Synthesis and Antiviral Activity of 5‑(4‑Chlorophenyl)-1,3,4-Thiadiazole Sulfonamides

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemistry



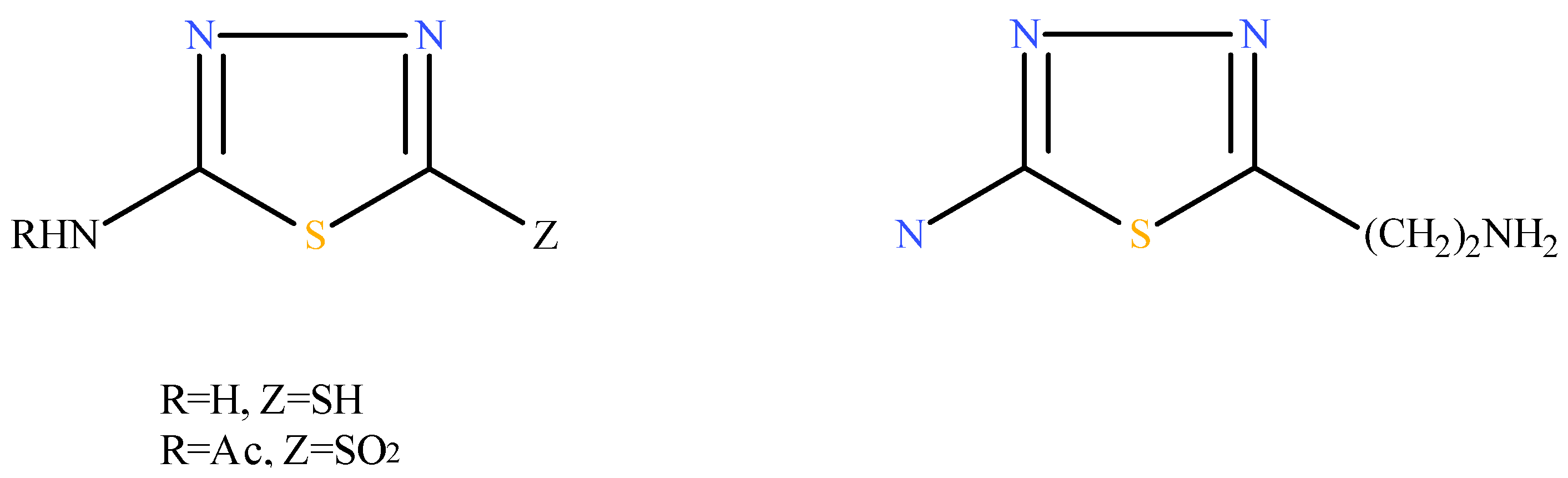{kind=link}
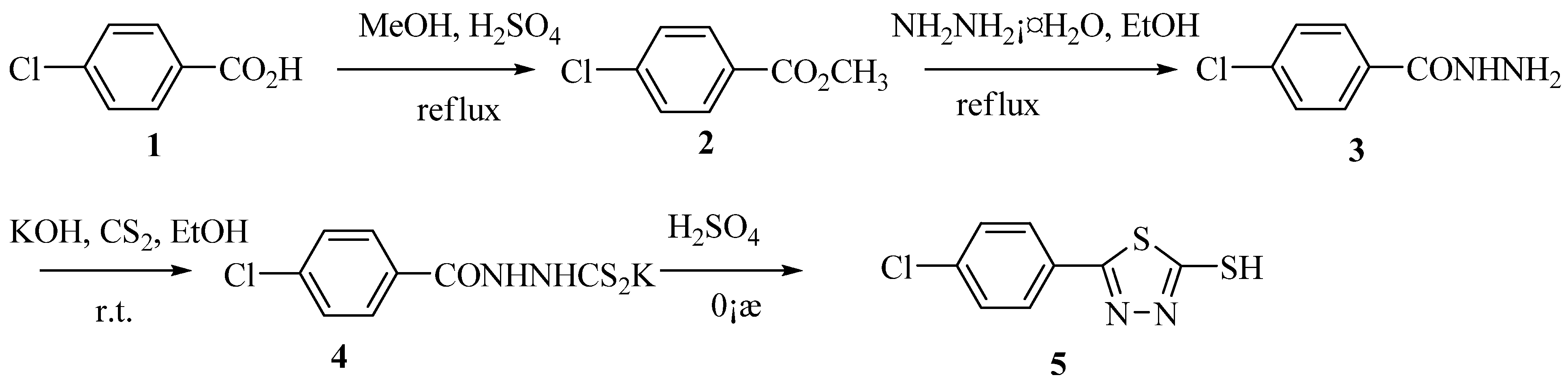{kind=link}
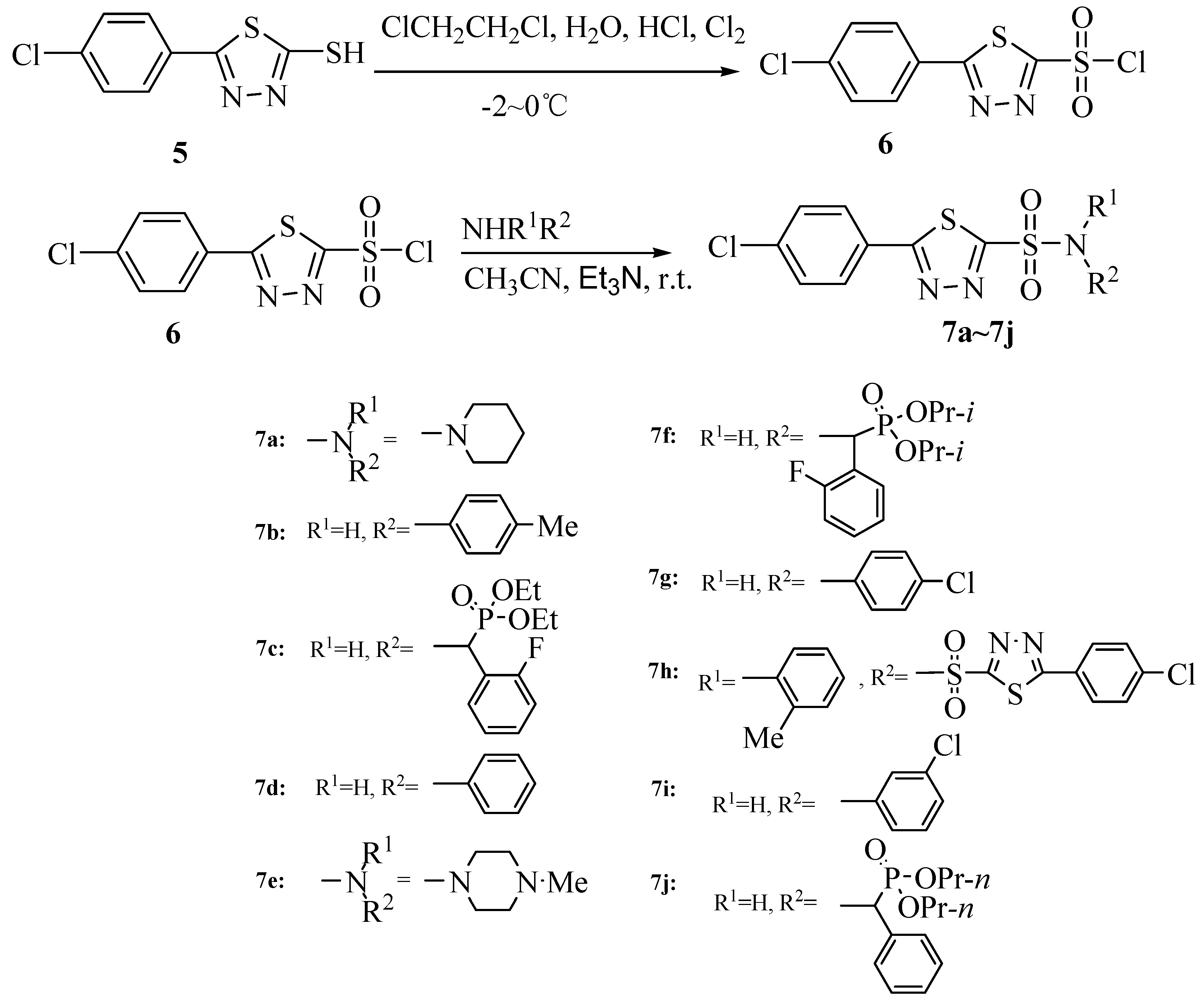{kind=link}
| Entry | Solvent | Time (h) | t (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | Acetonitrile | 5 | 25 | 32.8 |
| 2 | Dichloromethane | 5 | 25 | 17.5 |
| 3 | Acetone | 5 | 25 | trace |
| Entry | Solvent | Acid acceptor | t | Time | Yield |
|---|---|---|---|---|---|
| (V) | (V) | (°C) | (h) | (%) | |
| 1 | Acetonitrile | Triethylamine | 10 | 5 | 19.2 |
| (15 mL) | (0.15 mL) | ||||
| 2 | Acetonitrile | Triethylamine | r. t. | 5 | 48.0 |
| (15 mL) | (0.15 mL) | ||||
| 3 | Acetonitrile | Triethylamine | 40 | 5 | 41.0 |
| (15 mL) | (0.15 mL) |
2.2. Antiviral activity bioassay
| Compd | Concentration (mg/mL ) | Inhibition rate(%) |
|---|---|---|
| 7a | 0.5 | 38.42 |
| 7b | 0.5 | 42.00 |
| 7c | 0.5 | 31.55 |
| 7d | 0.5 | 34.70 |
| 7e | 0.5 | 25.00 |
| 7f | 0.5 | 39.60 |
| 7g | 0.5 | 31.60 |
| 7h | 0.5 | 36.80 |
| 7i | 0.5 | 42.49 |
| 7j | 0.5 | 33.92 |
| Ningnanmycin | 0.5 | 54.51 |
3. Experimental
3.1. General
3.2. Preparation of methyl 4-chlorobenzoate (2)
3.3. Preparation of 4-chlorobenzohydrazide (3)
3.4. Potassium N, p-chlorobenzoylhydrazinodithio formate (4)
3.5. 5-(4--Chlorophenyl)-1,3,4-thiadiazole-2-thiol (5)
3.6. 5-(4-Chlorophenyl)-1,3,4-thiadiazole-2-sulfonyl chloride (6)
3.7. 1-(5-(4-Chlorophenyl)-1,3,4-thiadiazol-2-ylsulfonyl)piperidine (7a)
