Spectral Data of Two New Asymmetric Sesquiterpene Alcohols: (14R)-β-Oplopenol and (14S)-β-Oplopenol

Abstract
:Introduction
Results and Discussion


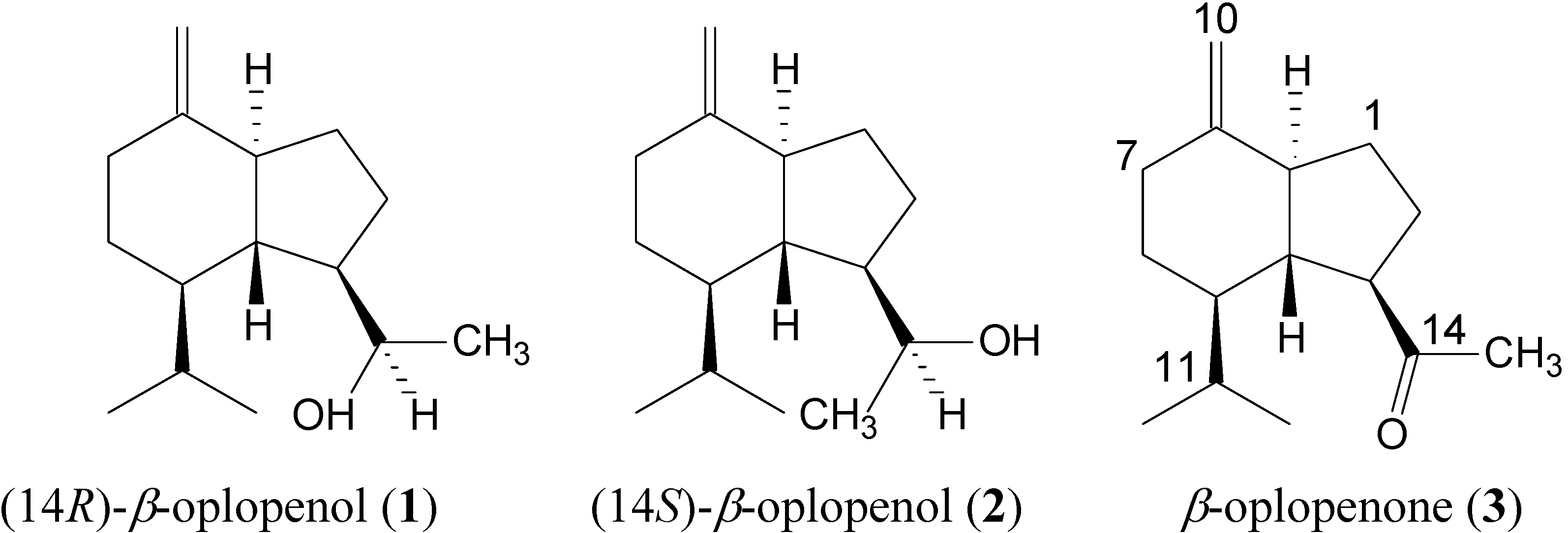{kind=link}
| C | ||||||
|---|---|---|---|---|---|---|
| 1 | 27.79 | CH2 | 1.34 | m | 9, 3, 2 | 2, 3, 9 |
| 1.43 | m | |||||
| 2 | 22.62 | CH2 | 1.62 | dtd (20.0; 7.4; 12.8) | 14, 9, 3, 1 | 1, 3, 4 |
| 1.81 | m | |||||
| 3 | 49.02 | CH | 1.77 | m | 14, 15, 6, 4, 2 | 2, 5, 1 |
| 4 | 50.23 | CH | 1.25 | m | 14, 11, 9, 6, 5, 3 | 3, 9, 1 |
| 5 | 50.09 | CH | 1.34 | m | 13, 12, 11, 7, 6, 4 | 6, 3 |
| 6 | 26.95 | CH2 | 1.10 | dq (4.4; 13.0) | 11, 7, 5, 4 | 5, 7 |
| 1.75 | m | |||||
| 7 | 35.32 | CH2 | 1.95 | m | 10, 6, 8 | 10, 6 |
| 2.35 | ddd (2.4; 4.3; 13.4) | 6, | ||||
| 8 | 151.98 | C | - | - | 9, 7, 6, 5, 1 | - |
| 9 | 52.50 | CH | 1.83 | m | 10, 7, 5, 4, 3, 2, 1 | 10, 4 |
| 10 | 102.70 | CH2 | 4.54 | q (1.7) | 9, 7 | 9 |
| 4.62 | q (1.7) | 7 | ||||
| 11 | 28.50 | CH | 1.95 | m | 13, 12, 6, 5 | 12, 13 |
| 12 | 21.95 | CH3 | 0.95 | d (6.9) | 13, 11, 5 | 11 |
| 13 | 15.85 | CH3 | 0.78 | d (6.9) | 12, 11, 5 | 11 |
| 14 | 68.71 | CH | 3.98 | q (6.3) | 15, 4, 3, 2 | 15 |
| 15 | 23.01 | CH3 | 1.18 | d (6.3) | 14, 4, 3, 2 | 14 |
| C | ||||||
|---|---|---|---|---|---|---|
| 1 | 28.12 | CH2 | 1.33 | m | 9, 2 | 9, 2 |
| 1.72 | m | 9, 2 | ||||
| 2 | 23.44 | CH2 | 1.65 | dtd (18.1; 7.0; 11.0) | 14, 1 | 3, 1 |
| 1.77 | m | |||||
| 3 | 48.79 | CH | 2.14 | ddt (7.0; 11.0; 3.6) | 15, 4, 2, 1 | 2,4,14, |
| 4 | 51.72 | CH | 0.87 | q (11.0) | 9, 6, 2, 1 | 9, 5, 3 |
| 5 | 49.94 | CH | 1.34 | m | 13, 12, 6, 4, 7 | 6, 4 |
| 6 | 26.95 | CH2 | 1.01 | dq (13.0; 4.5) | 11, 7 | 7, 5 |
| 1.73 | m | |||||
| 7 | 35.18 | CH2 | 1.96 | m | 10, 6 | 6, 10 6 |
| 2.34 | ddd (2.4; 4.5; 13.0) | |||||
| 8 | 152.03 | C | - | - | 9, 7, 1 | |
| 9 | 52.69 | CH | 1.86 | m | 10, 7, 5, 4, 1, 2, 6 | 10, 4 |
| 10 | 102.55 | CH2 | 4.54 | q (1.7) | 11, 7 | 9 |
| 4.61 | q (1.7) | 7 | ||||
| 11 | 28.69 | CH | 1.94 | m | 13, 12 | 13, 12, 6 |
| 12 | 21.95 | CH3 | 0.96 | d (6.8) | 13, 6 | 11 |
| 13 | 15.83 | CH3 | 0.70 | d (6.8) | 12, 4 | 11 |
| 14 | 69.56 | CH | 4.03 | dq (3.9; 6.3) | 15, 4, 2 | 3, 15 |
