Investigating Biological Activity Spectrum for Novel Styrylquinazoline Analogues


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
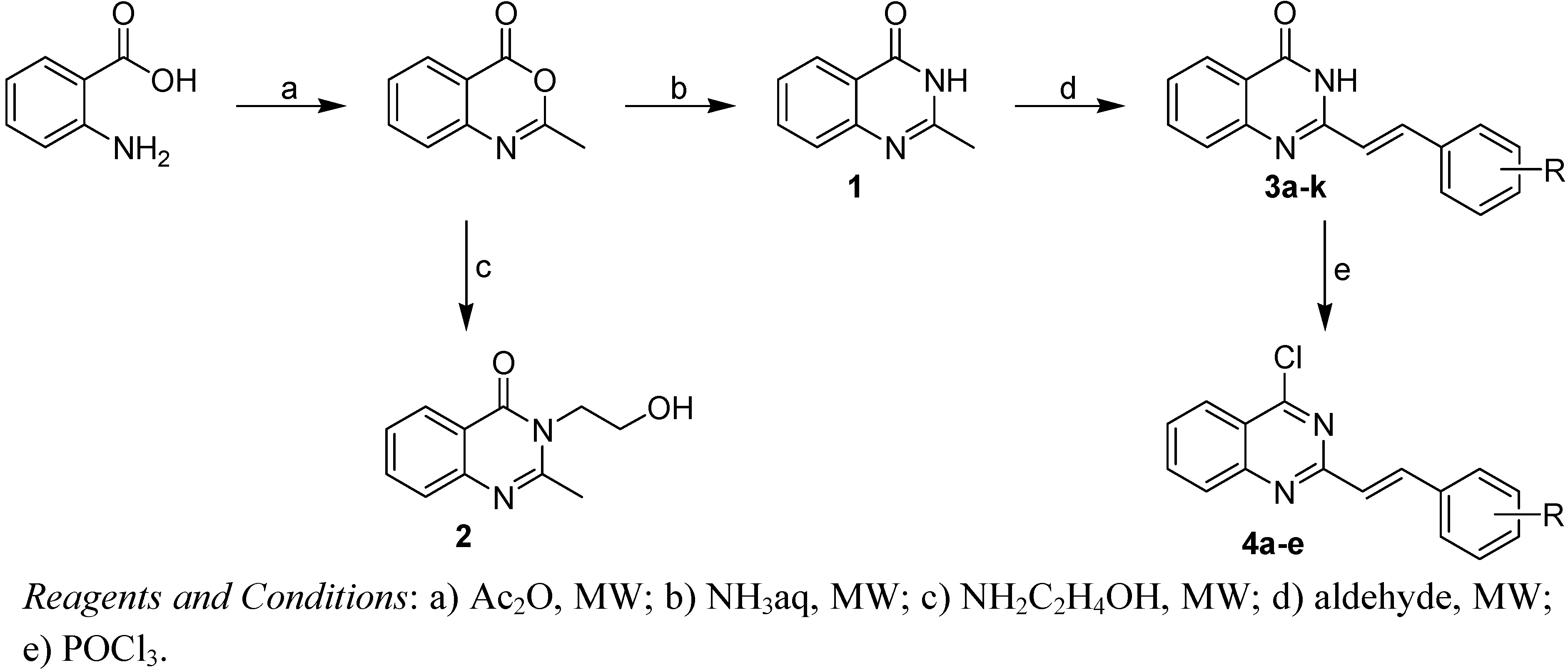
2.1. Chemistry

2.2. Lipophilicity

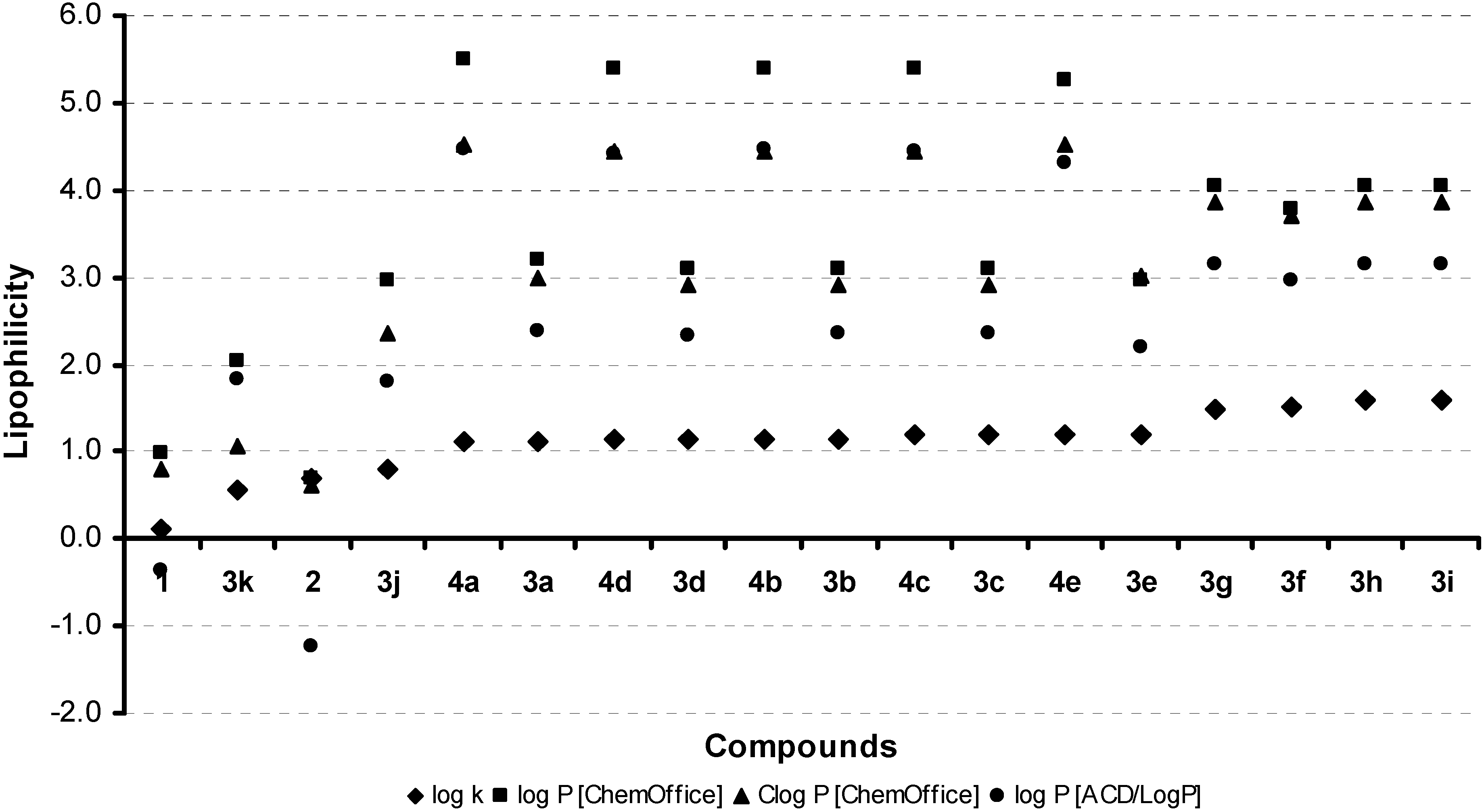{kind=link}
{kind=link}
| Comp. | R | log k | log P/Clog P ChemOffice | log P ACD/LogP | σ [60] | MR [60] |
| 1 |  | 0.1170 | 0.98 / 0.804 | -0.36 ± 0.59 | – | – |
| 2 |  | 0.7047 | 0.69 / 0.6149 | -1.23 ± 0.69 | – | – |
 | ||||||
| 3a | H | 1.1148 | 3.21 / 2.997 | 2.37 ± 0.61 | 0.0 | 0.0 |
| 3b | 2-OCH3 | 1.1509 | 3.09 / 2.916 | 2.35 ± 0.62 | -0.390 [61] | 6.5 |
| 3c | 3-OCH3 | 1.1837 | 3.09 / 2.916 | 2.35 ± 0.62 | 0.115 | 6.5 |
| 3d | 4-OCH3 | 1.1351 | 3.09 / 2.916 | 2.32 ± 0.62 | -0.268 | 6.5 |
| 3e | 2,4-OCH3 | 1.1982 | 2.96 / 3.005 | 2.20 ± 0.62 | -0.658 | 13.0 |
| 3f | 3-Cl | 1.5059 | 3.77 / 3.710 | 2.96 ± 0.61 | 0.373 | 4.8 |
| 3g | 2-Br | 1.4830 | 4.04 / 3.860 | 3.14 ± 0.64 | – | 7.6 |
| 3h | 3-Br | 1.5904 | 4.04 / 3.860 | 3.14 ± 0.64 | 0.391 | 7.6 |
| 3i | 4-Br | 1.5927 | 4.04 / 3.860 | 3.14 ± 0.64 | 0.232 | 7.6 |
| 3j | 4-CHO | 0.8005 | 2.96 / 2.350 | 1.79 ± 0.63 | 1.030 | 5.3 |
| 3k | 2,3,4-OH | 0.5510 | 2.05 / 1.066 | 1.84 ± 0.63 | – | 4.5 |
 | ||||||
| 4a | H | 1.1088 | 5.50 / 4.51522 | 4.47 ± 0.56 | 0.0 | 0.0 |
| 4b | 2-OCH3 | 1.1497 | 5.38 / 4.43422 | 4.47 ± 0.57 | -0.390 [61] | 6.5 |
| 4c | 3-OCH3 | 1.1827 | 5.38 / 4.43422 | 4.44 ± 0.57 | 0.115 | 6.5 |
| 4d | 4-OCH3 | 1.1337 | 5.38 / 4.43422 | 4.41 ± 0.57 | -0.268 | 6.5 |
| 4e | 2,4-OCH3 | 1.1953 | 5.25 / 4.52322 | 4.30 ± 0.57 | -0.658 | 13.0 |

2.3. Inhibition of photosynthetic electron transport (PET) in spinach chloroplasts
| Comp. | PET inhibition IC50 [μmol/L] | MIC/IC90 [µg/mL] | |||
|---|---|---|---|---|---|
| M. smegmatis | M. absessus | M. kansasii | M. avium complex | ||
| 1 | a | >300 | >300 | >300 | >300 |
| 2 | 362 | >300 | >300 | >300 | >300 |
| 3a | a | >300 | >300 | >300 | >300 |
| 3b | a | >100 | >100 | >100 | >100 |
| 3c | a | >100 | >100 | >100 | >100 |
