Recent Advances in the Discovery of Haem-Targeting Drugs for Malaria and Schistosomiasis

Abstract
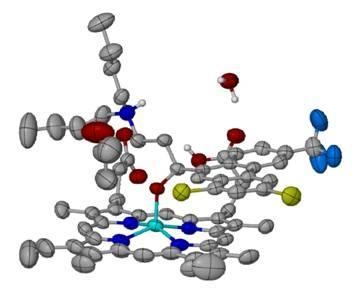
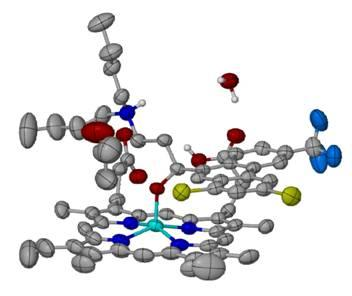
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction
Chemotherapy of Human Malaria and Schistosomiasis

Advances in High-Throughput Screening Methods



Interactions of Existing Antimalarials with Haem


Drug Discovery: Pharmacophore-Based Drug Design



Conclusions
References
- Egan, T.J.; Combrink, J.M.; Egan, J.; Hearne, G.R.; Marques, H.M.; Ntenteni, S.; Sewell, B.T.; Smith, P.J.; Taylor, D.; van Schalkwyk, D.A.; Walden, J.C. Fate of haem iron in the malaria parasite Plasmodium falciparum. Biochem. J. 2002, 365, 343–347. [Google Scholar] [CrossRef]
- Krogstad, D.J.; Schlesinger, P.H.; Gluzman, I.Y. Antimalarials increase vesicle pH in Plasmodium falciparum. J. Cell Biol. 1985, 101, 2302–2309. [Google Scholar] [CrossRef]
- Hayward, R.; Saliba, K.J.; Kirk, K. The pH of the digestive vacuole of Plasmodium falciparum is not associated with chloroquine resistance. J. Cell Sci. 2006, 119, 1016–1025. [Google Scholar] [CrossRef]
- Chou, A.C.; Fitch, C.D. Hemolysis of mouse erythrocytes by ferriprotoporphyrin IX and chlorquine. Chemotherapeutic implications. J. Clin. Invest. 1980, 66, 856–858. [Google Scholar] [CrossRef]
- Sugioka, Y.; Suzuki, M. The chemical basis for the ferriprotoporphyrin IX-chloroquine complex induced lipid peroxidation. Biochim. Biophys. Acta 1991, 1074, 19–24. [Google Scholar] [CrossRef]
- Loria, P.; Miller, S.; Foley, M.; Tilley, L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other antimalarials. Biochem. J. 1999, 339, 363–370. [Google Scholar] [CrossRef]
- Brown, W. Malaria pigment (so-called melanin): its nature and mode of production. J. Exp. Med. 1911, 13, 290–299. [Google Scholar] [CrossRef]
- Atamna, H.; Ginsburg, H. Heme degradation in the presence of glutathione - A proposed mechanism to account for the high levels on non-heme iron in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995, 270, 24876–24883. [Google Scholar] [CrossRef]
- Oliveira, M.F.; d'Avila, J.C.P.; Torres, C.R.; Oliveira, P.L.; Tempone, A.J.; Rumjanek, F.D.; Braga, C.M.S.; Silva, J.R.; Dansa-Petretski, M.; Oliveira, M.A.; De Souza, W.; Ferreira, S.T. Haemozoin in Schistosoma mansoni. Mol. Biochem. Parasitol. 2000, 111, 217–221. [Google Scholar] [CrossRef]