| 15 | 16.89 | CH3 | 1.13 | d (6.3) | 12 | 14 |
Experimental
General
Reduction
Acknowledgements
References
- Takeda, K.; Minato, H.; Ishikawa, M. Studies on sesquiterpenoids-XII- Structure and absolute configuration of oplopanone, a new sesquiterpene from oplopanax japonicus (Nakai). Tetrahedron Suppl. 1966, 7, 219–225. [Google Scholar] [CrossRef]
- David, J.R. The biology and chemistry of the Compositae; Heywood, V.H., Harborne, J.B., Turner, B.L, Eds.; Academic Press: London-New York-San Francisco, 1977; Vol. 2, pp. 831–850. [Google Scholar]
- Abdel Aal, M.; Bohlmann, F.; Sarg, T.; El-Domiaty, M.; Nordenstam, B. Oplopane derivatives from Acrisione denticulata. Phytochemistry 1988, 27, 2599–2602. [Google Scholar]
- Joseph-Nathan, P.; Villagómez, J.R.; Román, L.U.; Hernández, J.D. An oplopane from Senecio mexicanus. Phytochemistry 1989, 28, 1207–1209. [Google Scholar] [CrossRef]
- Joseph-Nathan, P.; Villagómez, J.R.; Román, L.U.; Hernández, J.D. Oplopanes from the leaves of Senecio mexicanus. Phytochemistry 1990, 29, 977–979. [Google Scholar] [CrossRef]
- Hayashi, K. Oplopane sesquiterpenes from Petasites palmatus. Phytochemistry 1989, 28, 3373–3376. [Google Scholar] [CrossRef]
- Arciniegas, A.; Pérez-Castorena, A.L.; Reyes, S.; Contreras, J.L.; De Vivar, A.R. New oplopane and eremophilane derivatives from Robinsonecio gerberifolius. J. Nat. Prod. 2003, 66, 225–229. [Google Scholar]
- Xie, W.D.; Zhang, Q.; Li, P.L.; Jia, Z.J. Two triterpenoids and other constituents from Petasites tricholobus. Phytochemistry 2005, 66, 2340–2345. [Google Scholar] [CrossRef]
- Paolini, J.; Nasica, E.; Desjobert, J.M.; Muselli, A.; Bernardini, A.F.; Costa, J. Analysis of volatile constituents isolated by hydrodistillation and headspace-solid phase microextraction from Adenostyles briquetii Gamisans. Phytochem. Ana 2007, in press. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C. Neue furanoeremophilane und andere sesquiterpene aus vertretern der gattung Euryops. Phytochemistry 1978, 17, 1135–1153. [Google Scholar] [CrossRef]
- Wratten, S.J.; Faulkner, D.J. Metabolites of the red alga Laurencia subopposita. J. Org. Chem. 1977, 42, 3343–3349. [Google Scholar] [CrossRef]
