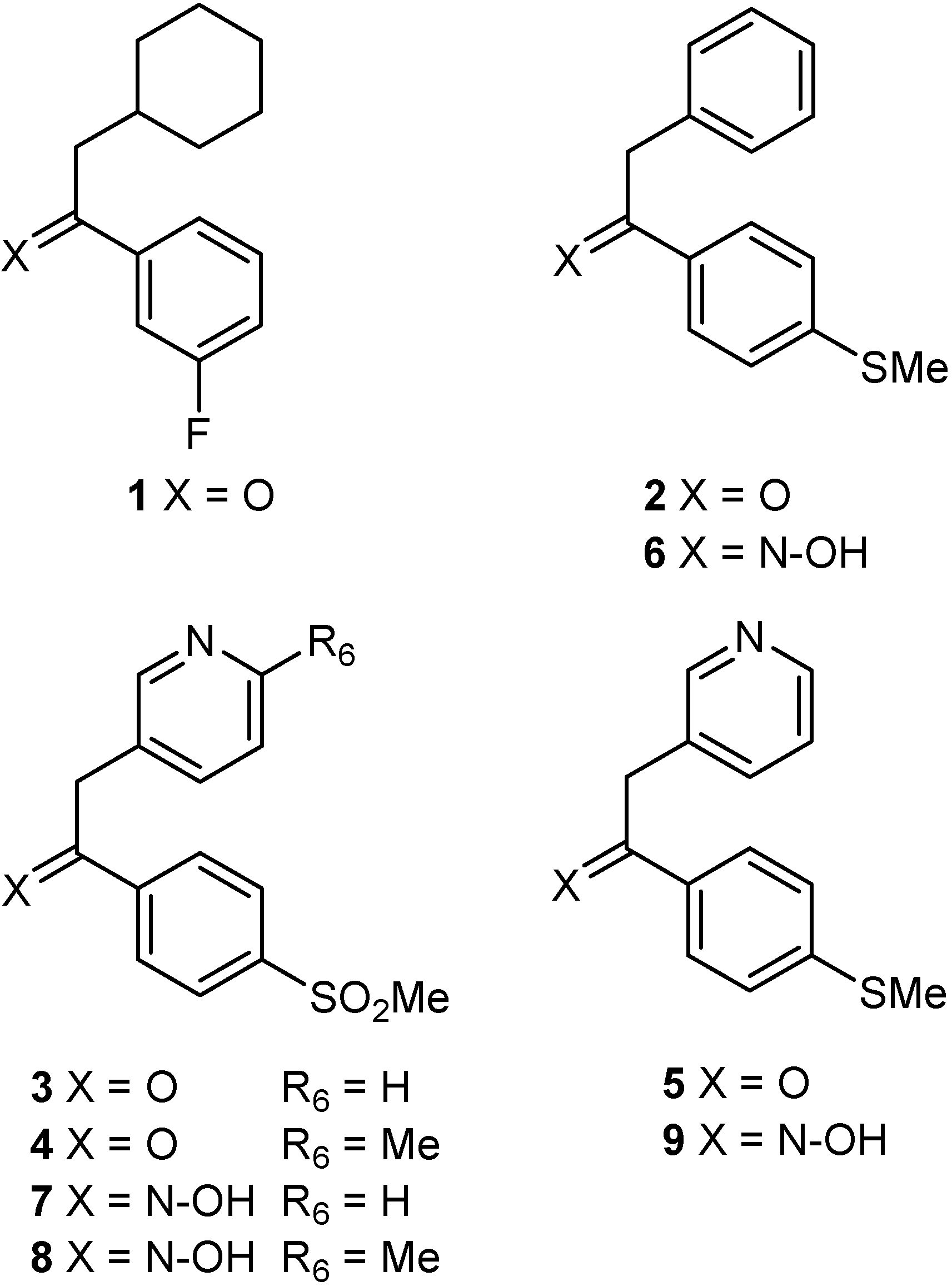
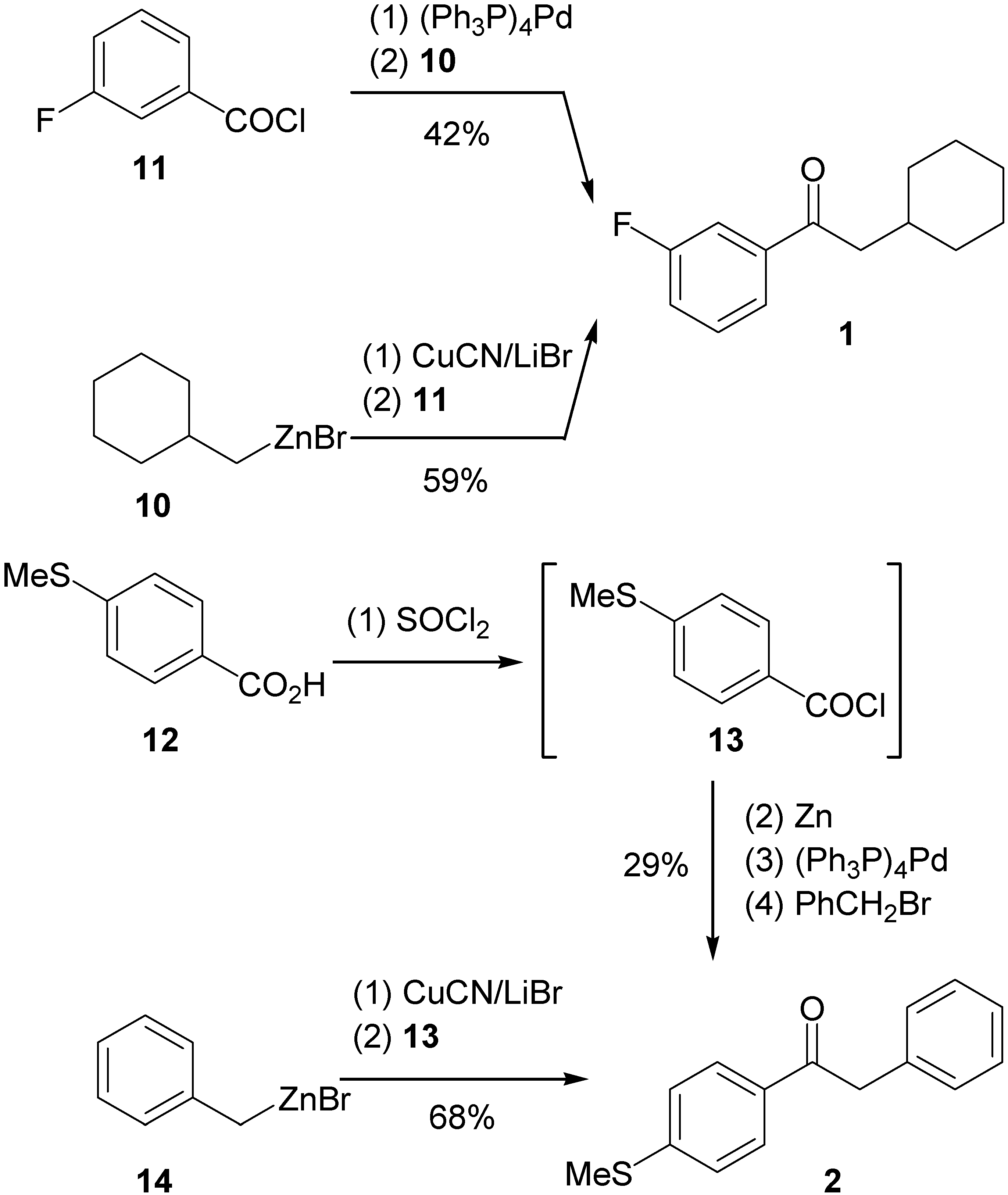
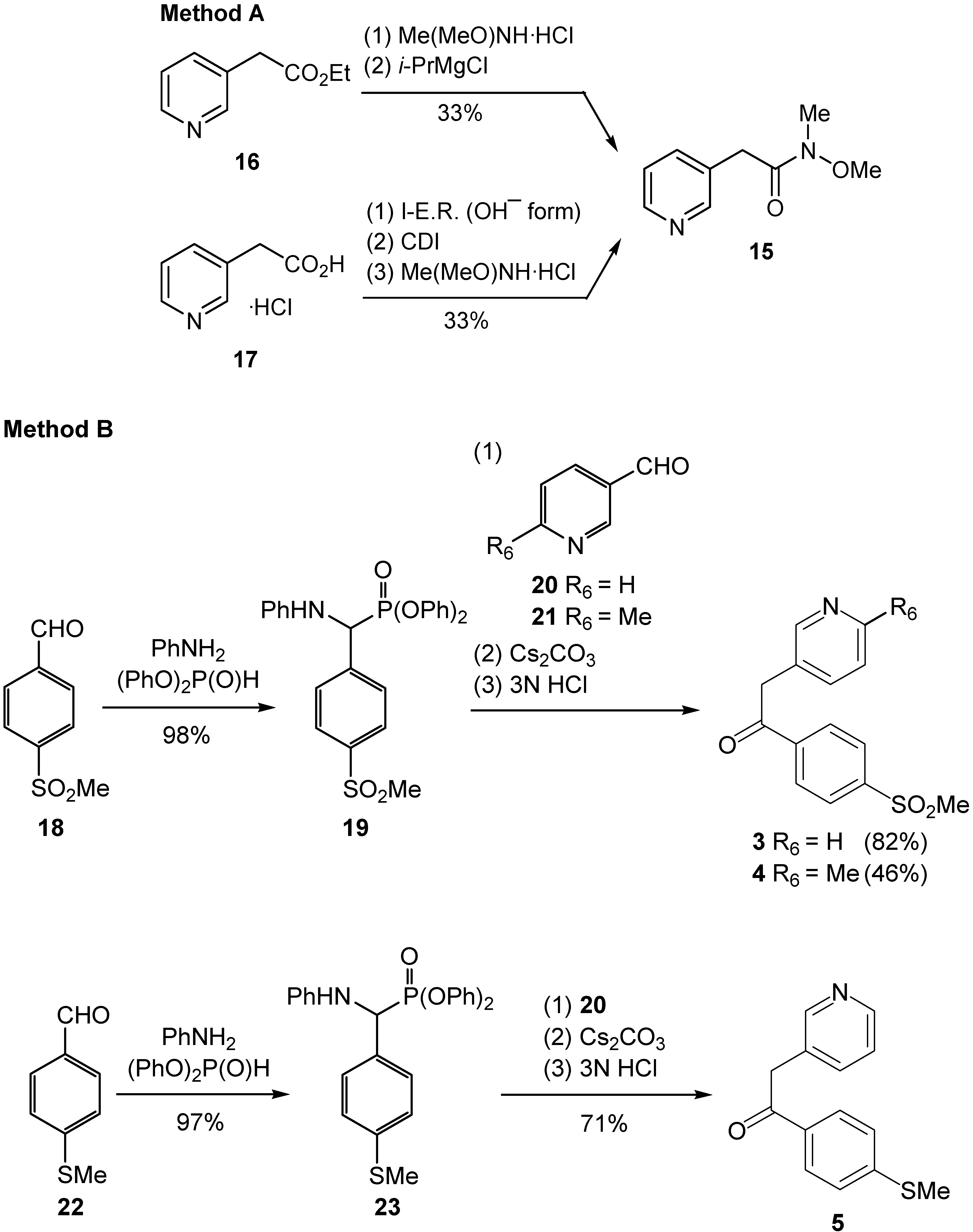
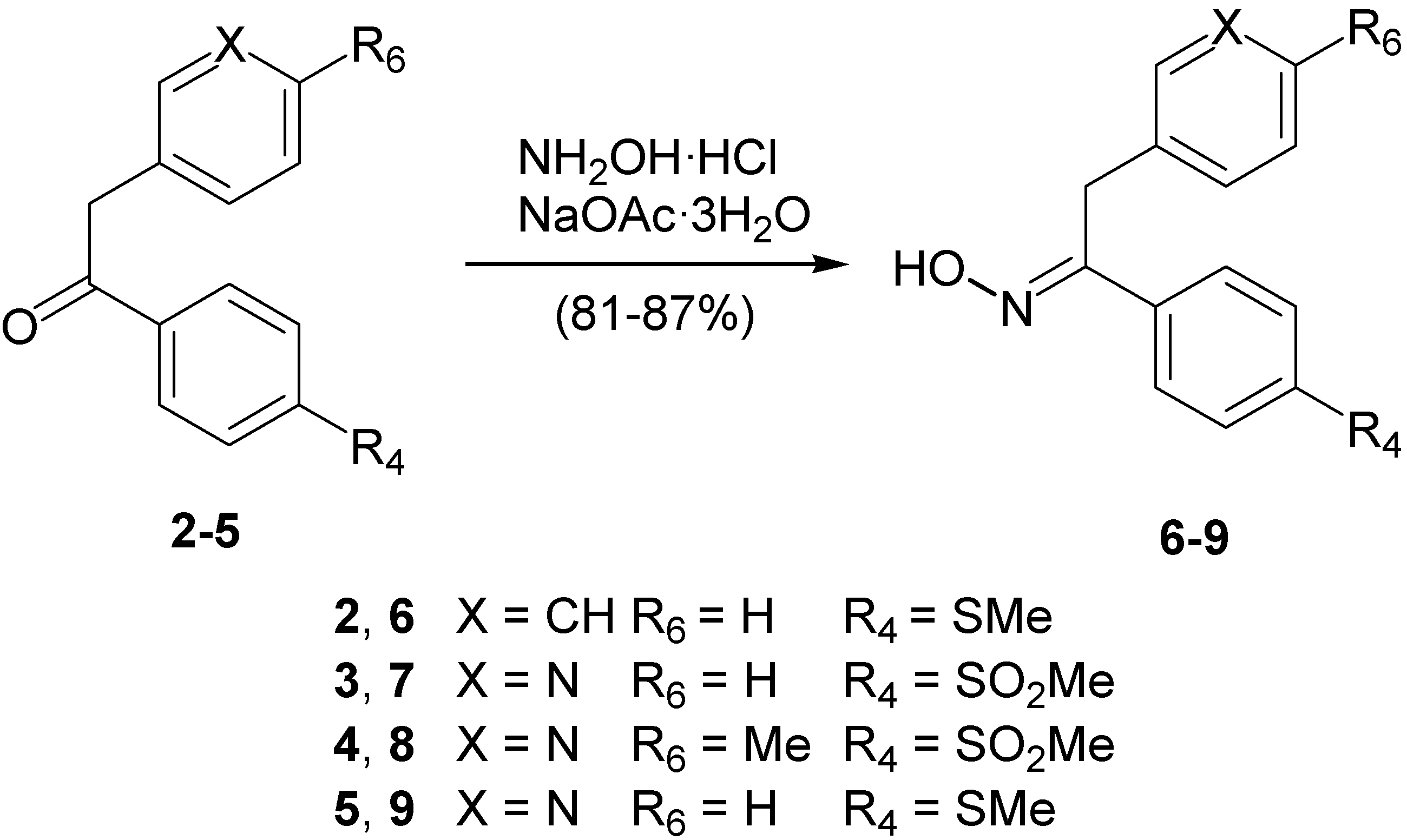
Synthesis
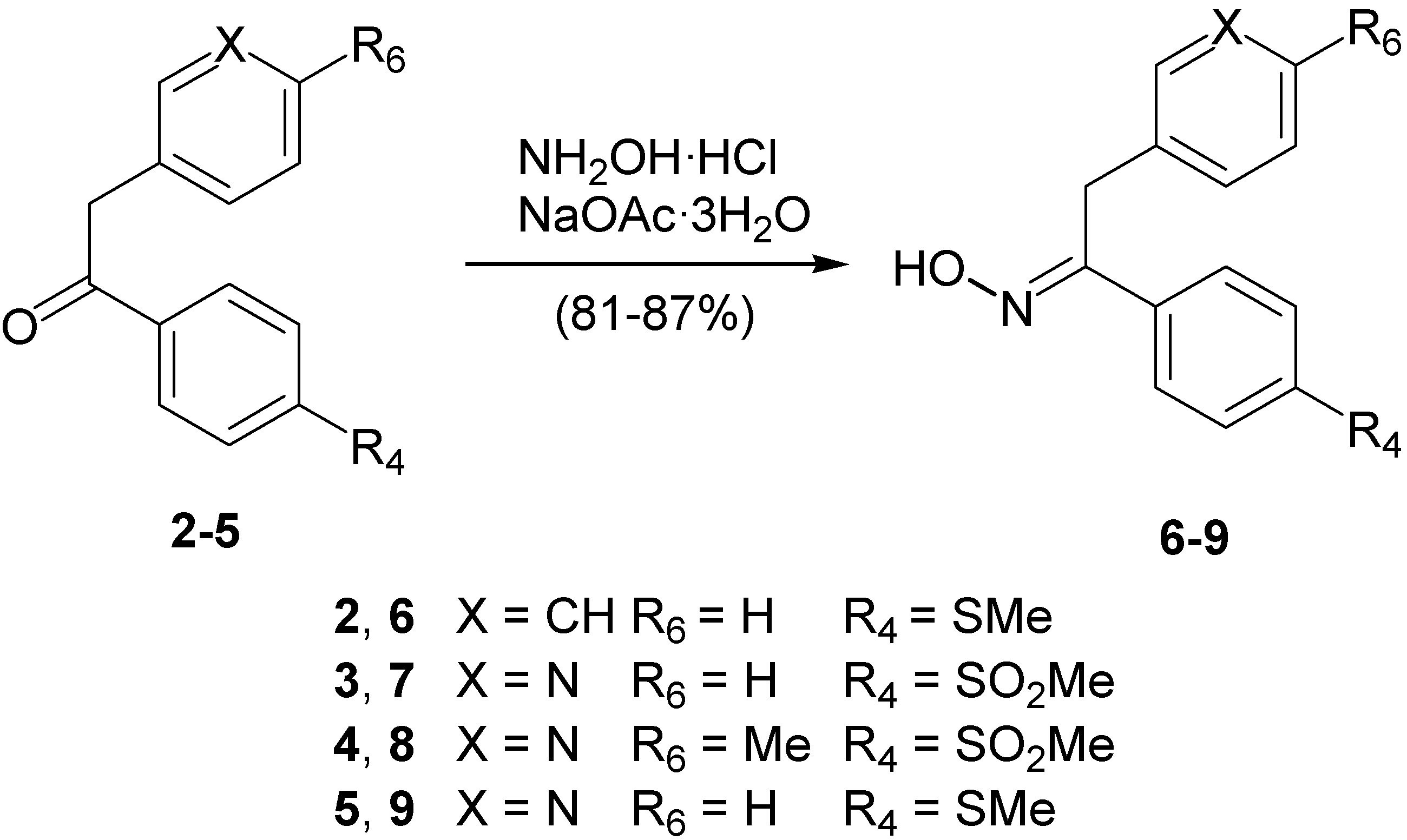
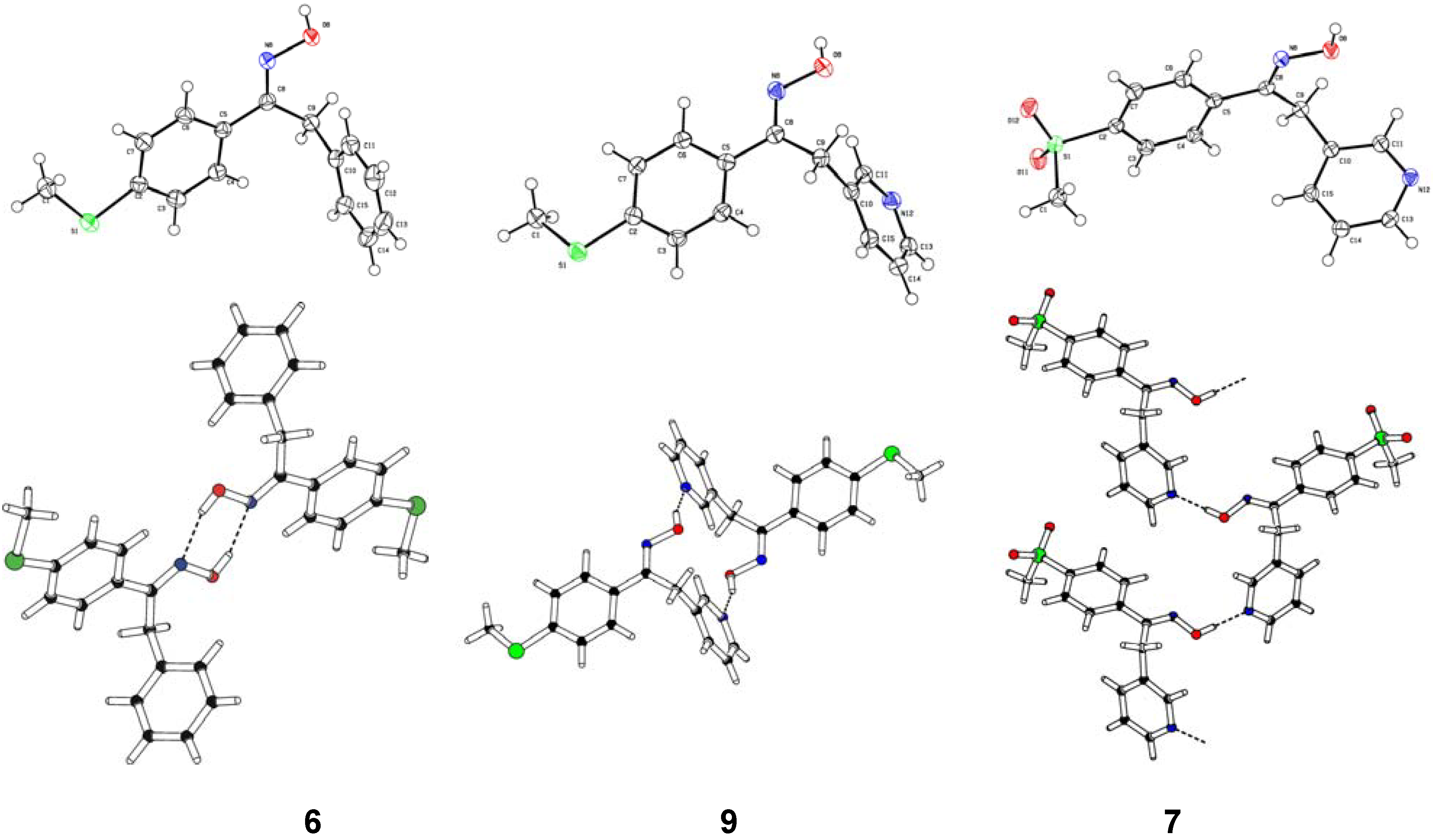
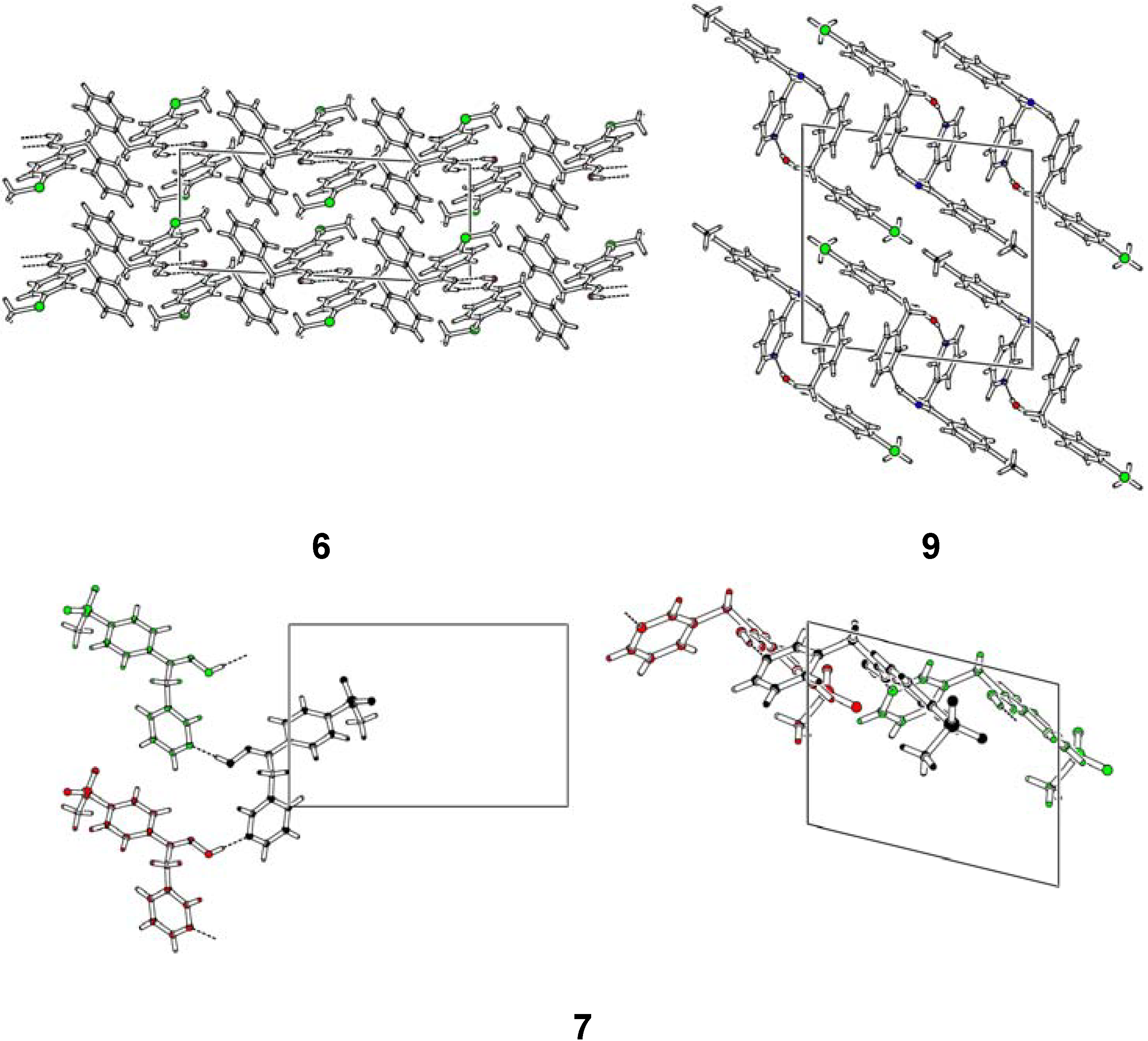
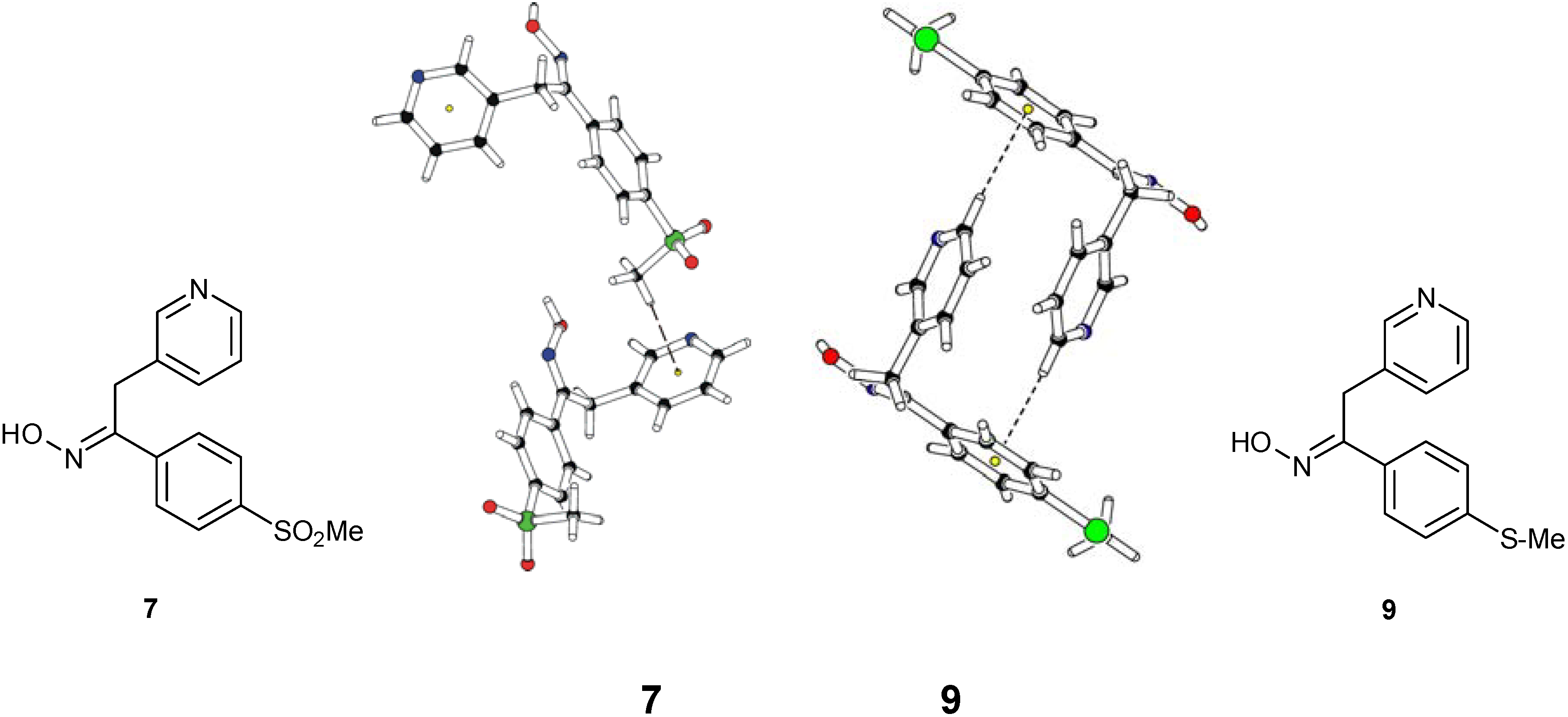
Structural Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Solvent | Conc. (mM) | C=NO-H | 2H-Py | 6H-Py | –CH2– | –CH3 |
|---|---|---|---|---|---|---|---|
| 6 | CD3CN | 9.33-103.62 | 9.07 | – | – | 4.15 | 2.46 |
| DMSO-d6 | 10.88-103.62 | 11.38a | – | – | 4.10a | 2.43 | |
| 7 | CD3CN | 10.36b | 9.65 | 8.47 | 8.39 | 4.22 | 3.04 |
| DMSO-d6c | 45.92 | 12.02a | 8.47 | 8.37 | 4.22a | 3.20 | |
| 9 | CD3CN | 11.87b | 9.20 | 8.46 | 8.36 | 4.15 | 2.46 |
| DMSO-d6 | 10.84-103.74 | 11.51a | 8.44 | 8.33 | 4.12a | 2.44 |
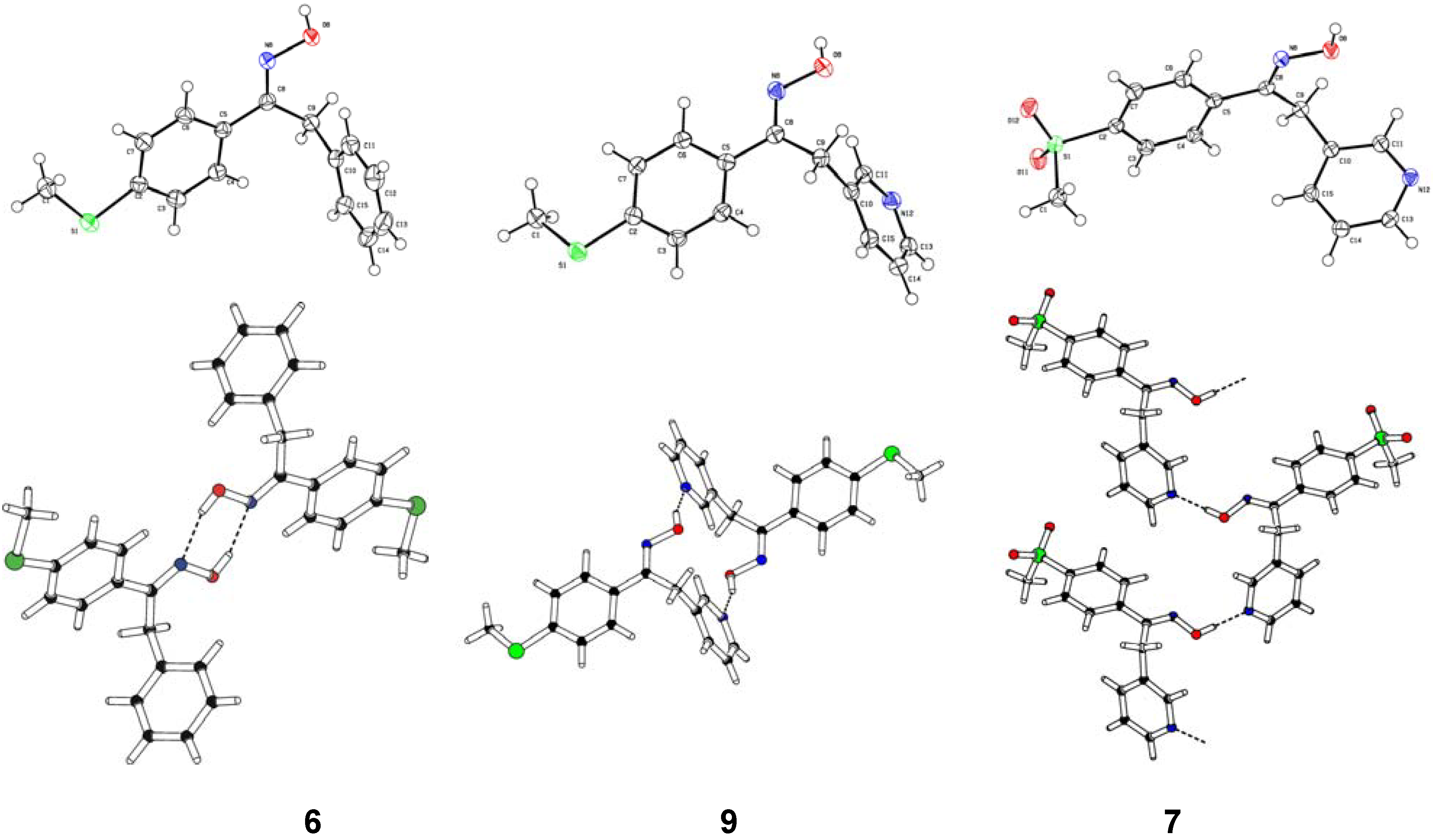
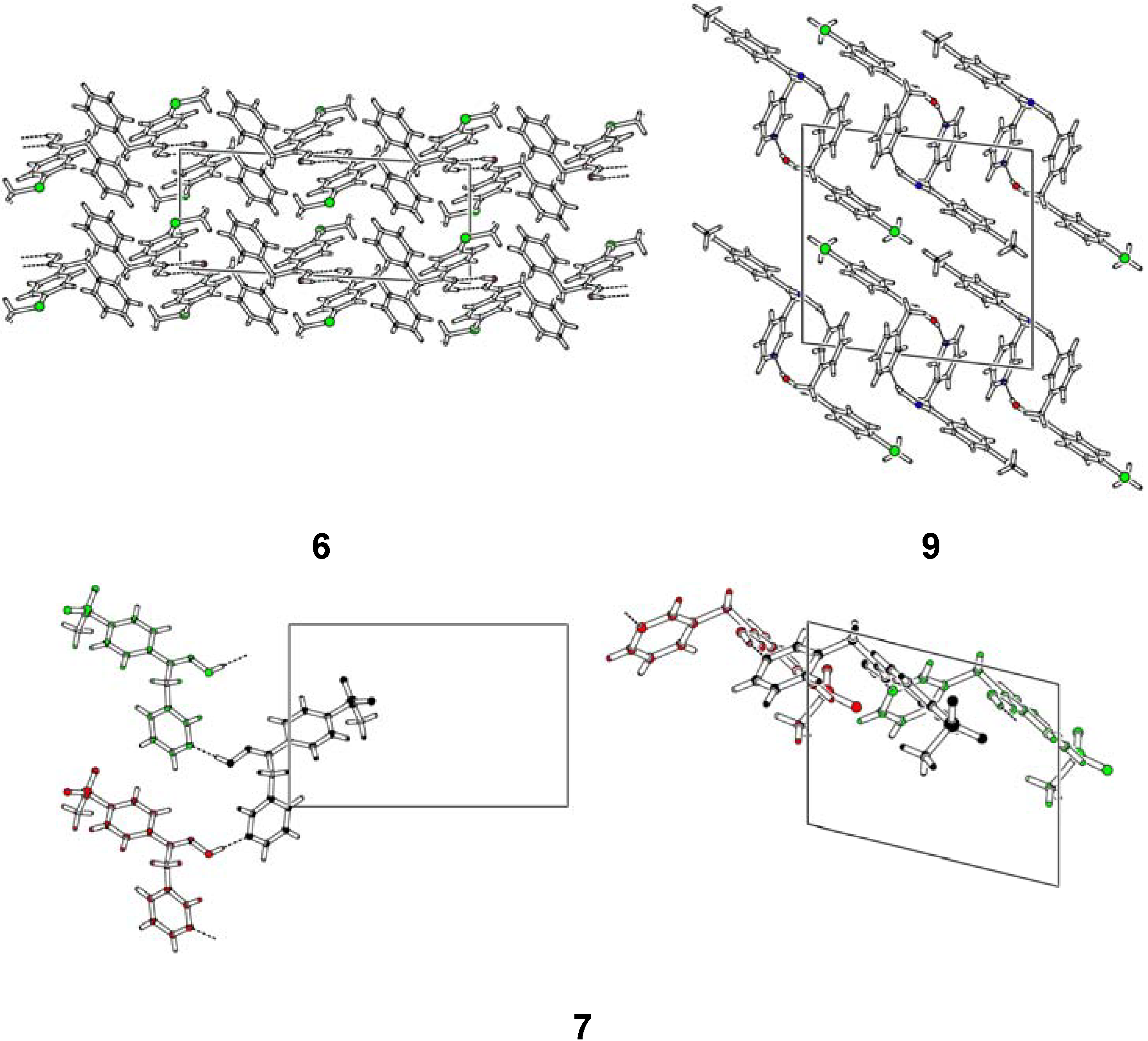
| Compd. | D–H | d(D–H) | d(H···A) | <DHA | d(D···A) | A | |||
|---|---|---|---|---|---|---|---|---|---|
| 6 | O8–H8 | 0.860 | 1.979 | [1] | 158.22 | [1] | 2.796 | [1] | N8 |
| 6 | C9–H9B | 0.944 | 2.306 | [2] | 102.83 | [2] | 2.679 | [2] | O8 |
| 9 | O8–H8 | 0.911 | 1.828 | [3] | 160.32 | [3] | 2.704 | [3] | N12 |
| 9 | C1–H1B | 0.980 | 2.567 | [4] | 158.38 | [4] | 3.497 | [4] | N8 |
| 7 | O8–H8 | 0.927 | 1.818 | [5] | 172.90 | [5] | 2.741 | [5] | N12 |
| 7 | C1–H1C | 0.980 | 2.66(2) | [6] | 164.86 | [6] | 3.577 | [6] | Fa |
| 9 | C13–H13 | 0.943 | 2.62(2) | [7] | 176.20 | [7] | 3.534 | [7] | Fb |
| 9 | C1–H1C | 0.980 | 2.63(2) | [8] | 144.14 | [8] | 3.463 | [8] | Fb |