- Oliveira, M.F.; Silva, J.R.; Dansa-Petretski, M.; de Souza, W.; Braga, C.M.S.; Masuda, H.; Oliveira, P.L. Haemozoin formation in the midgut of the blood-sucking insect Rhodnius prolixus. FEBS Lett. 2000, 477, 95–98. [Google Scholar] [CrossRef]
- Oliveira, M.F.; Kycia, S.W.; Gomez, A.; Kosar, A.J.; Bohle, D.S.; Hempelmann, E.; Menezes, D.; Vannier-Santos, M.A.; Oliveira, P.L.; Ferreira, S.T. Structural and morphological characterisation of hemozoin produced by Schistosoma mansoni and Rhodnius prolixus. FEBS Lett. 2005, 579, 6010–6016. [Google Scholar] [CrossRef]
- Pisciotta, J.M.; Coppens, I.; Tripathi, A.K.; Scholl, P.F.; Shuman, J.; Bajad, S.; Shulaev, V.; Sullivan, D.J.J. The role of neutral lipid nanospheres in Plasmodium falciparum heme crystallisation. Biochem. J. 2007, 402, 197–204. [Google Scholar] [CrossRef]
- Egan, T.J.; Chen, J.Y.-J.; de Villiers, K.A.; Mabotha, T.E.; Naidoo, K.J.; Ncokazi, K.K.; Langford, S.J.; McNaughton, D.; Pandiancherri, S.; Wood, B.R. Haemozoin (β-haematin) biomineralisation occurs by self assembly near the lipid/water interface. FEBS Lett. 2006, 580, 5105–5110. [Google Scholar] [CrossRef]
- Sullivan, D.J.J. Theories on malaria pigment formation and quinoline action. Int. J. Parasitol. 2002, 32, 1645–1653. [Google Scholar] [CrossRef]
- Egan, T.J. Haemozoin formation. Mol. Biochem. Parasitol. 2008, 157, 127–136. [Google Scholar] [CrossRef]
- Egan, T.J. Recent advances in understanding the mechanism of hemozoin (malaria pigment) formation. J. Inorg. Biochem. 2008, 102, 1288–1299. [Google Scholar] [CrossRef]
- Stocks, P.A.; Raynes, K.J.; Ward, S.A. Novel quinoline antimalarials. In Antimalarial chemotherapy: Mechanisms of action, resistance and new directions in drug discovery; Rosenthal, P.J., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2001; pp. 235–254. [Google Scholar]
- WHO, Drug alert: Halofantrine. Wkly. Epidemiol. Rec. 1993, 68, 269–270.
- Croft, A. Healthy people need safe drugs too: lessons from Lariam and Halfan. Available online: http://www.haiweb.org/medicineprices/2410/contributions/.
- Neondo, H. Coartem gets FDA approval. Available online: http://africasciencenews.org/asns/.
- Mutabingwa, T.K.; Anthony, D.; Heller, A.; Hallett, R.; Ahmed, J.; Drakeley, C.; Greenwood, B.M.; Whitty, C.J.M. Amodiaquine alone, amodiaquine+sulfadoxine-pyrimethamine, amodiaqu-ine+artesunate, and artemether-lumefantrine for outpatient treatment of malaria in Tanzanian children: a four-arm randomised effectiveness trial. The Lancet 2005, 365, 1474–1480. [Google Scholar] [CrossRef]