- De Pascual, J.; Feliciano, A.S.; Miguel de Corral, J.M.; Barrero, A.F. Terpenoids from Juniperus Sabina. Phytochemistry 1983, 22, 300–301. [Google Scholar] [CrossRef]
- Piers, E.; Gavai, A.V. Convenient (Z)-ethylidenecyclopentane annulation sequences. Total synthesis of (±)-oplopanone, (±)-8-epi-oplopanone, and (±)-anhydro-oplopanone. Tetrahedron Lett. 1986, 27, 313–316. [Google Scholar] [CrossRef]
- Joseph-Nathan, P.; Villagomez, J.R.; Rojas-Gardida, M.; Roman, L.U.; Hernandez, J.D. Minor oplopanes from Senecio mexicanus. Phytochemistry 1989, 28, 2397–2401. [Google Scholar]
- Piers, E.; Gavai, A.V. A (Z)-Ethylidenecyclopentane annulation method. Total syntheses of (±)-Anhydrooplopanone, (±)-Oplopanone, and (±)-8-epi-Oplopanone. J. Org. Chem. 1990, 55, 2380–2390. [Google Scholar] [CrossRef]
- König, W.A.; Hochmuth, D.H.; Joulain, D. Terpenoids and Related Constituents of Essential Oils; MassFinder 2.1 Library; Institute of Organic Chemistry: Hamburg, Germany, 2001. [Google Scholar]
- Karplus, M.J. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Karplus, M.J. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Van Den Dool, H.; Kratz, P.D. A generalization of the retention index system including linear temperature programmed gas-liquid partition chromatography. J. Chromatogr. 1963, 11, 463–471. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Paolini, J.; Desjobert, J.-M.; Muselli, A.; Costa, J. Spectral Data of Two New Asymmetric Sesquiterpene Alcohols: (14R)-β-Oplopenol and (14S)-β-Oplopenol. Molecules 2008, 13, 1004-1010. https://doi.org/10.3390/molecules13041004
Paolini J, Desjobert J-M, Muselli A, Costa J. Spectral Data of Two New Asymmetric Sesquiterpene Alcohols: (14R)-β-Oplopenol and (14S)-β-Oplopenol. Molecules. 2008; 13(4):1004-1010. https://doi.org/10.3390/molecules13041004
Chicago/Turabian StylePaolini, Julien, Jean-Marie Desjobert, Alain Muselli, and Jean Costa. 2008. "Spectral Data of Two New Asymmetric Sesquiterpene Alcohols: (14R)-β-Oplopenol and (14S)-β-Oplopenol" Molecules 13, no. 4: 1004-1010. https://doi.org/10.3390/molecules13041004