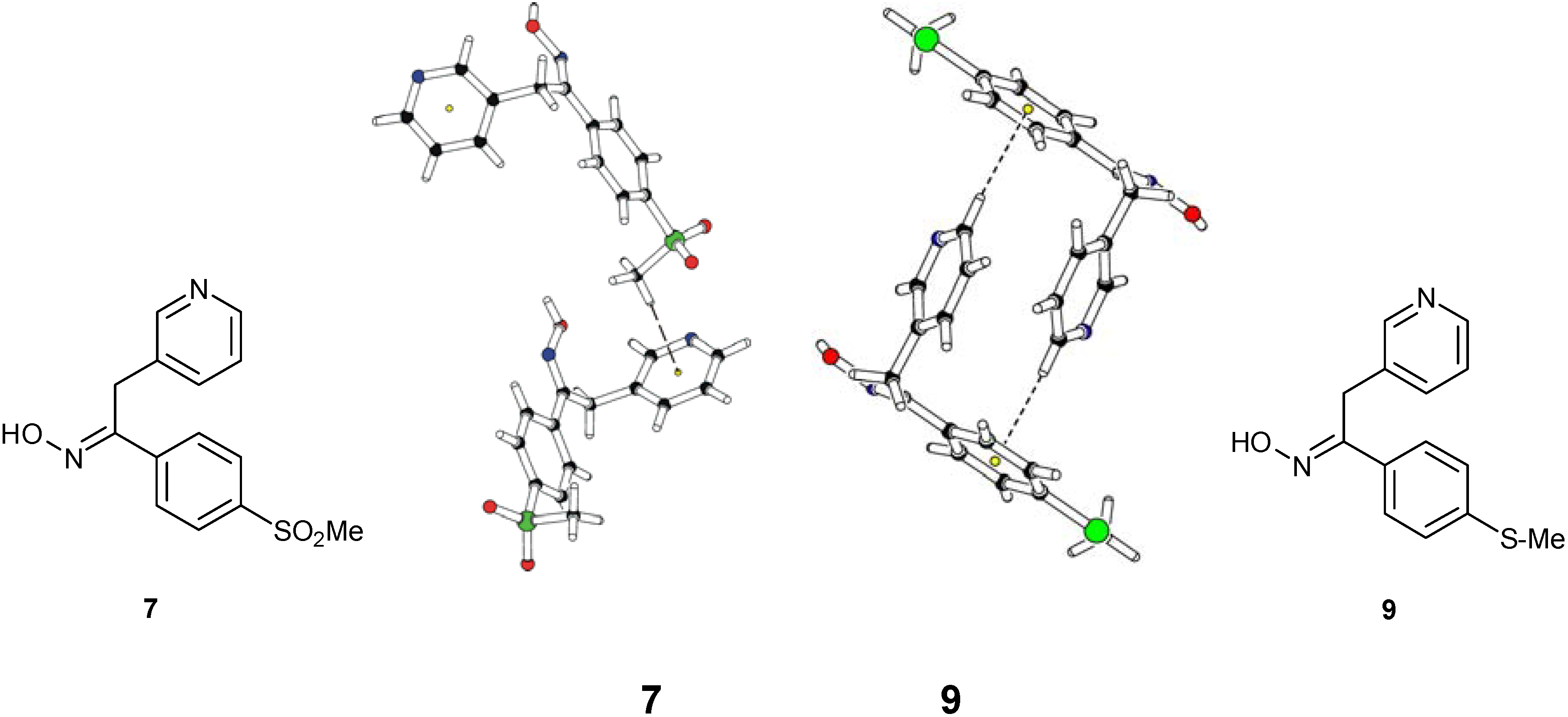
Conclusions
Experimental
General
Synthesis of 1-aryl-2-(aryl/alkyl)ethanones 1 and 2: 2-Cyclohexyl-1-(3-fluorophenyl)ethanone (1)
1-[4-(Methylthio)phenyl]-2-phenylethanone (2)
N-Methoxy-N-methyl-2-pyridin-3-ylacetamide (15)
Diphenyl {anilino[4-(methylsulfonyl)phenyl]methyl}phosphonate (19)
Diphenyl {anilino[4-(methylthio)phenyl]methyl}phosphonate (23)
Synthesis of 1-aryl-2-(3-pyridyl)ethanones 3–5. General procedure
Syntheis of ketoximes 6–9. General procedure
Single-crystal X-ray determination of ketoximes 6, 7 and 9
Supplementary Material
Acknowledgements
References and Notes
- Veeramaneni, V. R.; Pal, M.; Yeleswarapu, K. R. A high speed parallel synthesis of 1,2-diaryl-1-ethanones via a clean-chemistry C–C bond formation reaction. Tetrahedron 2003, 59, 3283–3290. [Google Scholar] [CrossRef] Katritzky, A. R.; Abdel-Fattah, A. A. A.; Akhmedova, R. G. A convenient access to 1-substituted-2-azinyl-1-ethanones via acylation of alkylated azines with N-acylbenzotriazoles. Arkivoc 2005, 329–338. [Google Scholar]
- Merkul, E.; Müller, J. J. A new consecutive three-component oxazole synthesis by an amidation–coupling–cycloisomerization (ACCI) sequence. Chem. Commun. 2006, 4817–4819. [Google Scholar] [CrossRef]
- Davies, I. W.; Marcoux, J. F.; Corley, E. G.; Journet, M.; Cai, D. -W.; Palucki, M.; Wu, J.; Larsen, R. D.; Rossen, K.; Pye, P. J.; DiMichele, L.; Dormer, P.; Reider, P. J. A Practical Synthesis of a COX-2-Specific Inhibitor. J. Org. Chem. 2000, 65, 8415–8420. [Google Scholar] [CrossRef]