- Orjih, A.U. Heme polymerase activity and the stage specificity of antimalarial action of chloroquine. J. Pharmacol. Exp. Ther. 1997, 282, 108–112. [Google Scholar]
- Macomber, P.B.; Sprinz, H.; Tousimis, A.J. Morphological effects of chloroquine on Plasmodium berghei in mice. Nature 1967, 214, 937–939. [Google Scholar] [CrossRef]
- Warhurst, D.C.; Hockley, D.J. Mode of action of chloroquine on Plasmodium berghei and P. cynomolgi. Nature 1967, 214, 935–936. [Google Scholar] [CrossRef]
- Chou, A.C.; Chevli, R.; Fitch, C.D. Ferriprotoporphyrin IX fulfills the criteria for identification as the chloroquine receptor of malaria parasites. Biochemistry 1980, 19, 1543–1549. [Google Scholar] [CrossRef]
- Krugliak, M.; Ginsburg, H. Studies on the antimalarial mode of action of quinoline-containing drugs: time-dependence and irreversibility of drug action, and interactions with compounds that alter the function of the parasite's food vacuole. Life Sci. 1991, 49, 1213–1219. [Google Scholar] [CrossRef]
- Mungthin, M.; Bray, P.G.; Ridley, R.G.; Ward, S.A. Central role of haemoglobin degradation in mechanisms of action of 4-aminoquinolines, quinoline methanols, and phenanthrene methanols. Antimicrob. Agents Chemother. 1998, 42, 2973–2977. [Google Scholar]
- Egan, T.J.; Ross, D.C.; Adams, P.A. Quinoline anti-malarials inhibit spontaneous formation of β-haematin (malaria pigment). FEBS Lett. 1994, 352, 54–57. [Google Scholar] [CrossRef]
- Slater, A.F.G.; Cerami, A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature 1992, 355, 167–169. [Google Scholar] [CrossRef]
- Karle, J.M.; Karle, I.L.; Gerena, L.; Milhous, W.K. Stereochemical evaluation of the relative activities of the cinchona alkaloids against Plasmodium falciparum. Antimicrob. Agents Chemother. 1992, 36, 1538–1544. [Google Scholar] [CrossRef]
- Sullivan, D.J.J.; Gluzman, I.Y.; Russel, D.G.; Goldberg, D.E. On the molecular mechanism of chloroquine's antimalarial action. Proc. Natl. Acad. Sci. USA 1996, 93, 11865–11870. [Google Scholar]
- Dorn, A.; Vippagunta, S.R.; Matile, H.; Jaquet, C.; Vennerstrom, J.L.; Ridley, R.G. An assessment of drug-haematin binding as a mechanism for inhibition of haem polymerisation by quinoline antimalarials. Biochem. Pharmacol. 1998, 55, 727–736. [Google Scholar] [CrossRef]