| 3d | 693 | >100 | >100 | >100 | >100 |
| 3e | 391 | >100 | 80 | 20 | 80 |
| 3f | 1034 | >300 | >300 | >300 | >300 |
| 3g | a | >100 | >100 | >100 | >100 |
| 3h | 561 | >100 | >100 | >100 | >100 |
| 3i | 665 | >100 | >100 | >100 | >100 |
| 4a | 285 | >100 | >100 | >100 | >100 |
| 4b | a | >100 | 80 | 60 | 80 |
| 4c | 303 | >100 | >100 | 60 | >100 |
| 4d | 390 | >100 | >100 | >100 | >100 |
| 4e | 508 | >100 | >100 | >100 | >100 |
| DCMU | 1.9 | – | – | – | – |
| INH | – | 39 | >100 | <10 | <10 |




2.4. In vitro antimycobacterial evaluation
2.5. In vitro antifungal susceptibility testing
3. Conclusions
4. Experimental
4.1. General
4.2. Synthesis
4.2.1. General procedures of synthesis of Compounds 3a-k
4.2.2. General procedures of synthesis of Compounds 4a-e
4.3. Lipophilicity HPLC determination (capacity factor k / calculated log k)
4.4. Lipophilicity calculations
4.5. Study of inhibition photosynthetic electron transport (PET) in spinach chloroplasts
4.6. In vitro antimycobacterial evaluation
4.7. In vitro antifungal susceptibility testing
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Roth, H.J.; Fenner, H. Arzneistoffe, 3rd ed; Deutscher Apotheker Verlag: Stuttgart, Germany, 2000; pp. 51–114. [Google Scholar]
- Harris, C.R.; Thorarensen, A. Advances in the discovery of novel antibacterial agents during the year 2002. Curr. Med. Chem. 2004, 11, 2213–2243. [Google Scholar]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; Williams, P.; de Chaffoy, D.; Huitric, E.; Hoffner, S.; Cambau, E.; Truffot-Pernot, C.; Lounis, N.; Jarlier, V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar]
- Vangapandu, S.; Jain, M.; Jain, R.; Kaur, S.; Singh, P.P. Ring-substituted quinolines as potential anti-tuberculosis agents. Bioorg. Med. Chem. 2004, 12, 2501–2508. [Google Scholar]
- Carta, A.; Piras, S.; Palomba, M.; Jabes, D.; Molicotti, P.; Zanetti, S. Anti-mycobacterial activity of quinolones. Triazoloquinolones a new class of potent anti-mycobacterial agents. Anti-Infective Agents Med. Chem. 2008, 7, 134–147. [Google Scholar]