- Hashimoto, H.; Imamura, K.; Haruta, J.; Wakitani, K. 4-(4-Cycloalkyl/aryl-oxazol-5-yl)benzenesulfonamides as Selective Cyclooxigenase-2 Inhibitors: Enhancement of the Selectivity by Introduction of a Fluorine Atom and Identification of a Potent, Highly Selective, and Orally Active COX-2 Inhibitor JTE-522. J. Med. Chem. 2002, 45, 1511–1517. [Google Scholar] [CrossRef] Talley, J. J.; Brown, D. L.; Carter, J. S.; Graneto, M. J.; Koboldt, C. M.; Masferrer, J. L.; Perkins, W. E.; Rogers, R. S.; Shaffer, A. F.; Zhang, Y. Y.; Zweifel, B. S.; Seibert, K. 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, Valdecoxib: A Potent and Selective Inhibitor of COX-2. J. Med. Chem. 2000, 43, 775–777. [Google Scholar] [CrossRef]
- Marcoux, J. F.; Corley, E. G.; Rossen, K.; Pye, P. J. M.; Wu, J.; Robbins, M. A.; Davies, I. W.; Larsen, R. D.; Reider, P. J. Annulation of Ketones with Vinamidinium Hexafluorophosphate Salts: An Efficient Preparation of Trisubstituted Pyridines. Org. Lett. 2000, 2, 2339–2341, and references cited therein. [Google Scholar] [CrossRef]
- Etter, M. C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 4–76. [Google Scholar] Steiner, T. C-H···O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Aakeröy, C. B.; Beatty, A. M.; Leinen, D. S. The Oxime Functionality: A Versatile Tool for Supramolecular Assembly of Metal-Containing Hydrogen-Bonded Architectures. J. Am. Chem. Soc. 1998, 120, 7383–7384, and references cited therein. [Google Scholar] [CrossRef] Aakeröy, C. B.; Beatty, A. M.; Leinen, D. S. New building blocks for crystal engineering. Syntheses and crystal structures of oxime-substituted pyridines. CrystEngComm. 