- Hawley, S.R.; Bray, P.G.; Mungthin, M.; Atkinson, J.D.; O'Neill, P.M.; Ward, S.A. Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerisation in Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 1998, 42, 682–686. [Google Scholar]
- Egan, T.J.; Hempelmann, E.; Mavuso, W.W. Characterisation of synthetic β-haematin and effects of the antimalarial drugs quinidine, halofantrine, desbutylhalofantrine and mefloquine on its formation. J. Inorg. Biochem. 1999, 73, 101–107. [Google Scholar] [CrossRef]
- Mavuso, W.W. Synthetic haemozoin: Characterisation, mechanism of formation from haematin and the effect of antimalarial drugs; University of Cape Town: Cape Town, South Africa, 2001. [Google Scholar]
- Caffrey, C.R. Chemotherapy of schistosomiasis: present and future. Curr. Opin. Chem. Biol. 2007, 11, 433–439. [Google Scholar] [CrossRef]
- Pax, R.; Bennett, J.L.; Fetterer, R. A benzodiazepine derivative and praziquantel: Effects on musculature of Schistosoma mansoni and Schistosoma japonicum. Naunyn-Schmiedeberg's Arch. Pharmacol. 1978, 304, 309–315. [Google Scholar] [CrossRef]
- Pica-Mattoccia, L.; Valle, C.; Basso, A.; Trioani, A.R.; Vigorosi, F.; Liberti, P.; Festucci, A.; Cioli, D. Cytochalasin D abolishes the schistosomicidal activity of praziquantel. Exp. Parasitol. 2007, 115, 344–351. [Google Scholar] [CrossRef]
- Oliveira, M.F.; d'Avila, J.C.P.; Tempone, A.J.; Correa Soares, J.B.R.; Rumjanek, F.D.; Ferreira-Pereira, A.; Ferreira, S.T.; Oliveira, P.L. Inhibition of heme aggregation by chloroquine reduces Schistosoma mansoni infection. J. Infect. Dis. 2004, 190, 843–852. [Google Scholar] [CrossRef]
- Van Nassauw, L.; Toovey, S.; Van Op den bosch, J.; Timmermans, J.-P.; Vercruysse, J. Schistosomicidal activity of the antimalarial drug, mefloquine, in Schistosoma mansoni-infected mice. Travel Med. Infect. Dis. 2008, 6, 253–258. [Google Scholar] [CrossRef]
- Ridley, R.G. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002, 415, 686–693. [Google Scholar] [CrossRef]
- Ginsburg, H. Should chloroquine be laid to rest? Acta Trop. 2005, 96, 16–23. [Google Scholar] [CrossRef]
- Fidock, D.A.; Nomura, T.; Talley, A.K.; Cooper, R.A.; Dzekunov, S.M.; Ferdig, M.T.; Ursos, L.M.B.; Sidhu, A.B.S.; Naude, B.; Deitsch, K.W.; Su, X.-Z.; Wootton, J.C.; Roepe, P.D.; Wellems, T.E. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 2000, 6, 861–871. [Google Scholar] [CrossRef]