- Sissi, C.; Palumbo, M. The quinolone family: From antibacterial to anticancer agents. Curr. Med. Chem. Anti-Canc. Agents 2003, 3, 439–450. [Google Scholar] [CrossRef]
- Bossu, E.; Agliano, A.M.; Desideri, N.; Sestili, I.; Porra, R.; Grandilone, M.; Quaglia, M.G. LTB4 as marker of 5-LO inhibitory activity of two new N-ethoxycarbonyl-4-quinolones. J. Pharm. Biomed. Anal. 1999, 19, 539–549. [Google Scholar]
- Ko, T.C.; Hour, M.J.; Lien, J.C.; Teng, C.M.; Lee, K.H.; Kuo, S.C.; Huang, L.J. Synthesis of 4-alkoxy-2-phenylquinoline derivatives as potent antiplatelet agents. Bioorg. Med. Chem. Lett. 2001, 11, 279–282. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Vichova, P.; Jun, D.; Raich, I.; O´Connor, R.; Clynes, M. Synthesis of (2E)-2-methyl-3-(4-{[4-(quinolin-2-ylmethoxy)phenyl]sulfanyl}phenyl)prop-2-enoic acid (VUFB 20609) and 2-methyl-3-(4-{[4-(quinolin-2-ylmethoxy)phenyl]sulfanyl}phenyl)propionic acid (VUFB 20584) as potential antileukotrienic agents. J. Pharm. Pharmacol. 2004, 56, 783–794. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Vichova, P.; Jun, D.; Raich, I.; O´Connor, R.; Clynes, M. Preparation of 2-(4-{[4-(quinolin-2-ylmethoxy)phenyl]sulfanyl}phenyl)propionic acid (VUFB 20615) and 2-methyl-2-(4-{[4-(quinolin-2-ylmethoxy)phenyl]sulfanyl}phenyl)propionic acid (VUFB 20623) as potential antileukotrienic agents. Curr. Org. Chem. 2004, 8, 1235–1243. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Opletalova, V.; Hartl, J. 5-Lipoxygenase, leukotrienes biosynthesis and potential antileukotrienic agents. Curr. Med. Chem. 2006, 13, 117–129. [Google Scholar] [CrossRef]
- Polanski, J.; Zouhiri, F.; Jeanson, L.; Desmaele, D.; d’Angelo, J.; Mouscadet, J.F.; Gieleciak, R.; Gasteiger, J.; Le Bret, M. Use of Kohonen neural network for rapid screening of ex vivo anti-HIV activity of styrylquinolines. J. Med. Chem. 2002, 45, 4647–4654. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Tabak, D.; Podeszwa, B.; Gieleciak, R.; Bak, A.; Palka, A.; Magdziarz, T. Analogues of the styrylquinoline and styrylquinazoline HIV-1 integrase inhibitors: Design and synthetic problems. Acta Poloniae Pharm. Drug Res. 2004, 61, 3–4. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Finster, J.; Mouscadet, J.F.; Le Bret, M. 5-Hydroxy-8-nitro-6-quinaldic acid as a novel molecular scaffold for HIV-1 integrase inhibitors. Lett. Drugs Des. Disc. 2006, 3, 175–178. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Mouscadet, J.F.; Le Bret, M. Fragment based approach for the investigation of HIV-1 integrase inhibition. Lett. Drugs Des. Disc. 2007, 4, 99–105. [Google Scholar]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Buchta, V.; Kralova, K. Quinaldine derivatives: Preparation and biological activity. Med. Chem. 2005, 1, 591–599. [Google Scholar]
- Musiol, R.; Jampilek, J.; Buchta, V.; Niedbala, H.; Podeszwa, B.; Palka, A.; Majerz-Maniecka, K.; Oleksyn, B.; Polanski, J. Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar]
- Musiol, R.; Jampilek, J.; Kralova, K.; Richardson, D.R.; Kalinowski, D.; Podeszwa, B.; Finster, J.; Niedbala, H.; Palka, A.; Polanski, J. Investigating biological activity spectrum fornovel quinoline analogues. Bioorg. Med. Chem. 2007, 15, 1280–1288. [Google Scholar]
- Musiol, R.; Tabak, D.; Niedbala, H.; Podeszwa, B.; Jampilek, J.; Kralova, K.; Dohnal, J.; Finster, J.; Mencel, A.; Polanski, J. Investigating biological activity spectrum for novel quinoline analogues 2: Hydroxyquinolinecarboxamides with photosynthesis inhibiting activity. Bioorg. Med. Chem. 2008, 16, 4490–4499. [Google Scholar]
- Jampilek, J.; Musiol, R.; Pesko, M.; Kralova, K.; Vejsova, M.; Carroll, J.; Coffey, A.; Finster, J.; Tabak, D.; Niedbala, H.; Kozik, V.; Polanski, J.; Csollei, J.; Dohnal, J. Ring-substituted 4-hydroxy-1H-quinolin-2-ones: Preparation and biological activity. Molecules 2009, 14, 1145–1159. [Google Scholar]