2000, 2, 1–6, and references cited therein. [Google Scholar] [CrossRef] Bruton, E. A.; Brammer, L.; Pigge, F. C.; Aakeröy, C. B.; Leinen, D. S. Hydrogen bond patterns in aromatic and aliphatic dioximes. New J. Chem. 2003, 27, 1084–1094. [Google Scholar] [CrossRef] Aakeröy, C. B.; Desper, J.; Scott, B. M. T. Balancing supramolecular reagents for reliable formation of co-crystals. ChemCommun. 2006, 1445–1447. [Google Scholar] Aakeröy, C. B.; Salmon, D. J.; Smith, M. M.; Desper, J. Cyanopheniloximes: Reliable and Versatile Tools for Hydrogen-Bond Directed Supramolecular Synthesis of Cocrystals. Crystal Growth & Desig 2006, 6, 1033–1042, and references cited therein. [Google Scholar] Aakeröy, C. B.; Schultheiss, N.; Desper, J. A pyridyl-functionalized cavitand. Starting point for hydrogen-bond driven assembly for heterodimeric capsules. CrystEngComm. 2006, 8, 502–506. [Google Scholar] [CrossRef]
- Mazik, M.; Bläser, D.; Boese, R. Programmed Construction of Discrete Self-Assembled Cyclic Aggregates. Chem. Eur. J. 2000, 6, 2865–2873. [Google Scholar] [CrossRef] Mazik, M.; Bläser, D.; Boese, R. α,β-Unsaturated Ketoximes Carrying a Terminal Pyridine or Quinoline Subunit as Building Blocks for Supramolecular Syntheses. J. Org. Chem. 2005, 70, 9115–9122. [Google Scholar] [CrossRef] Marsman, A. W.; van Walree, C. A.; Havenith, R. W. A.; Jenneskens, L. W.; Lutz, M.; Spek, A. L.; Egbertus, T. G.; Lutz, E. T. G.; van der Maas, J. H. Infinite, undulating chains of intermolecularly hydrogen bonded (E,E)-2,2-dimethylcyclohexane-1,3-dione dioximes in the solid state. A single crystal X-ray, charge density distribution and spectroscopic study. J. Chem. Soc., Perkin Trans. 2000, 501–510. [Google Scholar]