- Jiang, H.; Joy, D.A.; Furuya, T.; Su, X.-Z. Current understanding of the molecular basis of chloroquine-resistance in Plasmodium falciparum. J. Postgrad. Med. 2006, 52, 271–276. [Google Scholar]
- Cowman, A.F.; Galatis, D.; Thompson, J.K. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc. Natl. Acad. Sci. USA 1994, 91, 1143–1147. [Google Scholar] [CrossRef]
- Ridley, R.G. Plasmodium: Drug Discovery and Development - An Industrial Perspective. Exp. Parasitol. 1997, 87, 293–304. [Google Scholar] [CrossRef]
- Tekwani, B.L.; Walker, L.A. Targeting the hemozoin synthesis pathway for new antimalarial drug discovery: Technologies for In vitro beta-hematin formation assay. Comb. Chem. High Throughput Screen. 2005, 8, 63–79. [Google Scholar] [CrossRef]
- Carter, M.D.; Sandlin, R.D.; Wright, D.W. A high throughput screen for beta-hematin inhibitors. In 60th Southeast Regional Meeting of the American Chemical Society, Nashville, TN, USA, 12–15 November 2008; American Chemical Society: Washington D.C., USA, 2008. [Google Scholar]
- Jackson, K.E.; Klonis, N.; Ferguson, D.J.P.; Adisa, A.; Dogovski, C.; Tilley, L. Food vacuole-associated lipid bodies and heterogenous lipid environments in the malaria parasite, Plasmodium falciparum. Mol. Microbiol. 2004, 54, 109–122. [Google Scholar] [CrossRef]
- Kurosawa, Y.; Dorn, A.; Kitsuji-Shirane, M.; Shimada, H.; Satoh, T.; Matile, H.; Hofheinz, W.; Masciadri, R.; Kansy, M.; Ridley, R.G. Hematin polymerisation assay as a high-throughput screen for identification of new antimalarial pharmacophores. Antimicrob. Agents Chemother. 2000, 44, 2638–2644. [Google Scholar] [CrossRef]
- Rush, M.A.; Baniecki, M.L.; Mazitschek, R.; Cortese, J.F.; Wiegand, R.; ClardyJon; Wirth, D.F. Colorimetric high-throughput screen for detection of heme crystallisation inhibitors. Antimicrob. Agents Chemother. 2009, 53, 2564–2568. [Google Scholar] [CrossRef]
- Acharya, B.N.; Kaushik, M.P. Pharmacophore-based predictive model generation for potential antimalarials targeting haem detoxification pathway. Med. Chem. Res. 2007, 16, 213–229. [Google Scholar] [CrossRef]
- Li, H.; Sutter, J.; Hoffmann, R. HypoGen: An automated system for generating 3D pharmacophore models. In Pharmacophore Perception, Development and Use in Drug Design; Guner, O.F., Ed.; International University Line: La Jolla, CA, USA, 2000; pp. 171–189. [Google Scholar]