3.8. Antiviral biological assay


4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Ali, A.; Reddy, G.S.K.K.; Cao, H.; Anjum, S.G.; Nalam, M.N. L.; Schiffer, C.A.; Rana, T.M. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl oxazolidinone-5-carboxamides as novel p2 ligands. J. Med. Chem. 2006, 49, 7342–7356. [Google Scholar]
- McCarroll, A.J.; Bradshaw, T.D.; Westwell, A.D.; Matthews, C.S.; Stevens, M.F.G. Quinols as novel therapeutic agents. 7. Synthesis of antitumor 4-[1-(arylsulfonyl-H-indol-2-yl)]-4-hydroxy-cyclohexa-2,5-dien-1-ones by Sonogashira reactions. J. Med. Chem. 2007, 50, 1707–1710. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Bomaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Carbonic anhydrase inhibitors: Inhibition of isozymes I, II and IX with triazle-linked O-glycosides of benzene sulfonamides. J. Med. Chem. 2007, 50, 1651–1657. [Google Scholar]
- Liane, S.-U.; Maykel, P.G.; Isidro, G.C.; Rosario H.-G. Quantitative structure–activity relationship studies for the prediction of antifungal activity of N-arylbenzenesulfonamides against Botrytis cinerea. J. Mol. Graph. Mod. 2007, 25, 680–690. [Google Scholar] [CrossRef]
- Zhu, W.J.; Wu, P.; Liang, X.M.; Dong, Y.-H.; Zhang, J.-J.; Yuan, H.-Z.; Qi, S.-H.; Meng, X.-Q.; Wu, J.-P.; Chen, F.-H.; Wang, D.-Q. Design, synthesis, and fungicidal activity of macrolactones and macrolactams with a sulfonamide side chain. J. Agric. Food Chem. 2008, 56, 6547–6553. [Google Scholar]