- Chertanova, L.; Pascard, C. X-ray Investigation of Glyoxime Derivatives. V. Two Isomers of 2-(Thienyl)glyoxime. A Database Study of the Geometry and Hydrogen Bonding of the Oxime Group. Acta Crys. 1994, B50, 708–716. [Google Scholar] [CrossRef] Maurin, J. K. Oxime–Carboxyl Hydrogen Bonds: the Preferred Interaction Determining Crystal Packing of ‘Carboxyoximes’. Acta Crys. 1998, B54, 866–871. [Google Scholar] [CrossRef] Hoogesteger, F. J.; Jenneskensa, L. W.; Veldmanh, H. K. N.; Spekh, A. L. Self-Complementary Hydrogen Bonding of 1,1’-Bicyclohexylidene-4,4’-dione Dioxime. Formation of a Non-Covalent Polymer. Tetrahedron 1996, 52, 1773–1784. [Google Scholar] [CrossRef]
- Nishio, M. CH/π hydrogen bonds in crystals. CrystEngComm. 2004, 6, 130–158. [Google Scholar] [CrossRef] Nishio, M. CH/π hydrogen bonds in organic reactions. Tetrahedron 2005, 61, 6923–6950. [Google Scholar] [CrossRef]
- For examples of ethanones of type [Ar-CH2-CO-Ar′] [1,4] required for synthesis of selective COX-2 inhibitors, see: Singh, S. K.; Saibaba, V.; Ravikumar, V.; Rudrawar, S. V.; Daga, P.; Rao, C. S.; Akhila, V.; Hegde, P.; Rao, Y. K. Synthesis and biological evaluation of 2,3-diarylpyrazines and quinoxalines as selective COX-2 inhibitors. Bioorg. Med. Chem. 2004, 12, 1881–1893. [Google Scholar] [CrossRef] Baruah, B.; Dasu, K.; Vaitilingam, B.; Vanguri, A.; Casturi, S. R.; Yeleswarapu, K. R. 1,2-Diaryl-1-ethanone and pyrazolo [4,3-c] quinoline-4-one as novel selective cyclooxigenase-2-inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 445–448. [Google Scholar] [CrossRef]