- Dorn, A.; Vippagunta, S.R.; Matile, H.; Bubendorf, A.; Vennerstrom, J.L.; Ridley, R.G. A comparison and analysis of several ways to promote haematin (haem) polymerisation and an assessment of its initiation in vitro. Biochem. Pharmacol. 1998, 55, 737–747. [Google Scholar] [CrossRef]
- Ncokazi, K.K.; Egan, T.J. A colorimetric high-throughput β-haematin inhibition screening assay for use in the search for antimalarial compounds. Anal. Biochem. 2005, 338, 306–319. [Google Scholar] [CrossRef]
- Dorn, A.; Scovill, J.P.; Ellis, W.Y.; Matile, H.; Ridley, R.G.; Vennerstrom, J.L. Short report: Floxacrine analog WR 243251 inhibits hematin polymerisation. Am. J. Trop. Med. Hyg. 2001, 65, 19–20. [Google Scholar]
- Acharya, B.N.; Saraswat, D.; Kaushik, M.P. Pharmacophore based discovery of potential antimalarial agent targeting haem detoxification pathway. Eur. J. Med. Chem. 2008, 43, 2840–2852. [Google Scholar] [CrossRef]
- Egan, T.J. Interactions of quinoline antimalarials with hematin in solution. J. Inorg. Biochem. 2006, 100, 916–926. [Google Scholar] [CrossRef]
- Warhurst, D.C. The quinine-haemin interaction and its relationship to antimalarial activity. Biochem. Pharmacol. 1981, 30, 3323–3327. [Google Scholar] [CrossRef]
- Behere, D.V.; Goff, H.M. High-affinity binding of quinine to iron(III) porphyrins: novel formation of alkoxide complexes from alcohols and amines. J. Am. Chem. Soc. 1984, 106, 4945–4950. [Google Scholar] [CrossRef]
- Marques, H.M.; Voster, K.; Egan, T.J. The interaction of the heme-octapeptide, N-acetylmicroperoxidase-8 with antimalarial drugs: Solution studies and modeling by molecular mechanics methods. J. Inorg. Biochem. 1996, 64, 7–23. [Google Scholar] [CrossRef]
- Egan, T.J.; Mavuso, W.W.; Ross, D.C.; Marques, H.M. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J. Inorg. Biochem. 1997, 68, 137–145. [Google Scholar] [CrossRef]
- de Villiers, K.A.; Marques, H.M.; Egan, T.J. The crystal structure of halofantrine-ferriprotoporphyrin IX and the mechanism of action of arylmethanol antimalarials. J. Inorg. Biochem. 2008, 102, 1660–1667. [Google Scholar] [CrossRef]
- Pisciotta, J.M.; Sullivan, D.J.J. Haemozoin: Oil versus water. Parasitol. Int. 2008, 57, 89–96. [Google Scholar] [CrossRef]
- Buller, R.; Peterson, M.L.; Almarsson, Ö.; Leiserowitz, L. Quinoline binding site on malaria pigment crystal: A rational pathway for antimalarial drug design. Cryst. Growth Des. 2002, 2, 553–562. [Google Scholar] [CrossRef]