- Ezabadi, I.R.; Canoutsis, C.; Zoumpoulakis, P.; Geronikaki, A.; Sokovic, M.; Glamocilija, J.; Ciric, A. Sulfonamide-1,2,4-triazole derivatives as antifungal and antibacterial agents: Synthesis, biological evaluation, lipophilicity, and conformational studies. Bioorg. Med. Chem. 2008, 16, 1150–1161. [Google Scholar]
- Gonzalez, M.A.; Gorman, D.B.; Hamilton, C.T.; Roth, G.A. Process development for the sulfonamide herbicide pyroxsulm. Org. Process. Res. Dev. 2008, 12, 301–303. [Google Scholar] [CrossRef]
- Sharna, R.; Misra, G.P.; Sainy, J.; Chaturvedi, S.C. Synthesis and biological evaluation of 2-amino-5-sulfanyl-1,3,4-thiadiazole derivatives as antidepressant, anxiolytics and anticonvulsant agents. Med. Chem. Res. 2010. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.F.; Abdel-Aziz, H.A.; Ahmed, E.M. Synthesis and antimicrobial elevation of some 1,3-thiazole, 1,3,4-thiadiazole, 1,2,4-triazole, and 1,2,4-triazolo [3,4-b][1,3,4]-thiadiazine derivatives including a 5-(benzofuran-2-yl)-1-phenyl-pyrazole moity. Monatsh. Chem. 2009, 140, 601–605. [Google Scholar] [CrossRef]
- Aly, A.A.; El-Sayed, R. Synthesis and biological activity of new 1,3,4-thiadiazole derivatives. Chem. Pap. 2006, 60, 56–60. [Google Scholar] [CrossRef]
- Jatav, V.; Kashaw, S.; Mishra, P. Synthesis, antibacterial and antifugal activity of some novel 3-[5-(4-substituted phenyl)]1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Med. Chem. Res. 2008, 17, 169–181. [Google Scholar] [CrossRef]