- For key intermediates of type [Ar-CO-CH2-(3-Py)], see: Chen, H. Y.; Kim, S.; Wu, J. Y.; Birzin, E. T.; Chan, W.; Yang, Y. T.; Dahllund, J.; DiNinno, F.; Rohrer, S. P.; Schaeffer, J. M.; Hammond, M. L. Estrogen receptor ligands. Part 3: The SAR of dihydrobenzoxathiin SERMs. Bioorg. Med. Chem. Lett. 2004, 14, 2551–2554. [Google Scholar] [CrossRef] Biftu, T.; Feng, D.; Ponpipom, M.; Girotra, N.; Liang, G. -L.; Qian, X.; Bugianesi, R.; Simeone, J.; Chang, L.; Gurnett, A.; Liberator, P.; Dulski, P.; Leavitt, P. S.; Crumley, T.; Misura, A.; Murphy, T.; Rattray, S.; Samaras, S.; Tamas, T.; Mathew, J.; Brown, C.; Thompson, D.; Schmatz, D.; Fisher, M.; Wyvratt, M. Synthesis and SAR of 2,3-diarylpyrrole inhibitors of parasite cGMP-dependent protein kinase as novel anticoccidial agents. Bioorg. Med. Chem. Lett. 2005, 15, 3296–3301. [Google Scholar] [CrossRef]
- Wehmeyer, R. M.; Rieke, R. D. Direct formation of functionalized ketones via the coupling of functionalized organocopper reagents with acid chlorides. Tetrahedron Lett. 1988, 29, 4513–4516. [Google Scholar] [CrossRef] Zhu, L.; Wehmeyer, R. M.; Rieke, R. D. The Direct Formation of Functionalized Alkyl(aryl)zinc Halides by Oxidative Addition of Highly Reactive Zinc with Organic Halides and Their Reactions with Acid Chlorides, α,β-Unsaturated Ketones, and Allylic, Aryl, and Vinyl Halides. J. Org. Chem. 1991, 56, 1445–1453. [Google Scholar] [CrossRef] Rieke, R. D.; Hanson, M. V.; Brown, J. D. Direct Formation of Secondary and Tertiary Alkylzinc Bromides and Subsequent Cu(I)-Mediated Couplings. J. Org. Chem. 1996, 61, 2726–2730. [Google Scholar] [CrossRef]