- Chong, C.R.; Sullivan, D.J.J. Inhibition of heme crystal growth by antimalarials and other compounds: implications for drug discovery. Biochem. Pharmacol. 2003, 66, 2201–2212. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P.W.; Bohle, D.S.; Kosar, A.D.; Madsen, S.K. The structure of malaria pigment (β-haematin). Nature 2000, 404, 307–310. [Google Scholar] [CrossRef]
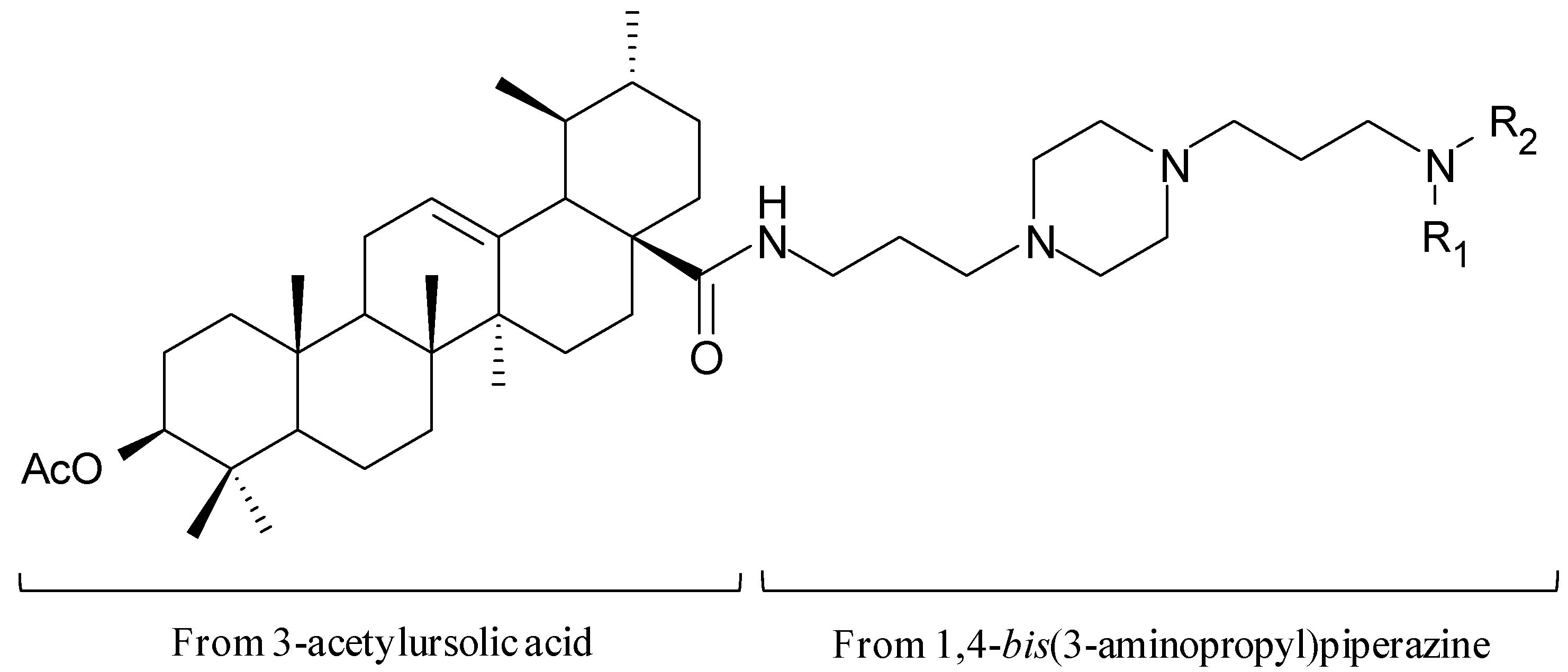
- Gnoatto, S.C.B.; Susplugas, S.; Vechia, L.D.; Ferreira, T.B.; Dassonville-Klimpt, A.; Zimmer, K.R.; Demailly, C.; Da Nascimento, S.; Guillon, J.; Grellier, P.; Verli, H.; Gosmann, G.; Sonnet, P. Pharmacomodulation on the 3-acetylursolic acid skeleton: design, synthesis, and biological evaluation of novel N-{3-[4-(3-aminopropyl)piperazinyl]propyl}-3-O-acetylursolamide derivati-ves as antimalarial agents. Bioorg. Med. Chem. 2008, 16, 771–782. [Google Scholar] [CrossRef]
- Pathak, A.; Singh, S.K.; Farooq Biabani, M.A.; Kulshreshtha, D.K.; Puri, S.K.; Srivastava, S.; Kundu, B. Synthesis of combinatorial libraries based on terpenoid scaffolds. Comb. Chem. High Throughput Screen. 2002, 5, 241–248. [Google Scholar] [CrossRef]
- Guillon, J.; Grellier, P.; Labaied, M.; Sonnet, P.; Lger, J.-M.; Dprez-Poulain, R.; Forfar-Bares, I.; Dallemagne, P.; Lematre, N.; Phourcq, F.; Rochette, J.; Sergheraert, C.; Jarry, C. Synthesis, antimalarial activity, and molecular modeling of new pyrrolo[1,2-a]quinoxalines, bispyrrolo[1,2-a]quinoxalines, bispyrido[3,2-e]pyrrolo[1,2-a]pyrazines, and bispyrrolo[1,2-a]thieno[3,2-e]pyrazines. J. Med. Chem. 2004, 47, 1997–2009. [Google Scholar] [CrossRef]
- Muller, S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2004, 53, 1291–1305. [Google Scholar] [CrossRef]
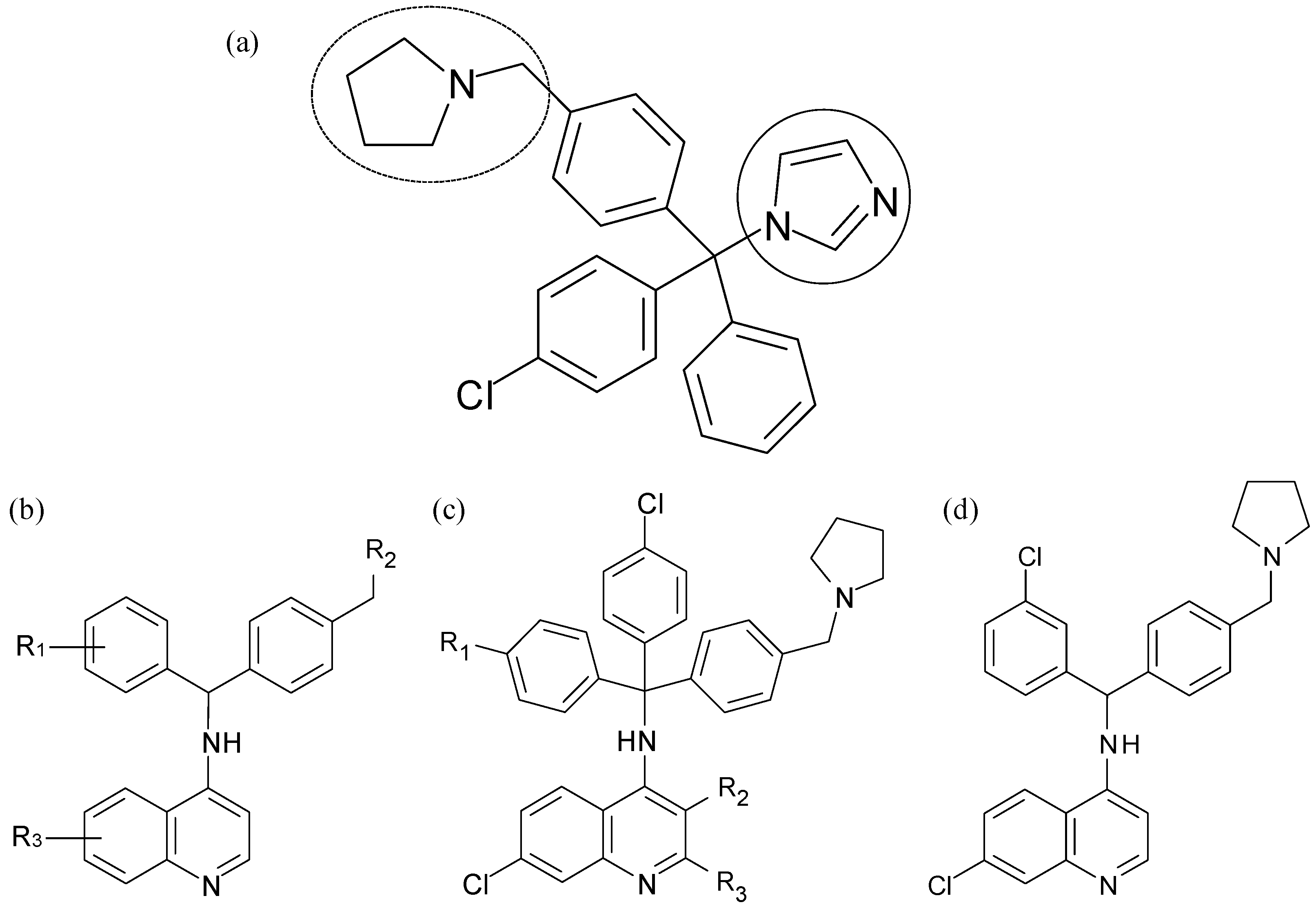
- Gemma, S.; Campiani, G.; Butini, S.; Joshi, B.P.; Kukreja, G.; Sanna Coccone, S.; Bernetti, M.; Persico, M.; Nacci, V.; Fiorini, I.; Novellino, E.; Taramelli, D.; Basilico, N.; Parapini, S.; Yardley, V.; Croft, S.; Keller-Maerki, S.; Rottmann, M.; Brun, R.; Coletta, M.; Marini, S.; Guiso, G.; Caccia, S.; Fattorusso, C. Combining 4-aminoquinoline- and clotrimazole-based pharmacophores toward innovative and potent hybrid antimalarials. J. Med. Chem. 2009, 52, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Gemma, S.; Campiani, G.; Butini, S.; Kukreja, G.; Sanna