- Podeszwa, B.; Niedbala, H.; Polanski, J.; Musiol, R.; Tabak, D.; Finster, J.; Serafin, K.; Wietrzyk, J.; Boryczka, S.; Mol, W.; Jampilek, J.; Dohnal, J.; Kalinowski, D.; Richardson, D.R. Investigating the antiproliferative activity of quinoline-5,8-dione analogues on tumour cell lines. Bioorg. Med. Chem. Lett. 2007, 17, 6138–6141. [Google Scholar]
- Draber, W.; Tietjen, K.; Kluth, J.F.; Trebst, A. Herbicides in photosynthesis research. Angew. Chem. 1991, 3, 1621–1633. [Google Scholar]
- Tischer, W.; Strotmann, H. Relationship between inhibitor binding by chloroplasts and inhibition of photosynthetic electron transport. Biochim. Biophys. Acta 1977, 460, 113–125. [Google Scholar] [CrossRef]
- Trebst, A.; Draber, W. Structure activity correlations of recent herbicides in photosynthetic reactions. In Advances in Pesticide Science; Greissbuehler, H., Ed.; Pergamon Press: Oxford, UK, 1979; pp. 223–234. [Google Scholar]
- Bowyer, J.R.; Camilleri, P.; Vermaas, W.F.J. Herbicides, Topics in Photosynthesis; Baker, N.R., Percival, M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 10, pp. 27–85. [Google Scholar]
- Kralova, K.; Sersen, F.; Kubicova, L.; Waisser, K. Inhibition of photosynthetic electron transport in spinach chloroplasts by 3-and 4-halogeno substituted benzanilides and thiobenzanilides. J. Trace Microprobe Techn. 2000, 18, 251–256. [Google Scholar]
- Kralova, K.; Sersen, F.; Miletin, M.; Dolezal, M. Inhibitory effects of substituted benzanilides on photosynthetic electron transport in spinach chloroplasts. Chem. Pap. 2002, 56, 214–217. [Google Scholar]
- Dolezal, M.; Miletin, M.; Kunes, J.; Kralova, K. Synthesis and biological evaluation of some amides of pyrazine-2-carboxylic acids. Molecules 2002, 7, 363–373. [Google Scholar] [CrossRef]
- Dolezal, M.; Palek, L.; Vinsova, J.; Buchta, V.; Jampilek, J.; Kralova, K. Substituted pyrazinecarboxamides: synthesis and biological evaluation. Molecules 2006, 11, 242–256. [Google Scholar] [CrossRef]
- Available online: http://www.who.int/tb/publications/global_report/2008/summary/en/index.html/ (21 September 2009).