- Singh, J.; Satyamurthi, N.; Aidhen, I. S. The Growing Synthetic Utility of Weinreb’s Amide. J. Prakt. Chem. 2000, 342, 340–347. [Google Scholar] [CrossRef]
- Maryanoff, B. E.; Reitz, A. B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–97. [Google Scholar] [CrossRef] Journet, M.; Cai, D.; Larsen, R. D.; Reider, P. J. An Improved and Practical Procedure for the Synthesis of Substituted Phenylacetylpyridines. Tetrahedron Lett. 1998, 39, 1717–1720, and references cited therein. [Google Scholar] [CrossRef] Piva, O.; Comesse, S. Tandem Michael-Wittig-Horner Reaction: One-Pot Synthesis of δ-Substituted α,β-Unsaturated Carboxylic Acid Derivatives - Application to a Concise Synthesis of (Z)- and (E)-Ochtoden-1-al. Eur. J. Org. Chem. 2000, 2417–2424. [Google Scholar] [CrossRef]
- Kolehmainen, E.; Gawinecki, R.; Osmialowski, B.; Trzebiatowska, K. NMR Spectral Assignment of Substituted Salicylaldoximes by Inverse Pulse Techniques with z-Gradient Selection: Correlation of NMR Parameters with Substituent Constants. Mag. Res. Chem. 1997, 35, 778–784, and references cited therein. [Google Scholar] [CrossRef]
- Oximes 6, 7 and 9: In all crystal structures -space group P21/c-, the asymmetric unit contained one independent oxime molecule.
- Zhu, L.; Rieke, R. D. A facile method for the preparation of functionalized 2,3-disubstituted-1,3-butadienes. Tetrahedron Letters 1991, 32, 2865–2866. [Google Scholar] [CrossRef]
- Velarde-Ortiz, R.; Guijarro, A.; Rieke, R. D. Electrophilic amination of organozinc halides. Tetrahedron Letters 1998, 38, 9157–9160. [Google Scholar] [CrossRef]
