Unexplored Nucleophilic Ring Opening of Aziridines †



Abstract
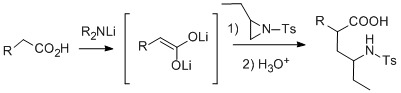
:
Introduction
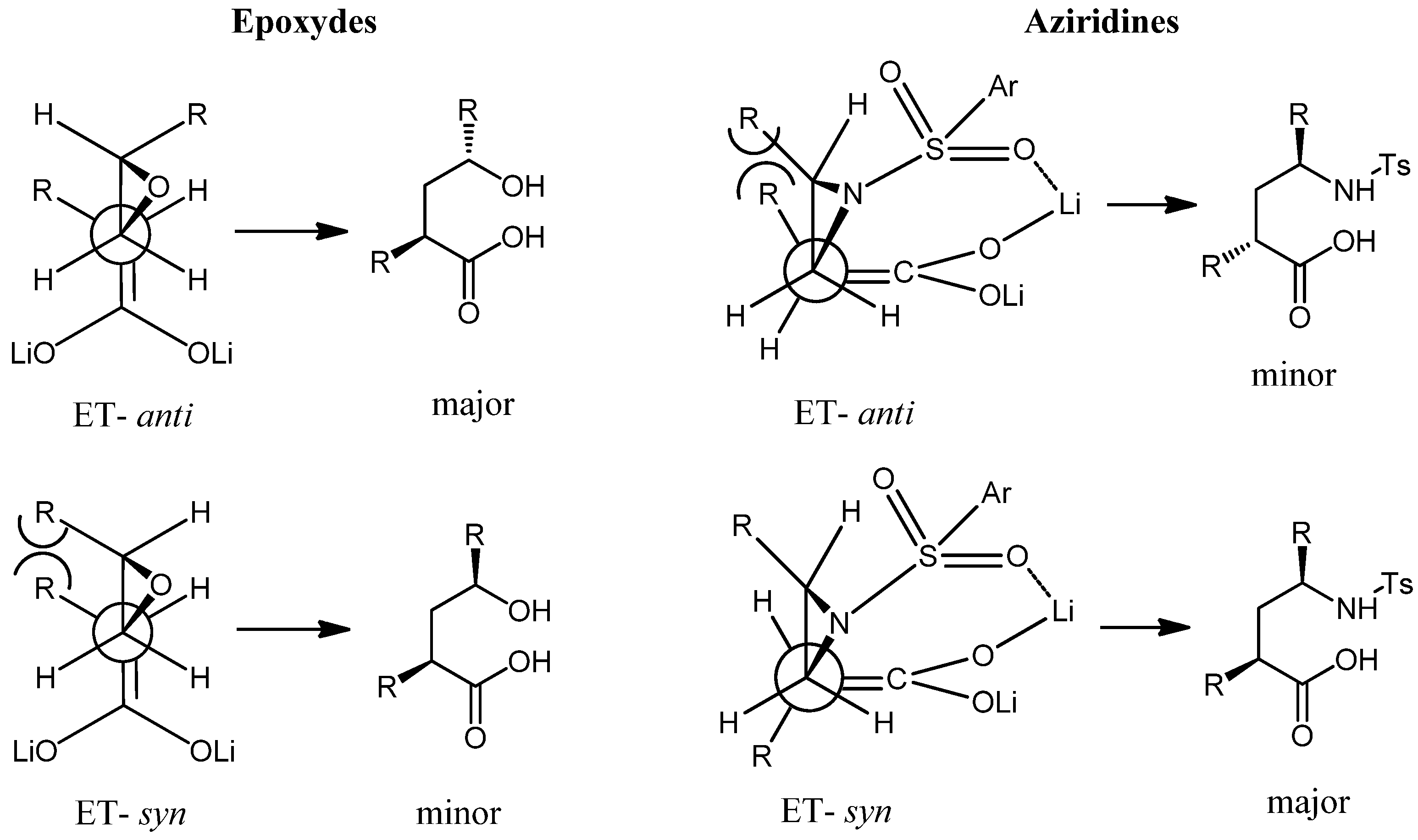
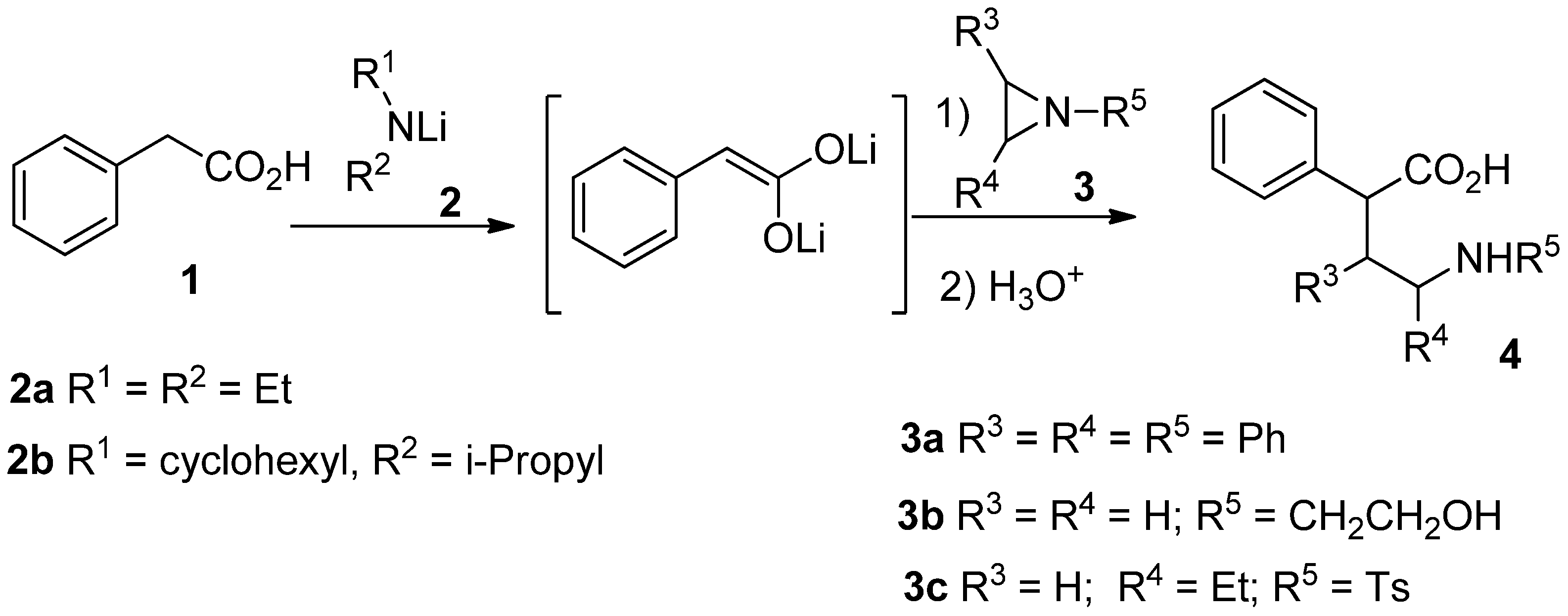
Results and Discussion
Conclusions
Experimental
General
General procedure for the reaction of lithium enediolates with N-tosylaziridine
Acknowledgements
References and Notes
- Lowden, P.A.S. Aziridines and Epoxides in Organic Synthesis; Yudin, A.K., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 399–442. [Google Scholar]
- Padwa, A.; Woolhouse, A.D. Comprehensive Heterocyclic Chemistry III; Lwowski, W., Ed.; Pergamon: Oxford, UK, 1984; pp. 47–93. [Google Scholar]
- Hu, X.E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar] [CrossRef]
- Pineschi, M. Asymmetric Ring-Opening of Epoxides and Aziridines with Carbon Nucleophiles. Eur. J. Org. Chem. 2006, 4979–4988. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Hu, Y.; Wang, D.-X.; Pan, J.; Huang, Z.-T.; Wang, M.-X. Unprecedented carbon-carbon bond cleavage in nucleophilic aziridine ring opening reaction, efficient ring transformation of aziridines to imidazolidin-4-ones. Chem. Commun. 2009, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Pandey, S.K.; Gandhi, S.; Singh, V.K. PPh3/halogenating agent-mediated highly efficient ring opening of activated and non-activated aziridines. Tetrahedron Lett. 2009, 50, 263–265. [Google Scholar] [CrossRef]
- Tarrade-Matha, A.; Valle, M.S.; Tercinier, P.; Dauban, P.; Dodd, R.H. Enantiospecific Synthesis of a Protected Equivalent of APTO, the beta-Amino Acid Fragment of Microsclerodermins C and D, by Aziridino-gamma-lactone Methodology. Eur. J. Org. Chem. 2009, 673–686. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M.; Rodriguez, P. A simple synthesis of γ-aminoacids. Tetrahedron Lett. 2007, 48, 3451–3453. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M.; Rodriguez, P. An efficient synthesis of γ-aminoacids. Attempts to drive it enantioselectively. Molecules 2008, 13, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.M. Dianion Chemistry in Organic Synthesis; CRC Press: Boca Raton, FL, USA, 1994; pp. 88–129. [Google Scholar]
- Gil, S.; Parra, M. Dienediolates of carboxylic acids in synthesis. Recent advances. Curr. Org. Chem. 2002, 6, 283–302. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M. Reactivity control of dianions of carboxylic acids. Synthetic applications. Recent Res. Devel. Org. Chem. 2002, 6, 449–481. [Google Scholar] [CrossRef]
- Clayden, I. Organolithiums: Selectivity for Synthesis; Pergamon Press: Oxford, UK, 2002; p. 73. [Google Scholar]
- Juaristi, E.; Beck, A.K.; Hansen, J.; Matt, T.; Mukhopadhyay, T.; Simson, M.; Seebach, D. Enantioselective Aldol and Michael Additions of Achiral Enolates in the presence of chiral lithium amides and amines. Synthesis 1993, 1271–1290. [Google Scholar] [CrossRef]
- Streitwieser, A.; Husemann, M.; Kim, Y.-J. Aggregation and reactivity of the dilithium and dicesium N-diolates of 1-naphthyl acetic acid. J. Org. Chem. 2003, 68, 7937–7942. [Google Scholar] [CrossRef] [PubMed]
- Eames, J.; Suggate, M.J. Recent developments in the transfer of chirality within enolate alkylation reactions. Angew. Chem. Int. Ed. 2005, 44, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Sott, R.; Granader, J.; Hilmersson, G. Solvent-dependent mixed complex formation–NMR studies and asymmetric addition reactions of lithiumacetonitrile to benzaldehyde mediated by chiral lithium amides. Chem. Eur. J. 2002, 8, 2081–2087. [Google Scholar] [CrossRef]