- Espinal, M.A. The global situation of MDR-TB. Tuberculosis 2003, 83, 44–51. [Google Scholar] [CrossRef]
- Field, S.K.; Cowie, R.L. Lung disease due to the more common nontuberculous mycobacteria. Chest 2006, 129, 1653–1672. [Google Scholar] [CrossRef]
- Wagner, D.; Young, L.S. Nontuberculous mycobacterial infections: A clinical review. Infection 2004, 32, 257–270. [Google Scholar] [CrossRef]
- Morrone, N.; Cruvinel, M.C.; Morrone, N., Jr.; Freire, J.A.; Oliveira, L.M.; Gonçalves, C. Pneumopatia causada por Mycobacterium kansasii. J. Pneumol. 2003, 29, 341–349. [Google Scholar] [CrossRef]
- Available online: http://www.doctorfungus.org/ (21 September 2009).
- Gershon, H.; Gershon, M.; Clarke, D.D. Synergistic mixtures of fungitoxic monochloro- and dichloro-8-quinolinols against five fungi. Mycopathologia 2004, 158, 131–135. [Google Scholar] [CrossRef]
- Dardari, Z.; Lemrani, M.; Bahloul, A.; Sebban, A.; Hassar, M.; Kitane, S.; Berrada, M.; Boudouma, M. Antileishmanial activity of a new 8-hydroxyquinoline derivative designed7-[5′-(3′-phenylisoxazolino)methyl]-8-hydroxyquinoline: preliminary study. Farmaco 2004, 59, 195–199. [Google Scholar] [CrossRef]
- Musiol, R.; Podeszwa, B.; Finster, J.; Niedbala, H.; Polanski, J. An efficient microwave-assisted synthesis of structurally diverse styrylquinolines. Monatsh. Chem. 2006, 137, 1211–1217. [Google Scholar]
- Musiol, R.; Niedbala, H.; Majerz-Maniecka, K.; Oleksyn, B.; Polanski, J. Synthesis and structure of styrylquinolines. Ann. Pol. Chem. Soc. 2005, 1, 118–122. [Google Scholar]
- Majerz-Maniecka, K.; Musiol, R.; Nitek, W.; Oleksyn, B.; Mouscadet, J.F.; Le Bret, M.; Polanski, J. Intermolecular interactions in the crystal structures of potential HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1005–1009. [Google Scholar]
- Kerns, E.H.; Li, D. Drug-like Properties: Concept, Structure Design and Methods; Elsevier: San Diego, CA, USA, 2008. [Google Scholar]
- Avdeef, A. Physicochemical profiling (permeability, solubility, charge state). Curr. Topics Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef]
- Pliska, V. Lipophilicity in drug action and toxicology. In Methods and Principles in Medicinal Chemistry, 1st; Pliska, V., Testa, B., van der Waterbeemd, H., Eds.; Wiley-VCH: Weinheim, DE, 1996; Volume 4, pp. 1–6. [Google Scholar]
- Valko, K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299–310. [Google Scholar]
- Valko, K.; Du, C.M.; Bevan, C.; Reynolds, D.P.; Abraham, M.H. Rapid method for the estimation of octanol/water partition coefficient (log Poct) from gradient RP-HPLC retention and a hydrogen bond acidity term (Σα2H). Curr. Med. Chem. 2001, 8, 1137–1146. [Google Scholar] [CrossRef]
- Cimpan, G.; Irimie, F.; Gocan, S.; Claessens, H.A. Role of stationary phase and eluent composition on the determination of log P values of N-hydroxyethylamide of aryloxyalkylen and pyridine carboxylic acids by reversed-phase high-performance liquid chromatography. J. Chromatogr. B 1998, 714, 247–261. [Google Scholar] [CrossRef]
- Gocan, S.; Cimpan, G.; Comer, J. Lipophilicity measurements by liquid chromatography. Adv. Chromatogr. 2006, 44, 79–176. [Google Scholar]