- Bruker. COLLECT data collection software; Bruker AXS: Delft, The Netherlands, 2004. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Blessing, R. H. An Empirical Correction for Absorption Anisotropy. Acta Cryst. 1995, A51, 33–38. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and Refinement of Crystal Structures with SIR92. J. Appl. Cryst. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G. M. SHELXL-97; University of Göttingen: Germany, 1997. [Google Scholar]
- Sample Availability: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Alcalde, E.; Mesquida, N.; Alvarez-Rúa, C.; Cuberes, R.; Frigola, J.; García-Granda, S. 1,2-Diaryl(3-pyridyl)ethanone Oximes. Intermolecular Hydrogen Bonding Networks Revealed by X-ray Diffraction. Molecules 2008, 13, 301-318. https://doi.org/10.3390/molecules13020301
Alcalde E, Mesquida N, Alvarez-Rúa C, Cuberes R, Frigola J, García-Granda S. 1,2-Diaryl(3-pyridyl)ethanone Oximes. Intermolecular Hydrogen Bonding Networks Revealed by X-ray Diffraction. Molecules. 2008; 13(2):301-318. https://doi.org/10.3390/molecules13020301
Chicago/Turabian StyleAlcalde, Ermitas, Neus Mesquida, Carmen Alvarez-Rúa, Rosa Cuberes, Jordi Frigola, and Santiago García-Granda. 2008. "1,2-Diaryl(3-pyridyl)ethanone Oximes. Intermolecular Hydrogen Bonding Networks Revealed by X-ray Diffraction" Molecules 13, no. 2: 301-318. https://doi.org/10.3390/molecules13020301
APA StyleAlcalde, E., Mesquida, N., Alvarez-Rúa, C., Cuberes, R., Frigola, J., & García-Granda, S. (2008). 1,2-Diaryl(3-pyridyl)ethanone Oximes. Intermolecular Hydrogen Bonding Networks Revealed by X-ray Diffraction. Molecules, 13(2), 301-318. https://doi.org/10.3390/molecules13020301