- Song, B.-A.; Chen, C.-J.; Yang, S.; Jin, L.-H.; Xue, W.; Zhang, S.-M.; Zou, Z.-H.; Hu, D.-Y.; Liu, G. Synthesis, structure and antitumor activity of 2-alkylthio-5-(3,4,5-trimethoxyphenyl)-1,3,4-thiadiazole compounds. Acta Chim. Sinica 2005, 63, 1720–1726. [Google Scholar]
- Song, B.A.; Yang, S.; Jin, L.H.; Bhadury, P.S. Environment Friendly Anti Plant Viral Agents, 1st ed; Chemical industry press: Beijing, China; Springer press: Heidelberg, Germany, 2010. [Google Scholar]
- Gooding, G.V., Jr.; Hebert, T.T. A simple technique for purification of tabacco mosaic virus in large quantities. Phytopathology 1967, 57, 1285–1290. [Google Scholar]
- Song, B.A.; Zhang, H.P.; Wang, H.; Yang, S.; Jin, L.H.; Hu, D.Y.; Pang, L.L.; Xue, W. Synthesis and antiviral activity of novel chiral cyanoacrylate derivatives. J. Agric. Food Chem. 2005, 53, 7886–7891. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, Z.; Xu, W.; Liu, K.; Yang, S.; Fan, H.; Bhadury, P.S.; Huang, D.-Y.; Zhang, Y. Synthesis and Antiviral Activity of 5‑(4‑Chlorophenyl)-1,3,4-Thiadiazole Sulfonamides. Molecules 2010, 15, 9046-9056. https://doi.org/10.3390/molecules15129046
Chen Z, Xu W, Liu K, Yang S, Fan H, Bhadury PS, Huang D-Y, Zhang Y. Synthesis and Antiviral Activity of 5‑(4‑Chlorophenyl)-1,3,4-Thiadiazole Sulfonamides. Molecules. 2010; 15(12):9046-9056. https://doi.org/10.3390/molecules15129046
Chicago/Turabian StyleChen, Zhuo, Weiming Xu, Keming Liu, Song Yang, Huitao Fan, Pinaki S. Bhadury, De-Yu Huang, and Yuping Zhang. 2010. "Synthesis and Antiviral Activity of 5‑(4‑Chlorophenyl)-1,3,4-Thiadiazole Sulfonamides" Molecules 15, no. 12: 9046-9056. https://doi.org/10.3390/molecules15129046