- Hartmann, T.; Schmitt, J. Lipophilicity – beyond octanol/water: A short comparison of modern technologies. Drug Discov. Today Technol. 2004, 1, 431–439. [Google Scholar] [CrossRef]
- Nasal, A.; Siluk, D.; Kaliszan, R. Chromatographic retention parameters in medicinal chemistry and molecular pharmacology. Curr. Med. Chem. 2003, 10, 381–426. [Google Scholar] [CrossRef]
- Piraprez, G.; Herent, M.F.; Collin, S. Determination of the lipophilicity of aroma compounds by RP-HPLC. Flavour Fragr. J. 1998, 13, 400–408. [Google Scholar] [CrossRef]
- Yamagami, C.; Iwasaki, K.; Ishikawa, A. Hydrophobicity parameters determined by reversed-phase liquid chromatography. XII. Comparison of capacity factors and octane/methanol-water partition coefficients for monosubstituted pyrazines, and effect of octanol added to both partitioning systems. Chem. Pharm. Bull. 1997, 45, 1653–1658. [Google Scholar] [CrossRef]
- Yamagami, C.; Araki, K.; Ohnishi, K.; Hanasato, K.; Inaba, H.; Aono, M.; Ohta, A. Measurement and prediction of hydrophobicity parameters for highly lipophilic compounds: Application of the HPLC column-switching technique to measurement of log P of diarylpyrazines. J. Pharm. Sci. 1999, 88, 1299–1304. [Google Scholar]
- Yamagami, C.; Kawase, K.; Iwaki, K. Hydrophobicity parameters determined by reversed-phase liquid chromatography. XV: Optimal conditions for prediction of log Poct by using RP-HPLC procedures. Chem. Pharm. Bull. 2002, 50, 1578–1583. [Google Scholar]
- Kucerova-Chlupacova, M.; Opletalova, V.; Jampilek, J.; Dolezel, J.; Dohnal, J.; Kunes, J.; Pour, M.; Kunes, J.; Vorisek, V. New hydrophobicity constants of substituents in pyrazine rings derived from RP-HPLC Study. Collect. Czech. Chem. Comm. 2008, 73, 1–18. [Google Scholar]
- Musiol, R.; Jampilek, J.; Podeszwa, B.; Finster, J.; Tabak, D.; Dohnal, J.; Polanski, J. RP-HPLC Determination of drug lipophilicity in series of quinoline derivatives. Cent. Eur. J. Chem. 2009, 7, 586–597. [Google Scholar] [CrossRef]
- Dolezal, M.; Jampilek, J.; Osicka, Z.; Kunes, J.; Buchta, V.; Vichova, P. Substituted 5-aroylpyrazine-2-carboxylic acid derivatives: Synthesis and biological activity. Farmaco 2003, 58, 1105–1111. [Google Scholar] [CrossRef]
- Jampilek, J.; Vinsova, J.; Dohnal, J. Synthesis and hydrophobic properties of benzoxazoles. In Proceedings of the 9th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-9), November 1–30, 2005; Seijas, J.A., Tato, M.P.V., Eds.; MDPI: Basel, Switzerland, 2005; p. a008. [Google Scholar]
- Jampilek, J.; Vinsova, J.; Dohnal, J. Synthesis and hydrophobic properties of substituted 2-aryl-5,7-di-tert-butylbenzoxazoles. In Proceedings of the 10th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-10), November 1–30, 2006; Seijas, J.A., Tato, M.P.V., Eds.; MDPI: Basel, Switzerland, 2006; p. a003. [Google Scholar]
- Vinsova, J.; Cermakova, K.; Tomeckova, A.; Ceckova, M.; Jampilek, J.; Cermak, P.; Kunes, J.; Dolezal, M.; Staud, F. Synthesis and antimicrobial evaluation of new 2-substituted 5,7-di-tert-butylbenzoxazoles. Bioorg. Med. Chem. 2006, 14, 5850–5865. [Google Scholar]
- Norrington, F.E.; Hyde, R.M.; Williams, S.G.; Wotton, R. Physicochemical-activity relations in practice. 1. Rational and self-consistent data bank. J. Med. Chem. 1975, 18, 604–607. [Google Scholar]