- Sotoca, E.; Bouillon, J.P.; Gil, S.; Parra, M.; Portella, C. Reaction of lithium enediolates with perfluoroketene dithioacetals. Synthesis of α-trifluoromethyl γ-dicarboxylic acid derivatives. Tetrahedron 2005, 61, 4395–4402. [Google Scholar] [CrossRef]
- Brun, E.M.; Casades, I.; Gil, S.; Mestres, R.; Parra, M. New conditions for the generation of dianions of carboxylic acids. Tetrahedron Lett. 1998, 39, 5443–5446. [Google Scholar] [CrossRef]
- Bieber, L.W.; de Araújo, M.C.F. Short and Efficient Synthesis of Optically Active N-Tosyl Aziridines from 2-Amino Alcohols. Molecules 2002, 7, 902–906. [Google Scholar] [CrossRef]
- Gil, S.; Torres, M.; Ortúzar, N.; Wincewicz, R.; Parra, M. Eficient addition of acid enediolates to epoxides. Eur. J. Org. Chem. 2004, 2160–2165. [Google Scholar] [CrossRef]
- Domingo, L.R.; Gil, S.; Parra, M.; Segura, J. Unusual Regioselectivity in the Opening of Epoxides by Carboxylic Acid Enediolates. Molecules 2008, 13, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.K. Reactions of epoxides with ester, ketone and amide enolates. Tetrahedron 2000, 56, 1149–1163. [Google Scholar] [CrossRef]
- Brun, E.M.; Gil, S.; Mestres, R.; Parra, M. Regioselective Alkylation of Lithium Dienediolates of α,β-unsaturated Carboxylic Acids. Synthesis-Stuttgart 2000, 1160–1165. [Google Scholar] [CrossRef]
- Brun, E.M.; Gil, S.; Mestres, R.; Parra, M. Lithium enediolates and dienediolates of carboxylic acids in synthesis: Alkylation with secondary halides. Tetrahedron 1998, 54, 15305–15320. [Google Scholar] [CrossRef]