Coccone, S.; Joshi, B.P.; Persico, M.; Nacci, V.; Fiorini, I.; Novellino, E.; Fattorusso, E.; Tagliatatela-Scafati, O.; Savini, L.; Taramelli, D.; Basilico, N.; Parapini, S.; Morace, G.; Yardley, V.; Croft, S.; Coletta, M.; Marini, S.; Fattorusso, C. Clotrimazole scaffold as an innovative pharmacophore towards potent antimalarial agents. Design, synthesis, biological and structure-activity relationship studies. J. Med. Chem. 2008, 51, 1278–1294. [Google Scholar] [CrossRef] [Green Version]
- Riscoe, M.K.; Kelly, J.X.; Winter, R. Xanthones as antimalarial agents: Discovery, mode of action, and optimization. Curr. Med. Chem. 2005, 12, 2539–2549. [Google Scholar] [CrossRef]
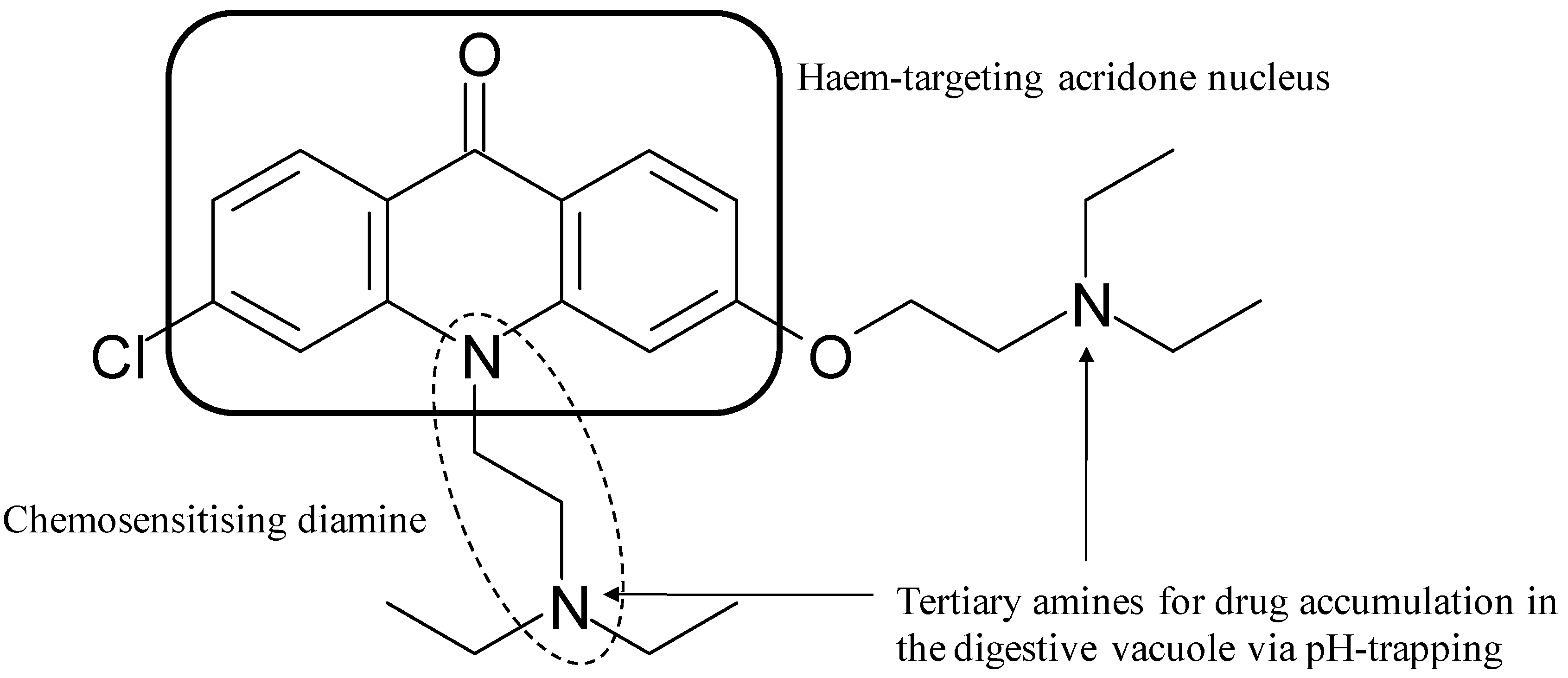
- Winter, R.; Kelly, J.X.; Smilkstein, M.J.; Dodean, R.A.; Bagby, G.C.; Rathbun, R.K.; Levin, J.I.; Hinrichs, D.J.; Riscoe, M.K. Evaluation and lead optimisation of anti-malarial acridones. Exp. Parasitol. 2006, 114, 47–56. [Google Scholar] [CrossRef]
- Kelly, J.X.; Smilkstein, M.J.; Cooper, R.A.; Lane, K.D.; Johnson, R.A.; Janowsky, A.; Dodean, R.A.; Hinrichs, D.J.; Winter, R.; Riscoe, M.K. Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitisers in Plasmodium falciparum. Antimicrob. Agents Chemother. 2007, 51, 4133–4140. [Google Scholar] [CrossRef]
- Kelly, J.X.; Smilkstein, M.J.; Brun, R.; Wittlin, S.; Cooper, R.A.; Lane, K.D.; Janowsky, A.; Johnson, R.A.; Dodean, R.A.; Winter, R.; Hinrichs, D.J.; Riscoe, M.K. Discovery of dual function acridones as a new antimalarial chemotype. Nature 2009, 459, 270–273. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Kyle, D.E.; Vennerstrom, J.L.; Milhous, W.K. A 3D QSAR pharmacophore model and quantum chemical structure-activity analysis of chloroquine (CQ)-resistance reversal. J. Chem. Inf. Comput. Sci. 2002, 42, 1212–1220. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Villiers, K.A.; Egan, T.J. Recent Advances in the Discovery of Haem-Targeting Drugs for Malaria and Schistosomiasis. Molecules 2009, 14, 2868-2887. https://doi.org/10.3390/molecules14082868
De Villiers KA, Egan TJ. Recent Advances in the Discovery of Haem-Targeting Drugs for Malaria and Schistosomiasis. Molecules. 2009; 14(8):2868-2887. https://doi.org/10.3390/molecules14082868
Chicago/Turabian StyleDe Villiers, Katherine A., and Timothy J. Egan. 2009. "Recent Advances in the Discovery of Haem-Targeting Drugs for Malaria and Schistosomiasis" Molecules 14, no. 8: 2868-2887. https://doi.org/10.3390/molecules14082868