- Takahata, Y.; Chong, D.P. Estimation of Hammett sigma constants of substituted benzenes through accurate density-functional calculation of core-electron binding energy shifts. Int. J. Quantum Chem. 2005, 103, 509–515. [Google Scholar] [CrossRef]
- Nielsen, K.E.; Pedersen, E.B. Phosphoramides. XII. Phosphorus pentaoxide – amine hydrochloride as reagents in the synthesis of 4-(3H)-quinazolinones and 4-quinazolinamines. Acta Chem. Scand. Ser. B 1980, 34, 637–642. [Google Scholar]
- Finster, J.; Kalinowski, D.; Musiol, R.; Mrozek, A.; Szurko, A.; Serafin, A.; Kamalapuram, S.K.; Kovacevic, Z.; Jampilek, J.; Ratuszna, A.; Rezeszowska-Wolny, J.; Richardson, D.R.; Polanski, J. Investigating anti-proliferative activity of styrylazanaftalenes and azanaftalenediones. Bioorg. Med. Chem. 2009. submitted. [Google Scholar]
- Kovalenko, S.; Belenichev, I.; Nikitin, V.; Karpenko, A. Search for substances with antioxidant and antiamnestic activities among 2-substituted 4-(3H)-quinazolones. Acta Pol. Pharm. Drug Design 2003, 60, 275–279. [Google Scholar]
- Botros, S.; Shaban, M. Synthesis of some 2-styrylquinazoline derivatives structurally related to certain chemotherapeutic agents. Pharmazie 1978, 33, 646–647. [Google Scholar]
- Masarovicova, E.; Kralova, K. Approaches to measuring plant photosynthesis activity. In Handbook of Photosynthesis, 2nd; Pessarakli, M., Ed.; Taylor & Francis Group: Boca Raton, London-New York-Singapore, 2005; pp. 617–656. [Google Scholar]
- Kralova, K.; Sersen, F.; Sidoova, E. Photosynthesis inhibition produced by 2-alkylthio-6-R-benzothiazoles. Chem. Pap. 1992, 46, 348–350. [Google Scholar]
- Fedke, C. Biochemistry and Physiology of Herbicide Action; Springer Verlag: Berlin-Heidelberg/New York, Germany/US, 1982. [Google Scholar]
- Sheehan, D.J.; Espinel-Ingroff, A.; Steele, M.; Webb, C.D. Antifungal susceptibility testing of yeasts: A brief overview. Clin. Infect. Dis. 1993, 17, 494–500. [Google Scholar]
- National Committee for Clinical Laboratory standards. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast. Approved Standard, NCCLS document M27-A; NCCLS: Villanova, PA, USA, 1997.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jampilek, J.; Musiol, R.; Finster, J.; Pesko, M.; Carroll, J.; Kralova, K.; Vejsova, M.; O'Mahony, J.; Coffey, A.; Dohnal, J.; et al. Investigating Biological Activity Spectrum for Novel Styrylquinazoline Analogues. Molecules 2009, 14, 4246-4265. https://doi.org/10.3390/molecules14104246
Jampilek J, Musiol R, Finster J, Pesko M, Carroll J, Kralova K, Vejsova M, O'Mahony J, Coffey A, Dohnal J, et al. Investigating Biological Activity Spectrum for Novel Styrylquinazoline Analogues. Molecules. 2009; 14(10):4246-4265. https://doi.org/10.3390/molecules14104246
Chicago/Turabian StyleJampilek, Josef, Robert Musiol, Jacek Finster, Matus Pesko, James Carroll, Katarina Kralova, Marcela Vejsova, Jim O'Mahony, Aidan Coffey, Jiri Dohnal, and et al. 2009. "Investigating Biological Activity Spectrum for Novel Styrylquinazoline Analogues" Molecules 14, no. 10: 4246-4265. https://doi.org/10.3390/molecules14104246