- Aurell, M.J.; Gil, S.; Mestres, R.; Parra, M.; Parra, L. Alkylation of lithium dienediolates of butenoic acids. Regioselectivity effects of structure and leaving group of the alkylating agent. Tetrahedron 1998, 54, 4357–4366. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4, 15, 16, 17, 20 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Entry | Aziridine | Amine | Time at r.t. | Yield (%) |
|---|---|---|---|---|
| 1 | 3a | 2a | 1h | 0 |
| 2 | 3b | 2a | 1h | 0 |
| 3 | 3c | 2a | 1h | 50 |
| 4 | 3c | 2a | 3h | 44 |
| 5 | 3c | 2a | 24h | 35 |
| 6 | 3c | 2a* | 1h | 46 |
| 7 | 3c | 2b | 1h | 67 |
| 8 | 3c | 2b | 1h | 58** |
| 9 | 3c | 2b | 1h | 71** |
| Entry | Acid | Product | Yield (%) | γ : α | Syn : Anti |
|---|---|---|---|---|---|
| 1 |  |  | 31 | 66 : 33 | |
| 2 |  |  | 67 | 64 :36 | |
| 3 |  |  | 59 | 67 : 33 | |
| 4 |  |  | 68 | 100 : 0 | |
| 5 |  |  | 60 | 41 : 59 | 69 : 31 |
| 6 |  |  | 25 | 50 : 50 | |
| 7 |  |  | 40 | ||
| 8 |  |  | 53 | ||
| 9 |  |  | 60 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Costero, A.M.; Gil, S.; Parra, M.; Rodríguez, P. Unexplored Nucleophilic Ring Opening of Aziridines. Molecules 2010, 15, 9135-9144. https://doi.org/10.3390/molecules15129135
Costero AM, Gil S, Parra M, Rodríguez P. Unexplored Nucleophilic Ring Opening of Aziridines. Molecules. 2010; 15(12):9135-9144. https://doi.org/10.3390/molecules15129135
Chicago/Turabian StyleCostero, Ana María, Salvador Gil, Margarita Parra, and Pablo Rodríguez. 2010. "Unexplored Nucleophilic Ring Opening of Aziridines" Molecules 15, no. 12: 9135-9144. https://doi.org/10.3390/molecules15129135
APA StyleCostero, A. M., Gil, S., Parra, M., & Rodríguez, P. (2010). Unexplored Nucleophilic Ring Opening of Aziridines. Molecules, 15(12), 9135-9144. https://doi.org/10.3390/molecules15129135